Pyruvate dehydrogenase E3-binding protein deficiency is a rare mild form of pyruvate dehydrogenase deficiency (PDHD, see this term) characterized by variable lactic acidosis and neurological dysfunction. ... The disorder is more frequent than PDHD E2 deficiency but less frequent than E1-alpha deficiency (see these terms). ... In contrast to other forms of pyruvate dehydrogenase deficiency, patients with E3 binding protein deficiency often survive well into childhood or adult life as there is some assembly of the pyruvate dehydrogenase complex even with complete deficiency of this protein. Thinning or agenesis of the corpus callosum is a common finding on MRI imaging of the brain. Etiology PDHD E3 binding protein deficiency is caused by a mutation in the PDHX gene (11p13) encoding the E3 binding protein subunit; also known as component X of the pyruvate dehydrogenase complex.
A number sign (#) is used with this entry because pyruvate dehydrogenase E3-binding protein deficiency is caused by homozygous or compound heterozygous mutation in the PDHX gene (608769) on chromosome 11p13. ... Clinical Features Robinson et al. (1990) described 2 patients who had decreased activity of the pyruvate dehydrogenase (PDH) complex without observable reduction in the activities of enzymes E1 (300502/179060), E2 (608770), or E3 (238331). Western blot analysis showed that 1 patient appeared to be missing the component X protein, while the other had 2 distinct bands. ... Immunoblotting of skin fibroblast mitochondrial extracts showed a specific deficiency in the protein X component of the PDH complex but normal E1, E2, and E3 components. Geoffroy et al. (1996) reported the clinical presentation, enzymatic analysis, and Western immunoblot analysis in a newborn girl with lactic acidemia as a result of a primary defect in the X component of the PDH complex. ... The index patients in each family had reduced PDC activity in cultured fibroblasts and no detectable immunoreactive E3 protein. Mine et al. (2007) reported a 25-year-old man with pyruvate dehydrogenase E3-binding protein (E3BP) deficiency confirmed by genetic analysis. ... In 2 unrelated patients with pyruvate dehydrogenase E3-binding protein deficiency, Brown et al. (2002) identified mutations in the PDHX gene (608769.0004 and 608769.0005).
While research establishing an official mortality rate may not exist, two recent studies of SMA syndrome patients, one published in 2006 looking at 22 cases [11] and one in 2012 looking at 80 cases, [17] show mortality rates of 0% [11] and 6.3%, [17] respectively. ... "Nutcracker and SMA syndromes: What is the normal SMA angle in children?". European Journal of Radiology . 81 (8): e854-61. doi : 10.1016/j.ejrad.2012.04.010 . ... "Late superior mesenteric artery syndrome in paraplegia: case report and review" . Spinal Cord . 40 (2): 88–91. doi : 10.1038/sj.sc.3101255 . ... Journal of Pediatric Surgery . 42 (10): e5–e8. doi : 10.1016/j.jpedsurg.2007.07.002 . ... "Case Report: A Pediatric AIDS Patient with Superior Mesenteric Artery Syndrome." http://www.liebertonline.com/doi/abs/10.1089/108729100318073 ^ Welsch T, Büchler MW, Kienle P (2007).
Superior mesenteric artery syndrome (SMAS) is a digestive condition that occurs when the duodenum (the first part of the small intestine) is compressed between two arteries (the aorta and the superior mesenteric artery). This compression causes partial or complete blockage of the duodenum. Symptoms vary based on severity, but can be severely debilitating. Symptoms may include abdominal pain, fullness, nausea, vomiting, and/or weight loss. SMAS typically is due to loss of the mesenteric fat pad (fatty tissue that surrounds the superior mesenteric artery). The most common cause is significant weight loss caused by medical disorders, psychological disorders, or surgery.
The pyruvate dehydrogenase complex is made up of multiple copies of several enzymes called E1, E2, and E3, each of which performs part of the chemical reaction that converts pyruvate to acetyl-CoA. In addition, other proteins included in the complex ensure its proper function. One of these proteins, E3 binding protein, attaches E3 to the complex and provides the correct structure for the complex to perform its function. ... Mutations in the gene that provides instructions for making E1 alpha, the PDHA1 gene, are the most common cause of pyruvate dehydrogenase deficiency, accounting for approximately 80 percent of cases. These mutations lead to a shortage of E1 alpha protein or result in an abnormal protein that cannot function properly. ... Mutations in the genes that provide instructions for E1 beta (the PDHB gene), the E2 enzyme (the DLAT gene), E3 binding protein (the PDHX gene), and pyruvate dehydrogenase phosphatase (the PDP1 gene) have been identified in people with this condition.
