To investigate whether CHEK2 variants confer susceptibility to breast cancer, Schutte et al. (2003) screened the full CHEK2 coding sequence in BRCA1/BRCA2-negative breast cancer cases from 89 pedigrees with 3 or more cases of breast cancer.
"Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis" . Mayo Clinic Proceedings . 89 (4): 484–92. doi : 10.1016/j.mayocp.2013.12.012 .
Hemophagocytic syndrome (HPS) is a rare immune disease (see this term) and a potentially life-threatening disorder characterized by cytokine storm and overwhelming inflammation causing fever, hepatosplenomegaly, cytopenia, hypertriglyceridemia, hyperferritinemia, and hemophagocytosis in bone marrow, liver, spleen or lymph nodes. It can be either primary due to a genetic defect (primary hemophagocytic lymphohistiocytosis ; see this term), or secondary to malignancies, to infections, most commonly with viruses such as Epstein-Barr virus or cytomegalovirus, human immunodeficiency virus, or to autoimmune disorders such as systemic lupus erythematosus or adult-onset Still disease (secondary hemophagocytic lymphohistiocytosis) (see these termes).
New York: McGraw Hill. pp. Chapter 87. ISBN 978-0-07-182852-9 . ^ Gaynes, Robert; Edwards, Jonathan R.; National Nosocomial Infections Surveillance System (2005-09-15).
Some hypotheses include the existence of significantly lower reproductive fitness in people with less melanin due to lethal skin cancer, lethal kidney disease due to excess vitamin D formation in the skin of people with less melanin, or simply natural selection due to mate preference and sexual selection. [17] When comparing the prevalence of albinism in Africa to its prevalence in other parts of the world, such as Europe and the United States, the potential evolutionary effects of skin cancer as a selective force due to its effect on these populations may not be insignificant. [17] It would follow, then, that there would be stronger selective forces acting on albino individuals in Africa than on albinoes in Europe and the US. [18] In two separate studies in Nigeria, very few people with albinism appear to survive to old age. One study found that 89% of people diagnosed with albinism are between 0 and 30 years of age, while the other found that 77% of albinos were under the age of 20. [18] Diagnosis [ edit ] This section needs additional citations for verification .
A number sign (#) is used with this entry because the disorder is caused by duplication or triplication of the gene encoding methyl-CpG-binding protein-2 (MECP2; 300005) on chromosome Xq28. The same gene is mutated in Rett syndrome (RTT; 312750). X-linked mental retardation with spasticity (300055) is an allelic disorder. Description MECP2 duplication syndrome is an X-linked neurodevelopmental disorder characterized by severe to profound mental retardation, infantile hypotonia, mild dysmorphic features, poor speech development, autistic features, seizures, progressive spasticity, and recurrent infections. Only males are affected, although female carriers may have some mild neuropsychiatric features, such as anxiety. Submicroscopic Xq28 duplications encompassing MECP2 are considered nonrecurrent events, because the breakpoint locations and rearrangement sizes vary among affected individuals (summary by Ramocki et al., 2010).
MECP2 duplication syndrome Other names X-linked intellectual disability-hypotonia-recurrent Infections syndrome This condition is due to MECP2 overexpression Specialty Medical genetics MECP2 duplication syndrome ( M2DS ) is a rare disease that is characterized by severe intellectual disability and impaired motor function. It is an X-linked genetic disorder caused by the overexpression of MeCP2 protein. Contents 1 Signs and symptoms 2 Cause 3 Diagnosis 4 Management 5 Epidemiology 6 History 7 References 8 Further reading 9 External links Signs and symptoms [ edit ] Symptoms of M2DS include infantile hypotonia and failure to thrive, delayed psychomotor development , impaired speech, abnormal or absent gait , epilepsy , spasticity , gastrointestinal motility problems, recurrent infections, and genitourinary abnormalities. [1] [2] [3] Many of those affected by M2DS also fit diagnostic criteria for autism . [4] M2DS can be associated with syndromic facies , namely an abnormally flat back of the head, underdevelopment of the midface, ear anomalies, deep-set eyes, prominent chin, pointed nose, and a flat nasal bridge. [4] Cause [ edit ] M2DS is one of the several types of X-linked intellectual disability . The cause of M2DS is a duplication of the MECP2 or Methyl CpG binding protein 2 gene located on the X chromosome (Xq28). [5] The MeCP2 protein plays a pivotal role in regulating brain function. Increased levels of MECP2 protein results in abnormal neural function and impaired immune system. [4] Mutations in the MECP2 gene are also commonly associated with Rett syndrome in females.
