Clinical Features In a male and female sib pair of Sikh origin and a male offspring of first-cousin Pakistani Muslims, Al-Gazali et al. (1988) described a combination of Hirschsprung disease (megacolon), hypoplastic nails, and minor dysmorphic facial features.
Hirschsprung disease-nail hypoplasia-dysmorphism syndrome is a fatal malformative disorder that is characterized by Hirschsprung disease, hypoplastic nails, distal limb hypoplasia and minor craniofacial dysmorphic features (flat facies, upward slanting palpebral fissures, narrow philtrum, narrow, high arched palate, micrognathia, low set ears with abnormal helices). Hydronephrosis has also been reported. There have been no further descriptions in the literature since 1988.
The 3 males were in 2 sibships, the offspring of 2 brothers married to 2 sisters to whom they were related as first cousins. Inheritance Consanguinity and occurrence in sibs suggest autosomal recessive inheritance of this disorder (Tiemann et al., 2005).
Clinical Features Longman et al. (2003) reported 3 sibs, offspring of phenotypically normal first-cousin Pakistani parents, who were variably affected by craniosynostosis, calcification of the basal ganglia, and mild facial dysmorphism comprising prominent eyes and a prominent nasal bridge.
Craniosynostosis-intracranial calcifications syndrome is a form of syndromic craniosynostosis characterized by pancraniosynostosis, head circumference below the mid-parental head circumference, mild facial dysmorphism (prominent supraorbital ridges, mild proptosis and maxillary hypoplasia) and calcification of the basal ganglia. The disease is associated with a favorable neurological outcome, normal intelligence and is inherited in an autosomal recessive manner.
Other findings included fibular hypoplasia, epiphyseal changes at multiple sites, and characteristic changes of the spine and pelvis. The parents were first cousins, suggesting autosomal recessive inheritance.
The son had a painless ulcer of the foot and osteomyelitis in bones of the feet. The mother was a first cousin of her husband and the husband had a sister with the same combination of cervical hypertrichosis and neuropathy.
In the offspring of Jewish first cousins, Currie (1970) described 4 sibs (3 brothers and a sister) out of 5 who suffered recurrent episodes of Bell palsy and external ophthalmoplegia.
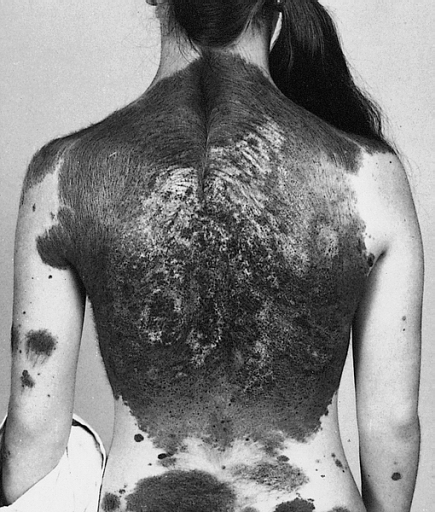
Labrune et al. (1992) reported the case of a boy, born of first-cousin Algerian parents, who had progressive vitiligo from the age of 12 years and retarded psychomotor development.
Although scratching is known to cause more nodules to appear, it is unclear what causes the itching to develop in the first place. Diagnosis of the disease is based on observing signs such as extremely itchy skin with the formation of nodules.
Azathioprine , also known by its brand name Imuran, is an immunosuppressive drug used in organ transplantation and autoimmune diseases and belongs to the chemical class of purine analogues. [ citation needed ] History [ edit ] Prurigo nodularis was first described by Hyde and Montgomery in 1909. [14] See also [ edit ] Pruritus Skin lesion Notes [ edit ] ^ Lockshin BN, Brogan B, Billings S, Billings S (2006).
Congenital hypertrophy of the lateral fold of the hallux Specialty Dermatology Congenital hypertrophy of the lateral fold of the hallux is a rare cutaneous condition of unknown pathology that present to newborns. The condition was "first described by Martinet et al. in 1984."
Clinical description Blistering occurs at first around nails, accompanied by nail dystrophy and shedding, and then affects the hands and feet and, to a lesser extent, the elbows, knees, along with atrophic scarring.