They cannot, however, supply ATP to these cells and, therefore, phenotype depends largely on the nature/severity of the mutation. [5] [6] More rarely, mutations occur in the E2 ( dihydrolipoyl transacetylase ) or the E3 ( dihydrolipoyl dehydrogenase ) subunits of the PDC enzymatic complex, DLAT and DLD genes respectively. ... The DLAT gene is responsible for the coding of the E2 subunit, and the PDP1 is responsible for producing the PDH phosphatase catalytic subunit that catalyzes PDH dephosphorylation. ... The final gene that could be responsible for this disease is the PDHX gene, which codes for the E3 binding protein which is responsible for binding E3 dimers to the E2 subunit of the complex [8] Epidemiology [ edit ] Pyruvate dehydrogenase deficiency is extremely rare, with ~500 reported cases in the medical literature.
INHERITANCE - Autosomal recessive SKIN, NAILS, & HAIR Skin - Large erythematous scales - Fine white scale on upper limbs and trunk (in some patients) - Large polygonal brown scales and lower limbs (in some patients) - Hyperkeratosis of elbows and knees - Palmoplantar hyperkeratosis - Hyperlinearity of palms and soles - Fungal infections of skin Skin Histology - Mild hypergranulosis - Marked hyperkeratosis Nails - Onychomycosis MISCELLANEOUS - Sparing of face and scalp in approximately two-thirds of patients - Severity decreases with age (in some patients) MOLECULAR BASIS - Caused by mutation in the short chain dehydrogenase/reductase family 9C, member 7 gene (SDR9C7, 609769.0001 ) ▲ Close
Etiology MSUD is due to mutations in the genes encoding subunits E1a, E1b, and E2 of the branched chain 2-ketoacid dehydrogenase (BCKAD) complex, involved in the second enzymatic step in the degradation of the branched chain amino acids (BCAAs): leucine, isoleucine and valine. BCKAD has four subunits: E1a, E1b, E2, and E3, which are encoded by the genes BCKDHA (19q13.1-q13.2), BCKDHB (6q14.1), DBT (1p31) and DLD (7q31-q32) respectively. Mutations in these genes lead to the accumulation of BCAAs (especially leucine) and their branched-chain alpha-ketoacids. Mutations in the E3 subunit gene ( DLD ) are not associated with MSUD but lead to pyruvate dehydrogenase E3 deficiency (see this term).
Molecular Genetic Testing Used in Maple Syrup Urine Disease View in own window Gene 1, 2 Proportion of MSUD Attributed to Pathogenic Variants in Gene Proportion of Pathogenic Variants 3 Detectable by Method Sequence analysis 4 Gene-targeted deletion/duplication analysis 5 BCKDHA 45% ~92% ~8% 6, 7 BCKDHB 35% ~93% ~7% DBT 20% ~86% ~14% 8 Unknown 9, 10 NA 1. ... Plasma leucine levels rise predictably as a result of net protein catabolism provoked by a variety of physiologic stresses, including (more...) ... Pathophysiology BCKD has four subunit components (E1a, E1b, E2, and E3). Pathogenic variants in both alleles encoding any subunit can result in decreased activity of the enzyme complex and the accumulation of BCAAs and corresponding BCKAs in tissues and plasma [Nellis et al 2003, Chuang et al 2004] (see Nomenclature). ... Note: Pathogenic variants in DLD , the gene encoding the E3 subunit, are associated with dihydrolipoamide dehydrogenase deficiency, which produces a different phenotype. ... Mean and 25th to 75th Percentile Range Nutrient Intakes (per kg-day) by Age Group View in own window Nutrient Age in Months (# of Persons) 0-2 (31) 3-5 (18) 6-8 (21) 9-12 (18) 13-18 (21) 19-24 (18) 25-36 (32) Mean Nutrient Intake per kg-day (25th to 75th %ile range) Leucine (mg) 72 (64-84) 58 (47-68) 44 (37-51) 35 (30-41) 33 (26-39) 27 (22-28) 21 (20-25) Energy (kcal) 111 (103-119) 99 (94-107) 89 (82-99) 78 (71-87) 67 (55-77) 57 (49-71) 38 (39-51) Total protein (g) 2.4 (2.1-2.6) 2.3 (1.9-2.6) 2.2 (1.8-2.5) 2.0 (1.5-2.5) 2.1 (1.8-2.4) 2 (1.7-2.3) 1.5 (1.5-2.9) Leucine/energy ratio (mg/kcal) 0.65 (0.57-0.72) 0.58 (0.48-0.66) 0.50 (0.43-0.56) 0.46 (0.37-0.53) 0.50 (0.43-0.58) 0.48 (0.38-0.58) 0.54 (0.44-0.55) Strauss et al [2010]; with permission from Elsevier Data derived from Mennonite children from birth to age 4 years with classic MSUD Mild illness.