MECP2 duplication syndrome is a severe neurological and developmental disorder. Signs and symptoms include low muscle tone (hypotonia) in infancy, developmental delay, severe intellectual disability, and progressive spasticity. Other signs and symptoms may include recurrent respiratory infections and seizures. Some people with MECP2 duplication syndrome may have autistic features, gastrointestinal problems, and/or mildly distinctive facial features. The syndrome is caused by having an extra copy (duplication) of the MECP2 gene, and inheritance is X-linked.
MECP2 duplication syndrome is a condition that occurs almost exclusively in males and is characterized by moderate to severe intellectual disability. Most people with this condition also have weak muscle tone in infancy, feeding difficulties, poor or absent speech, or muscle stiffness (rigidity). Individuals with MECP2 duplication syndrome have delayed development of motor skills such as sitting and walking. About half of individuals have seizures, often of the tonic-clonic type. This type of seizure involves a loss of consciousness, muscle rigidity, and convulsions and may not respond to medication.
A number sign (#) is used with this entry because this disorder is caused by copy number increase of a small region on distal chromosome Xq28. One report has identified a 0.3-Mb region of Xq28 (chrX:153.2-153.5 Mb, NCBI36) containing at least 11 genes and including the GDI1 gene (300104), which is mutated in MRX41 (300849). Two additional reports have identified a more distal 0.5-Mb region (chrX:153.7-154.2 Mb (NCBI36/hg19)) containing at least 8 genes and including the RAB39B gene (300774), which is mutated in MRX72 (300271). Clinical Features Vandewalle et al. (2009) reported 4 families with X-linked mental retardation. The first family was of Belgian origin and had 4 affected males in 2 generations.
Distal Xq duplications refer to chromosomal disorders resulting from involvement of the long arm of the X chromosome (Xq). Clinical manifestations vary widely depending on the gender of the patient and on the gene content of the duplicated segment. The prevalence of Xq duplications remains unknown. Epidemiology About 40 cases of Xq28 functional disomy due to cytogenetically visible rearrangements, and about 50 cases of cryptic duplications encompassing the MECP2 gene have been reported (intellectual deficit-progressive spasticity, X-linked; see this term). Clinical description The most frequently reported distal duplications involve the Xq28 segment and yield a recognizable phenotype including distinctive facial features (premature closure of the fontanels or a ridged metopic suture, a broad face with full cheeks, epicanthal folds, large ears, a small and open mouth, ear anomalies, a pointed nose, an abnormal palate and facial hypotonia), major axial hypotonia, severe developmental delay, severe feeding difficulties, abnormal genitalia and susceptibility to infections. Etiology Xq duplications may be caused either by an intrachromosomal duplication or by an unbalanced X/Y or X/autosome translocation.
The data informing this article came from three sources: Analysis of emails reporting cases of FCKS sent to the owner of a website giving information about the condition and methods of treatment, 1999-2019 [87 of these kittens were examined in person by the author] Data gathered by the THINK project 2005-2015 who published an online questionnaire (the project is now defunct) Directly solicited reports requested from 27 individual breeders of Abyssinian (4 responded) and 35 breeders of British Shorthair (2 responded) in the UK The 20-year analysis was compared with the Governing Council of the Cat Fancy in the UK (GCCF) analysis of numbers of kittens registered during the same period. ... Journal of the American Veterinary Medical Association 195: 91–97 Hosgood, G., Hoskins J. D. (1998) Small Animal Paediatric Medicine and Surgery.
Animals are confined to houses, apartments, or trailer-homes. [34] In one case, 306 cats were removed from a home, 87 of which were dead. Corpses were found embedded in the chimney and living room furniture. [3] In addition to lack of living space, overcrowding facilitates the spread of diseases among animals. [35] Furthermore, in cases where more than one species is confined to the same living space, animals can pose a danger to one other due to inter-species aggression. [36] Owner neglect [ edit ] Various other health problems arise from hoarders' neglect of the animals and inability to provide basic care for them. ... Retrieved 8 April 2014 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag Hayes, Victoria (2010). "Detailed Discussion of Animal Hoarding" . ... American Journal of Public Health. 97 (2007-09): 1671. ^ a b c d e f g Frost, Randy (2000).