It is believed to be caused by heat-induced vascular disruption near the fourth week of embryonic development . The first known case was reported in the early 18th century by a member of the prominent De Jussieu family in France and cases to this day remain rare. [1] References [ edit ] ^ Gupta, SR (2012).
Tornwaldt's disease Specialty ENT surgery Tornwaldt's disease is the inflammation or abscess of the embryonic cyst of pharyngeal bursa . It is located in the midline of the posterior wall of the nasopharynx . It is covered anteriorly by mucosa in the adenoid mass. It is bounded posteriorly by longus muscle . [1] Contents 1 Signs and symptoms 2 Cause 3 Pathogenesis 4 Diagnosis 5 Treatment 6 History 7 See also 8 References 9 External links Signs and symptoms [ edit ] The symptoms usually appear when there is inflammation of pharyngeal bursa causing Tornwaldt's cyst. This is caused by spontaneous drainage in the nasopharyngeal cavity or can also be caused because of involvement of nervous plexus. The symptoms are occipital headache, cough, middle ear effusion, cervical myalgia, halitosis ie bad breath.
Propriospinal myoclonus (PSM) is a rare movement disorder first described in 1991. It is characterized by painless, repetitive jerking of the trunk, neck, hips, and knees.
Neurological features typically present in the first or second year. Intracranial hypertension is the most common presentation, along with seizures, decreased alertness, and cranial nerve dysfunction.The underlying cause, while not completely understood, is believed to be a primary defect in the neural crest.
None had neurologic complications at the time of referral. The mean age at first MRI was 2.46 years. MRI abnormalities were found in 22 (18%) of patients and were significantly associated with projected adult size of the CMN, particularly greater than 40 cm. ... Kinsler et al. (2013) studied tissue samples from 15 unrelated patients with congenital melanocytic nevi who ranged in age from 2 to 23 years. Routine brain imaging in the first year of life showed 7 patients with parenchymal neuromelanosis, 1 with frontal lobe meningioma, and 5 with normal findings. ... INHERITANCE - Somatic mutation SKIN, NAILS, & HAIR Skin - Numerous congenital melanocytic nevi - Giant pigmented nevi, often in lumbosacral region NEUROLOGIC Central Nervous System - Parenchymal neuromelanosis - Dandy-Walker malformation (in some patients) - Delayed development (in some patients) - Seizures (in some patients) - Hydrocephalus (in some patients) - Leptomeningeal melanocytosis (in some patients) - Choroid plexus papilloma (in some patients) - Meningioma (in some patients) - Spinal cysts (in some patients) - Arachnoid cysts (in some patients) - Syringomyelia (in some patients) NEOPLASIA - Susceptibility to malignant melanoma MISCELLANEOUS - Onset in first years of life - Some patient may be asymptomatic MOLECULAR BASIS - Caused by somatic mutation in the NRAS proto-oncogene gene, GTPase gene (NRAS, 164790.0002 ) ▲ Close
The median survival time for these patients is 6.5 months after becoming symptomatic. [8] History [ edit ] Neurocutaneous melanosis was first described in 1861 by Rokitansky. [13] It was first named by Van Bongaert in 1948. [14] Premortem detection is difficult without the use of MRI.
Neurocutaneous melanocytosis (NCM) is a rare congenital neurological disorder characterized by abnormal aggregations of nevomelanocytes within the central nervous system (leptomeningeal melanocytosis) associated with large or giant congenital melanocytic nevi (CMN; see this term). NCM can be asymptomatic or present as variably severe and progressive neurological impairment, sometimes resulting in death. Epidemiology Prevalence is estimated at 1/50,000-1/200,000. The incidence of symptomatic NCM appears to be approximately a third to a half of these. Clinical description A large, or giant, CMN is a pigmented skin lesion of more than 20 cm projected adult diameter (40 for "giant"), composed of aggregated melanocytes in a delimited area of the body, and presenting with an elevated risk of malignant transformation. Leptomeningeal melanocytosis nearly always presents with CMN, though not conversely; a case with no pigmented lesions and another with only café-au-lait spots have been reported.