At age 21, she worked as a nurse and had an IQ of 90. Brain MRI was normal, but blood levels of branched-chain amino and keto acids remained mildly increased.
Maple syrup urine disease (MSUD) occurs when the body is unable to breakdown certain parts of proteins. This leads to the build-up of toxic substances that can cause organ and brain damage. There are several forms of MSUD. The most common is the classic or infantile form. Symptoms of the classic form of MSUD start in early infancy and include poor feeding, irritability, extra sleepiness, and muscle spasms. If untreated, respiratory failure (lack of oxygen getting to the blood) may occur.
Yamada et al. (1996) reported a lack of association between the E4 allele and cerebral amyloid angiopathy in elderly Japanese patients. Nicoll et al. (1996, 1997) did not find an association between the E4 allele and CAA-related hemorrhage. However, they did find a high frequency of the E2 allele in patients with CAA-related hemorrhage, regardless of the presence of AD. ... The authors suggested that apolipoprotein E2 and E4 might promote hemorrhage through separate mechanisms: E4 by enhancing amyloid deposition and E2 by promoting rupture. ... O'Donnell et al. (2000) identified a specific apolipoprotein E genotype as a risk factor for early recurrence: carriers of the E2 (107741.0001) or E4 (107741.0016) allele had an increased risk for early recurrence compared to individuals with the E3/E3 (107741.0015) genotype.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by age of onset between 50-66 years of age, memory impairment, myoclonic jerks, expressive dysphagia, short-stepped gait, personality changes, and lobar intracerebral hemorrhages. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
Hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D) is a form of HCHWA (see this term), a group of familial central nervous system disorders, characterized by severe cerebral amyloid angiopathy (CAA), hemorrhagic and non-hemorrhagic strokes and dementia. Epidemiology The prevalence is unknown. It has been seen in three Dutch families, that are most likely related (before 17th century), to date. Approximately 200-300 patients are known to be affected. Clinical description HCHWA-D presents with hemorrhagic stroke between the ages of 39-76 years (average age 50 years). In one third of cases, the initial stroke is fatal, recurrent strokes are seen in those who survive and patients often suffer from serious disability. Cognitive decline can be a presenting symptom in some cases but often occurs after the initial or recurrent strokes.
Hereditary cerebral amyloid angiopathy is a condition that can cause a progressive loss of intellectual function (dementia), stroke, and other neurological problems starting in mid-adulthood. Due to neurological decline, this condition is typically fatal in one's sixties, although there is variation depending on the severity of the signs and symptoms. Most affected individuals die within a decade after signs and symptoms first appear, although some people with the disease have survived longer. There are many different types of hereditary cerebral amyloid angiopathy. The different types are distinguished by their genetic cause and the signs and symptoms that occur.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset between 50-70 years of age, recurrent lobar intracerebral hemorrhages and cognitive decline. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset of 45 years of age, progressive Alzheimer's disease-like dementia, and lobar intracerebral hemorrhage in some patients. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset of 54-61 years, progressive Alzheimer's disease-like dementia, and absence of intracerebral hemorrhages. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
A form of hereditary cerebral hemorrhage with amyloidosis characterized by an age of onset of 50 years of age, dementia and lobar intracerebral hemorrhage. This subtype is due to a mutation in the APP gene (21q21.2), encoding the beta-amyloid precursor protein. This mutation causes an increased accumulation of amyloid-beta protein in the walls of the arteries and capillaries of the meninges, cerebellar cortex and cerebral cortex, leading to the weakening and eventual rupture of these vessels.
PMID 27188519 . ^ Pereira PC, Miranda DM, Oliveira EA, Silva AC (March 2009). "Molecular pathophysiology of renal tubular acidosis" . ... A defect in bicarbonate reabsorption with normal urinary acidification" . Pediatric Research . 1 (2): 81–98. doi : 10.1203/00006450-196703000-00001 . ... Journal of the American Society of Nephrology . 13 (8): 2160–70. doi : 10.1097/01.ASN.0000023430.92674.E5 . PMID 12138150 . ^ de Jong PE, Koomans HA, Weening JJ (2000). ... "Carbonic anhydrase II (CA II) deficiency in Maghrebian patients: evidence for founder effect and genomic recombination at the CA II locus". Human Genetics . 99 (5): 634–7. doi : 10.1007/s004390050419 . ... The Journal of Pediatrics . 8 (4): 489–99. doi : 10.1016/s0022-3476(36)80111-5 . ^ Baines AM, Barelay JA, Cooke WT (1945).