The Canadian Journal of Cardiology . 30 (1): 87–95. doi : 10.1016/j.cjca.2013.11.008 . ... Journal of the American College of Cardiology . 58 (5): e9. doi : 10.1016/j.jacc.2010.11.077 . ... "Ebstein's anomaly: a complex congenital heart defect". AORN Journal . 89 (6): 1098–110, quiz 1111–4. doi : 10.1016/j.aorn.2009.03.003 . ... "Preoperative secundum atrial septal defect with coexisting sinus node and atrioventricular node dysfunction" . Circulation . 65 (5): 976–80. doi : 10.1161/01.CIR.65.5.976 . PMID 7074763 . ^ a b c "UOTW #54 - Ultrasound of the Week" .
A number sign (#) is used with this entry because of evidence that atrial septal defect-6 (ASD6) is caused by heterozygous mutation in the TLL1 gene (606742) on chromosome 4q32. For a general phenotypic description and discussion of genetic heterogeneity in atrial septal defect, see ASD1 (108800). Molecular Genetics Based on data from mouse models of incomplete heart septation associated with inactivation of mouse Tll1, Stanczak et al. (2009) analyzed the candidate gene TLL1 in 19 unrelated patients with atrial septal defect and identified heterozygosity for 3 missense mutations in 3 patients (606742.0001-606742.0003, respectively). One of the patients had an isolated ostium primum defect; the other 2 patients, who had ostium secundum defects, displayed additional features including interatrial aneurysm and cardiac arrhythmias. INHERITANCE - Autosomal dominant CARDIOVASCULAR Heart - Atrial septal defect, type I or II - Aneurysm of interatrial septum (in some patients) - Atrial fibrillation (in some patients) - Bradycardia (in some patients) MOLECULAR BASIS - Caused by mutation in the tolloid-like 1 gene (TLL1, 606742.0001 ) ▲ Close
A number sign (#) is used with this entry because of evidence that atrial septal defect-5 (ASD5) is caused by heterozygous mutation in the ACTC1 gene (102540) on chromosome 15q14. For a phenotypic description and discussion of genetic heterogeneity in atrial septal defect, see ASD1 (108800). Mapping Matsson et al. (2008) studied 2 large Swedish families segregating autosomal dominant isolated secundum atrial septal defect (ASD) with variable clinical expression. Genotyping with microsatellite markers in 'family 1' revealed a specific haplotype in all affected individuals spanning a 15.1-cM region of chromosome 15q13-q21; analysis of 'family 2' identified a minimal haplotype with significant linkage to ASD consisting of markers GT44248, GATA12322, and ACTC. All affected individuals genotyped had identical allele sizes for the marker haplotype, suggesting a shared ancestral mutation for the 2 families.
A number sign (#) is used with this entry because atrial septal defect-2 (ASD2) is caused by heterozygous mutation in the GATA4 gene (600576) on chromosome 8p23. For discussion of genetic heterogeneity in atrial septal defect, see ASD1 (108800). Clinical Features Garg et al. (2003) identified a large kindred spanning 5 generations in which 16 individuals had congenital heart defects. Detailed clinical evaluations reviewed for all available family members demonstrated an autosomal dominant pattern of inheritance. All affected family members had atrial septal defects. Eight individuals had additional congenital heart defects, including ventricular septal defects (VSD), atrioventricular septal defects (AVSD), pulmonary valve thickening, or insufficiency of the cardiac valves.
Description Secundum atrial septal defect (ASD) is a common congenital heart malformation that occurs as an isolated anomaly in 10% of individuals with congenital heart disease. Uncorrected ASD can cause pulmonary overcirculation, right heart volume overload, and premature death (summary by Benson et al., 1998). Genetic Heterogeneity of Atrial Septal Defect The ASD1 locus has been mapped to chromosome 5p. Other forms of atrial septal defect that are associated with other congenital heart disease but no conduction defects or noncardiac abnormalities include ASD2 (607941), caused by mutation in the GATA4 gene (600576), and ASD4 (611363), caused by mutation in the TBX20 gene (606061). ASD3 (614089) and ASD5 (612794), in which atrial septal defect is not associated with other cardiac abnormalities, are caused by mutation in the MYH6 (160710) and ACTC1 (102540) genes, respectively.