Sugarman syndrome is the common name of autosomal recessive oral-facial-digital syndrome type III, one of ten distinct genetic disorders that involve developmental defects to the mouth. [1] Alternative names for this condition include: Brachydactyly of the hands and feet with duplication of the first toes , Sugarman brachydactyly and Brachydactyly with major proximal phalangeal shortening. [2] References [ edit ] ^ "Oral-Facial-Digital Syndrome" .
Oral-facial-digital syndrome, type 3 is characterized by anomalies of the mouth, eyes and digits, associated with severe intellectual deficit. Epidemiology Five cases in two families have been reported (two males and three females). Clinical description Main clinical features include prominent forehead and occiput, round face with full cheeks, hypertelorism, esotropia, downslanting palpebral fissures, myoclonic twitching of the eyelids, conjugate deviation of the eyes, bifid uvula, pectus excavatum, short sternum, kyphosis, postaxial polydactyly, normal stature and severe spasticity. Etiology The causative gene has not yet been identified. Genetic counseling Autosomal recessive inheritance has been suggested.
Clinical Features Sugarman et al. (1971) reported a new form of oral-facial-digital syndrome in 2 sisters. Features were mental retardation, eye abnormalities, lobulated hamartomatous tongue, dental abnormalities, bifid uvula, postaxial hexadactyly of hands and feet, pectus excavatum, short sternum, and kyphosis. One of the sibs showed ceaseless 'see-saw winking' of the eyes. The parents were not related. We have observed a family in which 3 of 4 sibs (2 males, 1 female) were affected. None had 'see-saw' winking. However, all had myoclonic jerks, affected lids, extraocular muscles, arms, etc.
The fourth sib, who had a different father, was normal. The first infant developed erythematous and vesicular lesions of the face after a 6-hour exposure to sun at the age of 2 weeks and died at the age of 6 months.
Cutaneous photosensitivity and lethal colitisis is a rare inflammatory bowel disease (see this term) characterized by early cutaneous photosensitivity manifesting by sun-induced facial erythematous and vesicular lesions and severe recurent colitis which lead to untreatable diarrhea. There have been no further descriptions in the literature since 1991.
Kirby et al. (2005) described 3 nondysmorphic sibs from a consanguineous Pakistani family who had severe mental retardation, keratoconus resulting in significant vision failure, and febrile seizures throughout the first few years of life. Two of the sibs developed sinoatrial heart block, 1 of whom required insertion of a cardiac pacemaker.
Deficiency was identified in the activity of the pyruvate dehydrogenase complex although not in the thiamine-dependent first enzyme of that complex. The patient was thought to have a partial genetic defect affecting the tricarboxylic acid cycle.
Hepatoblastoma is a rare malignant (cancerous) tumor of the liver that usually occurs in the first 3 years of life. In early stages of the condition, there may be no concerning signs or symptoms.
A malignant hepatic tumor, typically affecting the pediatric population, arising mostly in an otherwise healthy liver. The most common signs are addominal distension and abdominal mass. Sometimes patients present with anorexia, weight loss, fatigue. Most HBLs are sporadic, but some cases are associated with genetic factors, especially overgrowth syndromes, such as Beckwith-Wiedemann syndrome (BWS) or hemihypertrophy, and familial adenomatous polyposis (FAP). Epidemiology Hepatoblastoma (HB) accounts for about 0,5-2% of all pediatric tumors and for 2/3 of primary hepatic tumors in children. Its incidence is estimated to be 1/1,000,000 in Europe and 1-1,5/1,000,000 in USA.
The disease is most commonly diagnosed during a child's first three years of life. [1] Alpha-fetoprotein (AFP) levels are commonly elevated, but when AFP is not elevated at diagnosis the prognosis is poor. [2] Contents 1 Signs and symptoms 2 Pathophysiology 3 Diagnosis 4 Treatment 5 References 6 External links Signs and symptoms [ edit ] Patients are usually asymptomatic [3] at diagnosis. ... "Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group" .
Sommerfelt et al. (1991) described a seemingly new disorder in 4 sibs, the offspring of first-cousin parents: hereditary spastic paraplegia with epileptic myoclonus.