JAK2-positive polycythaemia vera - diagnosis requires both criteria to be present: Criteria Notes A1 High erythrocyte volume fraction (EVF or haematocrit) (>0.52 in men, >0.48 in women) OR raised red cell mass (>25% above predicted) A2 Mutation in JAK2 JAK2-negative polycythemia vera - diagnosis requires A1 + A2 + A3 + either another A or two B criteria: Criteria Notes A1 Raised red cell mass (>25% above predicted) OR haematocrit >0.60 in men, >0.56 in women A2 Absence of mutation in JAK2 A3 No cause of secondary erythrocytosis A4 Palpable splenomegaly A5 Presence of an acquired genetic abnormality (excluding BCR-ABL) in the haematopoietic cells B1 Thrombocytosis (platelet count >450 * 109/l) B2 Neutrophil leucocytosis (neutrophil count > 10 * 109/l in non-smokers; > 12.5*109/l in smokers) B3 Radiological evidence of splenomegaly B4 Endogenous erythroid colonies or low serum erythropoietin Treatment [ edit ] Untreated, polycythemia vera can be fatal. [16] [17] Research has found that the "1.5-3 years of median survival in the absence of therapy has been extended to at least 10-20 years because of new therapeutic tools." [18] As the condition cannot be cured, treatment focuses on treating symptoms and reducing thrombotic complications by reducing the erythrocyte levels. ... "Increased thromboxane biosynthesis in patients with polycythemia vera: evidence for aspirin-suppressible platelet activation in vivo" . Blood . 80 (8): 1965–71. doi : 10.1182/blood.V80.8.1965.1965 . ... "Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis". Cancer Cell . 7 (4): 387–97. doi : 10.1016/j.ccr.2005.03.023 . ... "Polycythemia vera in young patients: a study on the long-term risk of thrombosis, myelofibrosis and leukemia". Haematologica . 88 (1): 13–8. PMID 12551821 . ^ a b Anía B, Suman V, Sobell J, Codd M, Silverstein M, Melton L (1994). "Trends in the incidence of polycythemia vera among Olmsted County, Minnesota residents, 1935-1989". Am J Hematol . 47 (2): 89–93. doi : 10.1002/ajh.2830470205 .
Baxter et al. (2005) and Kralovics et al. (2005) found that 97% (71 of 73) and 65% (83 of 128) of patients with polycythemia vera, respectively, carried a val617-to-phe mutation (V617F; 147796.0001) in the JAK2 gene. James et al. (2005) found that 88% (40 of 45) of patients with polycythemia vera carried the V617F mutation in JAK2.
Polycythemia vera (PV) is an acquired myeloproliferative disorder characterized by an elevated absolute red blood cell mass caused by uncontrolled red blood cell production, frequently associated with uncontrolled white blood cell and platelet production. Epidemiology Annual incidence is estimated at approximately 1/36,000-1/100,000 and prevalence at 1/3,300. PV occurs at all ages but is most common in those aged 50-70 years. Clinical description Symptoms are often insidious at onset and may include headache, dizziness, vertigo, tinnitus, visual disturbances, and pruritus after bathing, a ruddy complexion that manifests in the face, palms, nailbeds, mucosa and conjunctiva. Complications may be associated including arterial thrombosis (in cerebrovascular, myocardial or peripheral territories), angina pectoris or intermittent claudications, or venous thromboses including deep vein thrombosis, pulmonary embolism, splanchnic thrombosis (portal vein thrombosis and Budd-Chiari syndrome; see these terms) and bleeding including gum bleeding, ecchymoses and gastrointestinal bleeding. PV may also be characterized by splenomegaly. Myelofibrosis and acute leukemia or a myelodysplastic syndrome occur in a minority of patients, usually late in the disease.
Polycythemia vera is a condition characterized by an increased number of red blood cells in the bloodstream (erythrocytosis). Affected people may also have excess white blood cells and platelets. Conditions where the body makes too many of these cells are known as myeloproliferative neoplasms. These extra cells cause the blood to be thicker than normal, increasing the risk for blood clots that can block blood flow in arteries and veins. If a blood clot occurs in the veins deep in the arms and the legs, it is known as deep vein thrombosis (DVT).
Polycythemia vera is a condition characterized by an increased number of red blood cells in the bloodstream. Affected individuals may also have excess white blood cells and blood clotting cells called platelets. These extra cells and platelets cause the blood to be thicker than normal. As a result, abnormal blood clots are more likely to form and block the flow of blood through arteries and veins. Individuals with polycythemia vera have an increased risk of deep vein thrombosis (DVT), a type of blood clot that occurs in the deep veins of the arms or legs.