A congenital cardiac malformation characterized by a communication between the atrial chambers of the heart. Epidemiology Overall, openings between the atrial chambers account for about 6 to 8% of all congenitally malformed hearts. As a group, the prevalence at birth is between 6 to 9/10000; however, this is likely an underestimated due to the asymptomatic nature of the disease. The female-to-male ratio is 2-4:1. Clinical description There are four types of defects, named according to their position relative to the atrial septum. The ostium secundum defect is the most common type, accounting for three-quarters of all cases, located to the region of the oval fossa, most commonly due to a deficiency of the primary atrial septum (septum primum) but deficiency of the septum secundum (superior interatrial fold) may also contribute.
A number sign (#) is used with this entry because of evidence that atrial septal defect-8 (ASD8) can be caused by heterozygous mutation in the CITED2 gene (602937) on chromosome 6q23.3. For discussion of genetic heterogeneity of atrial septal defect, see ASD1 (108800). Molecular Genetics Sperling et al. (2005) screened a cohort of 392 patients with congenital heart defects and 192 controls for mutations in the CITED2 gene and identified a 27-bp insertion (602937.0002) in a patient with a secundum atrial septal defect, and a 6-bp deletion (602937.0003) in a patient with a sinus venosus atrial septal defect and abnormal pulmonary venous return to the right atria. Functional analysis of the mutations, which were not found in controls, revealed that both significantly reduced the capacity of CITED2 to transrepress HIF1A (603348). INHERITANCE - Autosomal dominant CARDIOVASCULAR Heart - Atrial septal defect, secundum type (in some patients) - Atrial septal defect, sinus venosus type (in some patients) Vascular - Abnormal pulmonary venous return to right atria (in some patients) MOLECULAR BASIS - Caused by mutation in the CBP/p300-interacting transactivator, with glu/asp-rich C-terminal domain, 2 gene (CITED2, 602937.0001 ) ▲ Close
Overview An atrial septal defect (ASD) is a hole in the heart between the upper chambers (atria). The hole increases the amount of blood that flows through the lungs. The condition is present at birth (congenital heart defect). Small atrial septal defects might be found by chance and never cause a concern. Others close during infancy or early childhood. A large, long-term atrial septal defect can damage the heart and lungs. Surgery may be needed to repair an atrial septal defect and to prevent complications.
A number sign (#) is used with this entry because of evidence that atrial septal defect-9 (ASD9) is caused by heterozygous mutation in the GATA6 gene (601656) on chromosome 18q11. For discussion of genetic heterogeneity of atrial septal defect (ASD), see ASD1 (108800). Molecular Genetics Lin et al. (2010) analyzed the GATA6 gene in 270 unrelated Chinese patients with congenital heart defects and identified heterozygosity for a missense mutation in the GATA6 gene (S184N; 601656.0005) in 2 Chinese children with atrial septal defect. One of the ASD patients was a 3-year-old girl with an ostium secundum ASD and mild pulmonary arterial hypertension, whereas the other was a 4-year-old boy with an ostium secundum ASD and mild tricuspid valve disease and pulmonary valve replacement. The S184N mutation was detected in the unaffected father of the girl as well as in the clinically unaffected mother of the boy; the latter parent was found to have bicuspid aortic valve on echocardiography.
The male proband of the second family presented with arrhythmias at 31 years of age and developed slowly progressive weakness of the proximal leg muscles starting at age 52. Overall, 13 (87%) of 15 presumably affected family members presented with cardiac problems between the ages of 25 and 60 years, consisting mainly of cardiac conduction defects.