Overview Polycythemia vera (pol-e-sy-THEE-me-uh VEER-uh) is a type of blood cancer. It causes your bone marrow to make too many red blood cells. These excess cells thicken your blood, slowing its flow, which may cause serious problems, such as blood clots. Polycythemia vera is rare. It usually develops slowly, and you might have it for years without knowing. Often the condition is found during a blood test done for another reason. Without treatment, polycythemia vera can be life-threatening. But proper medical care can help ease signs, symptoms and complications of this disease.
Treatment [ edit ] Anti-IgLON5 disease is mainly treated with immunosuppressants (80%), mostly cycles of IV corticosteroids (58%) in combination with IV immunoglobulins (IVIg−36%) and/or TPE (27%). ... "Clinical manifestations of the anti-IgLON5 disease" . Neurology . 88 (18): 1736–1743. doi : 10.1212/WNL.0000000000003887 . ... The Lancet. Neurology . 13 (6): 575–86. doi : 10.1016/S1474-4422(14)70051-1 . ... The Lancet. Neurology . 13 (6): 575–86. doi : 10.1016/S1474-4422(14)70051-1 . ... "IgLON5: A case with predominant cerebellar tau deposits and leptomeningeal inflammation". Neurology . 91 (4): 180–182. doi : 10.1212/WNL.0000000000005859 .
A rare neurologic disorder characterized by a unique non-REM and REM parasomnia with sleep breathing dysfunction, gait instability and repetitive episodes of respiratory insufficiency, as well as autoantibodies against IgLON5. Patients may present stridor, chorea, limb ataxia, abnormal ocular movements, and bulbar symptoms (i.e. dysphagia, dysarthria, episodic central hypoventilation) with normal brain MRI. Excessive day sleepiness and cognitive deterioration have also been reported.
Thymomas are currently divided according to the World Health Organization (WHO) into four histological subclasses: types A, B, AB, and C. Type A tumors are composed of spindle or oval epithelial cells. Type B tumors consist of round or polygonal epithelial cells and are divided into three subtypes (B1, B2, and B3) based on their various proportions of background lymphocytes and increase in cytologic atypia of the epithelial cells. Type AB tumors consist of type A thymoma features with a variably dense lymphocytic component. ... In advanced stages (stage II according to Masaoka staging system) and in high-risk histologic subtypes (B3), surgery is accompanied by additional adjuvant therapy (consisting of post-operative radiotherapy) or neoadjuvant therapy.
In a series of 274 thymic epithelial tumors, Petrini et al. (2014) detected the GTF2I mutation in 82% of type A and 74% of type AB thymomas but rarely in the aggressive subtypes, where recurrent mutations of known cancer genes have been identified.
Histopathological image of thymoma type B1. Anterior mediastinal mass surgically resected. ... Histopathological image of thymoma type B1. Anterior mediastinal mass surgically resected. ... Histopathological image representing a noninvasive thymoma type B1, surgically resected. Hematoxylin & eosin. ... "Thymoma associated with autoimmune diseases: 85 cases and literature review". Autoimmunity Reviews . 15 (1): 82–92. doi : 10.1016/j.autrev.2015.09.005 . ... "Follow-up study of thymomas with special reference to their clinical stages" . Cancer . 48 (11): 2485–92. doi : 10.1002/1097-0142(19811201)48:11<2485::AID-CNCR2820481123>3.0.CO;2-R .
Общая психопатология: Учебное пособие для студентов высших учебных заведений [ General psychopathology: Textbook for students of higher educational institutions ] (in Russian). Moscow: Academia. pp. 80–82. ISBN 5-7695-0838-8 . ^ a b Kaptsan, A; Miodownick, C; Lerner, V (2000). ... The Israel Journal of Psychiatry and Related Sciences . 37 (4): 278–85. PMID 11201932 . ^ a b Shorter, E. (2005). ... ISBN 978-0-19-517668-1 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab Жариков, Н. М.; Тюльпин, Ю. Г. (2002). ... Journal of Psychiatry & Neuroscience . Joule Inc. 35 (4): E5–6. doi : 10.1503/jpn.100087 . ISSN 1180-4882 .
Six subtypes related to the affected subunit of the PDH complex have been recognized with significant clinical overlap: PDHD due to E1-alpha, E1-beta, E2 and E3 deficiency, PDHD due to E3-binding protein deficiency, and PDH phosphatase deficiency (see these terms). ... Mutations in the genes for the other subunits have been described, but are far less frequent: E1-beta and E2 subunits ( PDHB , DLAT ); E3 binding protein ( PDHX gene); and E3 and PDH phosphatase ( DLD and PDP1 ).