Desmin-related myofibrillar myopathy Specialty Rheumatology Desmin-related myofibrillar myopathy , also called Helmer’s myopathy, is a subgroup of the myofibrillar myopathy diseases and is the result of a mutation in the gene that codes for desmin which prevents it from forming protein filaments , instead forming aggregates of desmin and other proteins throughout the cell. [1] [2] Contents 1 Presentation 2 Genetics 3 Pathophysiology 4 Diagnosis 5 Treatment 6 Prognosis 7 References 8 External links Presentation [ edit ] Common symptoms of the disease are weakness and atrophy in the distal muscles of the lower limbs which progresses to the hands and arms, then to the trunk, neck and face. Respiratory impairment often follows. Genetics [ edit ] There are three major types of inheritance for this disease: Autosomal dominant , autosomal recessive and de novo. The most severe form is autosomal recessive and it also has the earliest onset. [3] It usually involves all three muscle tissues and leads to cardiac and respiratory failure as well as intestinal obstruction. [3] Autosomal Dominant inheritance shows a later onset and slower progression. It usually involves only one or two of the muscle tissues. [3] De novo diseases occur when a new mutation arises in the person that was not inherited through either parent. This form has a wide range of symptoms and varies depending on the mutation made. [3] Pathophysiology [ edit ] The sarcomeres become misaligned and result in the disorganization of muscle fibers. [1] This mutation also results in muscle cell death by apoptosis and necrosis. [1] The muscle cell may also be disorganized because the aggregates may interrupt other filament structures and/or normal cellular function. [3] Desminopathies are very rare diseases and As of 2004 [update] only 60 patients have been diagnosed, however this number probably does not accurately represent the population due to frequent mis or under diagnosis. [3] Diagnosis [ edit ] Desminopathies are diagnosed by genetic analysis.
A rare genetic skeletal muscle disease characterized by abnormal chimeric aggregates of desmin and other cytoskeletal proteins and granulofilamentous material at the ultrastructural level in muscle biopsies and variable clinical myopathological features, age of disease onset and rate of disease progression. Patients present with bilateral skeletal muscle weakness that starts in distal leg muscles and spreads proximally, sometimes involving trunk, neck flexors and facial muscles and often cardiomyopathy manifested by conduction blocks, arrhythmias, chronic heart failure, and sometimes tachyarrhythmia. Weakness eventually leads to wheelchair dependence. Respiratory insufficiency can be a major cause of disability and death, beginning with nocturnal hypoventilation with oxygen desaturation and progressing to daytime respiratory failure.
Myofibrillar myopathy is part of a group of disorders called muscular dystrophies that affect muscle function and cause weakness. Myofibrillar myopathy primarily affects skeletal muscles, which are muscles that the body uses for movement. In some cases, the heart (cardiac) muscle is also affected. The signs and symptoms of myofibrillar myopathy vary widely among affected individuals, typically depending on the condition's genetic cause. Most people with this disorder begin to develop muscle weakness (myopathy) in mid-adulthood. However, features of this condition can appear anytime between infancy and late adulthood.
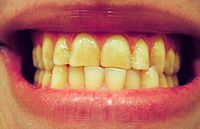
Awake bruxism is thought to be usually semivoluntary, and often associated with stress caused by family responsibilities or work pressures. [5] Some suggest that in children, bruxism may occasionally represent a response to earache or teething. [20] Awake bruxism usually involves clenching [5] (sometimes the term "awake clenching" is used instead of awake bruxism), [21] but also possibly grinding, [4] and is often associated with other semivoluntary oral habits such as cheek biting, nail biting , chewing on a pen or pencil absent mindedly, or tongue thrusting (where the tongue is pushed against the front teeth forcefully). [4] There is evidence that sleep bruxism is caused by mechanisms related to the central nervous system , involving sleep arousal and neurotransmitter abnormalities. [1] Underlying these factors may be psychosocial factors including daytime stress which is disrupting peaceful sleep. [1] Sleep bruxism is mainly characterized by "rhythmic masticatory muscle activity" (RMMA) at a frequency of about once per second, and also with occasional tooth grinding. [22] It has been shown that the majority (86%) of sleep bruxism episodes occur during periods of sleep arousal. [22] One study reported that sleep arousals which were experimentally induced with sensory stimulation in sleeping bruxists triggered episodes