A number sign (#) is used with this entry because of evidence that pyruvate dehydrogenase phosphatase deficiency is caused by homozygous mutation in the PDP1 gene (605993) on chromosome 8q22. For a general phenotypic description and a discussion of genetic heterogeneity of pyruvate dehydrogenase (PDH) deficiency, see 312170. Clinical Features Robinson and Sherwood (1975) reported a male infant who presented on the first day of life with lactic acidosis and died at age 6 months. Activity of the pyruvate dehydrogenase complex in tissue homogenates preincubated with ATP was reduced by 60 to 75% in liver of the patient and controls because of the inactivation of the enzyme by pyruvate dehydrogenase kinase (see, e.g., PDK1, 602524). Addition of calcium and magnesium to the inactivated enzyme caused a prompt return of the activity to normal in controls, but not in the patient's cells.
The pyruvate dehydrogenase complex showed about 30% activity in kidney. The second (E2) and third (E3) components of the complex were normal. ... The X-inactivation pattern in the fibroblasts was 80 to 20. This was the third child of consanguineous parents; a previous male infant had died with severe developmental delay and lactic acidosis.
A number sign (#) is used with this entry because of evidence that pyruvate dehydrogenase E2 deficiency is caused by homozygous mutation in the DLAT gene (608770) on chromosome 11q23. ... Control of blood lactates was achieved by carbohydrate restriction and bicarbonate supplementation, but at age 3.5 years she had profound psychomotor retardation and was microcephalic. Deficiency in the E2 dihydrolipoyl transacetylase activity of the pyruvate dehydrogenase complex was demonstrated enzymatically, and a very low E2 protein component was found on Western blotting of fibroblast proteins. ... Head et al. (2005) reported 2 unrelated patients with pyruvate dehydrogenase E2 deficiency. Both patients were born of first-cousin parents, indicating autosomal recessive inheritance. ... Friedman et al. (2017) reported a patient with pyruvate dehydrogenase E2 deficiency who also had paroxysmal exercise-induced dyskinesia. ... Molecular Genetics In 2 unrelated patients with pyruvate dehydrogenase E2 deficiency, Head et al. (2005) identified 2 different homozygous mutations in the DLAT gene (c.361del3, 608770.0001 and F576L, 608770.0002).
Pyruvate dehydrogenase (E1) and dihydrolipoamide acetyltransferase (E2) activities were normal in the patient's muscle, but E3 activity was not detectable in either muscle or liver specimens obtained at autopsy. ... These findings suggested that a partial reduction in E3 is not rate-limiting for KGDC activity in fibroblasts. ... All patients had decreased levels of the E3 protein (range, 35-68% of controls) and decreased E3 activity (8 to 33% of controls). ... Clinical Variability Shaag et al. (1999) studied 13 patients with E3 deficiency originating from 7 Ashkenazi Jewish families. ... In 4 patients tested, the E3 protein in muscle was reduced to 20 to 60% of control.
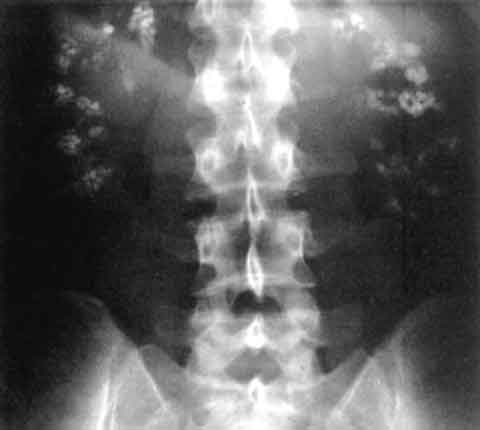
A disorder that is the most frequent form of pyruvate dehydrogenase deficiency (PDHD) characterized by variable lactic acidosis, impaired psychomotor development, hypotonia and neurological dysfunction. Epidemiology Prevalence is unknown. Over 200 patients have been reported and while there are approximately equal numbers of males and females, male patients are generally more severely affected. Clinical description Patients present with a range of classic signs and symptoms of PDHD, including lactic acidosis, poor feeding, lethargy, tachypnea, developmental delay, growth retardation, poor acquisition or loss of motor milestones, hypotonia, seizures, ataxia and dystonia. Structural brain lesions including cortical atrophy, dilated ventricles, and incomplete corpus callosum, absence of the medullary pyramids and ectopia of the olivary nuclei are commonly observed, especially in female patients heterozygous for the disease-causing mutations that result in complete deficiency of E1-alpha subunit protein in cells expressing the gene mutation. Etiology The disease is caused by deficiency of the E1-alpha subunit of the PDH complex related to mutations in the PDHA1 gene (Xp22.1).