of sleep bruxism. [23] Sleep arousals are a sudden change in the depth of the sleep stage, and may also be accompanied by increased heart rate, respiratory changes and muscular activity, such as leg movements. [5] Initial reports have suggested that episodes of sleep bruxism may be accompanied by gastroesophageal reflux, decreased esophageal pH (acidity), swallowing, [23] and decreased salivary flow. [10] Another report suggested a link between episodes of sleep bruxism and a supine sleeping position (lying face up). [23] Disturbance of the dopaminergic system in the central nervous system has also been suggested to be involved in the etiology of bruxism. [10] Evidence for this comes from observations of the modifying effect of medications which alter dopamine release on bruxing activity, such as levodopa, amphetamines or nicotine. ... E.g. several studies use self-reported bruxism as a measure of bruxism, and since many people with bruxism are not aware of their habit, self-reported tooth grinding and clenching habits may be a poor measure of the true prevalence. [5] The ICSD-R states that 85–90% of the general population grind their teeth to a degree at some point during their life, although only 5% will develop a clinical condition. [27] Some studies have reported that awake bruxism affects females more commonly than males, [5] while in sleep bruxism, males and females are affected equally. [27] [26] Children are reported to brux as commonly as adults. ... Behavioral approaches in research declined from over 60% of publications in the period 1966–86 to about 10% in the period 1997–2007. [6] In the 1960s, a periodontist named Sigurd Peder Ramfjord championed the theory that occlusal factors were responsible for bruxism. [57] Generations of dentists were educated by this ideology in the prominent textbook on occlusion of the time, however therapy centered around removal of occlusal interference remained unsatisfactory.
Overview Bruxism (BRUK-siz-um) is a condition in which you grind, gnash or clench your teeth. If you have bruxism, you may unconsciously clench your teeth when you're awake (awake bruxism) or clench or grind them during sleep (sleep bruxism). Sleep bruxism is considered a sleep-related movement disorder. People who clench or grind their teeth (brux) during sleep are more likely to have other sleep disorders, such as snoring and pauses in breathing (sleep apnea). Mild bruxism may not require treatment. However, in some people, bruxism can be frequent and severe enough to lead to jaw disorders, headaches, damaged teeth and other problems. Because you may have sleep bruxism and be unaware of it until complications develop, it's important to know the signs and symptoms of bruxism and to seek regular dental care.
Mold grows best in warm temperatures, 77 to 86 °F (25 to 30 °C), although growth may occur between 32 and 95 °F (0 and 35 °C). [ citation needed ] Removing one of the three requirements for mold reduces (or eliminates) new mold growth: moisture; food for the mold spores (for example, dust or dander ); and warmth since mold generally does not grow in cold environments.
The SATB2-associated syndrome (SAS) is a recently described condition, characterized by developmental delay, intellectual disability with absent or limited language skills, palatal and dental abnormalities, behavioral problems, and unusual facial features. The main symptoms can be remembered using the acronym S.A.T.B.2 ( S , S evere speech anomalies; A , A bnormalities of the palate; T , T eeth anomalies; B , B ehavioral issues with or without B one or B rain anomalies, and age of onset before 2 years of age). Other features may include osteopenia and Rett-like problems . The SATB 2 gene is located in chromosome 2q32 (the region designated as q32 on the long ("q") arm of chromosome 2), and many of the features are similar to the " 2q33.1 microdeletion syndrome ". This gene is important for the development of the face, brain and bone. Alterations to the SATB 2 gene can result from different mechanisms, such as contiguous deletions (missing pieces of the chromosome 2 that include the SATB2 gene and other genes that are close together) , duplications (extra pieces of genetic material) translocations (rearrangements involving the gene), or point mutations (a mutation that only affects a single nucleotide of the DNA) .
"The Physiological Basis of Decompression" . 38th Undersea and Hyperbaric Medical Society Workshop . 75(Phys)6–1–89: 437 . Retrieved 15 May 2010 . ^ Brubakk, Alf O; Neuman. ... Journal of Applied Physiology . 104 (4): 986–90. CiteSeerX 10.1.1.528.4523 . doi : 10.1152/japplphysiol.00641.2007 . ... "Pulmonary Oedema of Immersion". Sports Medicine . 35 (3): 183–90. doi : 10.2165/00007256-200535030-00001 . ... US Navy Experimental Diving Unit Technical Report . NEDU-1-90 . Retrieved 11 June 2008 . ^ Stinton, R.