Pyruvate dehydrogenase complex (PDC) deficiency is a type of metabolic disease. This means that the body is not able to efficiently break down nutrients in food to be used for energy. Symptoms of PDC deficiency include signs of metabolic dysfunction such as extreme tiredness (lethargy), poor feeding, and rapid breathing ( tachypnea ). Other symptoms may include signs of neurological dysfunction such as developmental delay, periods of uncontrolled movements (ataxia), low muscle tone (hypotonia), abnormal eye movements, and seizures. Symptoms usually begin in infancy, but signs can first appear at birth or later in childhood.
A number sign (#) is used with this entry because pyruvate dehydrogenase E1-beta deficiency is caused by homozygous mutation in the PDHB gene (179060) on chromosome 3p14. For a general phenotypic description and a discussion of genetic heterogeneity of pyruvate dehydrogenase deficiency, see 312170. Clinical Features Brown et al. (2004) reported 2 unrelated patients with pyruvate dehydrogenase deficiency. The first patient, the son of first-cousin parents, was investigated at age 3 months because of lactic acidosis and hypotonia. Two previous sibs had died early, one with lactic acidosis. The patient developed metabolic acidosis on day 1.
A chest X-ray or an echocardiogram may also be performed to look for signs of heart enlargement or damage to the heart. [23] Classification in adults [ edit ] Classification in adults (Persons with systolic and diastolic in different categories are assigned to the higher category. [7] ) Category Systolic , mmHg Diastolic , mmHg Hypotension < 90 < 60 Normal 90–119 [7] 90–129 [83] 60–79 [7] 60–84 [83] Prehypertension (high normal, elevated [7] ) 120–129 [7] 130–139 [83] [84] 60–79 [7] 85–89 [83] [84] Stage 1 hypertension 130-139 [7] 140–159 [83] 80-89 [7] 90–99 [83] Stage 2 hypertension >140 [7] 160–179 [83] >90 [7] 100–109 [83] Hypertensive crises ≥ 180 [7] ≥ 120 [7] Isolated systolic hypertension ≥ 140 [7] < 90 [7] Isolated diastolic hypertension [85] [86] < 140 ≥ 90 In people aged 18 years or older hypertension is defined as either a systolic or a diastolic blood pressure measurement consistently higher than an accepted normal value (this is above 129 or 139 mmHg systolic, 89 mmHg diastolic depending on the guideline). [5] [7] Other thresholds are used (135 mmHg systolic or 85 mmHg diastolic) if measurements are derived from 24-hour ambulatory or home monitoring. [73] Recent international hypertension guidelines have also created categories below the hypertensive range to indicate a continuum of risk with higher blood pressures in the normal range. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure (JNC7) published in 2003 [27] uses the term prehypertension for blood pressure in the range 120–139 mmHg systolic or 80–89 mmHg diastolic, while European Society of Hypertension Guidelines (2007) [87] and British Hypertension Society (BHS) IV (2004) [88] use optimal, normal and high normal categories to subdivide pressures below 140 mmHg systolic and 90 mmHg diastolic. ... Hypertension is classified as "resistant" if medications do not reduce blood pressure to normal levels. [27] In November 2017, the American Heart Association and American College of Cardiology published a joint guideline which updates the recommendations of the JNC7 report. [89] The 2020 International Society of Hypertension guidelines define hypertension based on office blood pressure ≥140/90 mmHg or home monitoring blood pressure ≥135/85 mmHg, or 24-hour ambulatory blood pressure average ≥130/80 mmHg (daytime average ≥135/85 mmHg or nighttime average BP ≥120/70 mmHg). [90] Children [ edit ] Hypertension occurs in around 0.2 to 3% of newborns; however, blood pressure is not measured routinely in healthy newborns. [33] Hypertension is more common in high risk newborns. ... High blood pressure must be confirmed on repeated visits however before characterizing a child as having hypertension. [91] Prehypertension in children has been defined as average systolic or diastolic blood pressure that is greater than or equal to the 90th percentile, but less than the 95th percentile. [91] In adolescents, it has been proposed that hypertension and pre-hypertension are diagnosed and classified using the same criteria as in adults. [91] The value of routine screening for hypertension in children over the age of 3 years is debated. [92] [93] In 2004 the National High Blood Pressure Education Program recommended that children aged 3 years and older have blood pressure measurement at least once at every health care visit [91] and the National Heart, Lung, and Blood Institute and American Academy of Pediatrics made a similar recommendation. [94] However, the American Academy of Family Physicians [95] supports the view of the U.S. Preventive Services Task Force that the available evidence is insufficient to determine the balance of benefits and harms of screening for hypertension in children and adolescents who do not have symptoms. [96] [97] Prevention [ edit ] Much of the disease burden of high blood pressure is experienced by people who are not labeled as hypertensive. [88] Consequently, population strategies are required to reduce the consequences of high blood pressure and reduce the need for antihypertensive medications.
Overview Intermittent explosive disorder involves repeated, sudden episodes of impulsive, aggressive, violent behavior or angry verbal outbursts in which you react grossly out of proportion to the situation. Road rage, domestic abuse, throwing or breaking objects, or other temper tantrums may be signs of intermittent explosive disorder. These intermittent, explosive outbursts cause you significant distress, negatively impact your relationships, work and school, and they can have legal and financial consequences. Intermittent explosive disorder is a chronic disorder that can continue for years, although the severity of outbursts may decrease with age. Treatment involves medications and psychotherapy to help you control your aggressive impulses.
The structural polyprotein is cleaved into six chains: capsid protein (C), p62, E3 protein or spike glycoprotein E3, E2 envelope glycoprotein or spike glycoprotein E2, 6K protein, and E1 envelope glycoprotein known also as spike glycoprotein E1. [7] [8] The envelope lipid component is critical for virus particle stability and infectivity in mammalian cells [9] Once the virus enters into the host cell, the genomic RNA is released into the cytoplasm, where the two ORFs are translated into proteins and the synthesis of negative-stranded RNA starts. ... The amplicon used for phylogenetic analysis includes E1 and E2 glycoprotein genes and the 3' NCR. ... Journal of Virology . 86 (24): 13609–20. doi : 10.1128/JVI.01805-12 . PMC 3503070 . PMID 23035234 . ^ Firth AE, Chung BY, Fleeton MN, Atkins JF (2008). ... The American Journal of Tropical Medicine and Hygiene . 85 (4): 750–7. doi : 10.4269/ajtmh.2011.11-0359 .
Among women who experience a third or fourth degree tear, 60-80% are asymptomatic after 12 months. [21] Faecal incontinence, faecal urgency, chronic perineal pain, pain with sex, and fistula formation occur in a minority of people, but may be permanent. [22] The symptoms associated with perineal tear are not always due to the tear itself, since there are often other injuries, such as avulsion of pelvic floor muscles, that are not evident on examination. [23] There are claims that sometimes the perineum is excessively repaired after childbirth, using a so-called " husband stitch " and that this can increase vaginal tightness or result in pain during intercourse. [24] References [ edit ] ^ a b Elharmeel, Suzan MA; Chaudhary, Yasmin; Tan, Stephanie; Scheermeyer, Elly; Hanafy, Ashraf; van Driel, Mieke L (2011-08-10). ... American Journal of Obstetrics and Gynecology . 200 (5): 573.e1–573.e7. doi : 10.1016/j.ajog.2008.11.022 . ... "Antenatal perineal massage to prevent birth trauma". American Family Physician . 89 (5): 335–6. PMID 24695503 . ^ Wang, H; Jayasekara, R; Warland, J (12 March 2015). ... Third degree obstetric anal sphincter tears: risk factors and outcome of primary repair" . BMJ . 308 (6933): 887–91. doi : 10.1136/bmj.308.6933.887 . ... Obstetrics and Gynecology . 107 (6): 1261–8. doi : 10.1097/01.aog.0000218693.24144.bd . PMID 16738150 . S2CID 23901136 . ^ Lammers, K; Prokop, M; Vierhout, ME; Kluivers, KB; Fütterer, JJ (August 2013).
Upsala Journal of Medical Sciences . 121 (2): 96–112. doi : 10.3109/03009734.2016.1165317 . ... The Journal of Clinical Endocrinology and Metabolism . 94 (11): 4284–4291. doi : 10.1210/jc.2009-1231 . ... Molecular Syndromology . 4 (1–2): 74–86. doi : 10.1159/000345205 . ISSN 1661-8769 . ... Part A, Clinical and Molecular Teratology . 88 (8): 601–611. doi : 10.1002/bdra.20680 . ... Part A, Clinical and Molecular Teratology . 88 (10): 791–803. doi : 10.1002/bdra.20734 .
Bronchopulmonary dysplasia is a chronic respiratory disease that results from complications related to lung injury during the treatment of infant acute respiratory distress syndrome (see these terms) in low-birth-weight premature infants or from abnormal lung development in older infants. Clinical signs are tachypnea, tachycardia and signs of respiratory distress such as intercostal recession, grunting and nasal flaring.
Stroke (Sixth ed.). Elsevier. pp. 413–448.e7. doi : 10.1016/B978-0-323-29544-4.00026-8 . ... The movements may involve the Eustachian tube and make a click that the patient can hear. ^ Zadikoff C, Lang AE, Klein C. (29 November 2005). "The 'essentials' of essential palatal tremor: a reappraisal of the nosology" .