Treatment/prognosis [ edit ] The overall 5-year survival is estimated to be approximately 90%, [19] [20] but for individuals the prognosis is highly dependent on individual staging and treatment . ... Stage [23] Histopathology [23] 4 Year relapse-free survival (RFS) or event-free survival (EFS) [23] 4 Year overall survival (OS) [23] Treatment [23] Stage I [23] Favorable histology in children younger than 24 months or tumor weight less than 550g 85% 98% Surgery only (should be done only within the context of a clinical trial) Favorable histology in children older than 24 months or tumor weight more than 550g 94% RFS 98% Nephrectomy + lymph node sampling followed by regimen EE-4A Diffuse anaplastic 68% EFS80% Nephrectomy + lymph node sampling followed by regimen EE-4A and radiotherapy Stage II [23] Favorable histology 86% RFS 98% Nephrectomy + lymph node sampling followed by regimen EE-4A Focal anaplastic 80% EFS80% Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen DD-4A Diffuse anaplastic 83% EFS 82% Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen I Stage III [23] Favorable histology 87% RFS 94% Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen DD-4A Focal anaplastic 88% RFS 100% (8 people in study) Nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen DD-4A Focal anaplastic (preoperative treatment) 71% RFS 71% Preoperative treatment with regimen DD-4A followed by nephrectomy + lymph node sampling and abdominal radiotherapy Diffuse anaplastic 46% EFS 53% Preoperative treatment with regimen I followed by nephrectomy + lymph node sampling and abdominal radiotherapy Diffuse anaplastic 65% EFS 67% Immediate nephrectomy + lymph node sampling followed by abdominal radiotherapy and regimen I Stage IV [23] Favorable histology 76% RFS 86% Nephrectomy + lymph node sampling, followed by abdominal radiotherapy, bilateral pulmonary radiotherapy, and regimen DD-4A Focal anaplastic 61% EFS 72% Nephrectomy + lymph node sampling, followed by abdominal radiotherapy, bilateral pulmonary radiotherapy, and regimen DD-4A Diffuse anaplastic 33% EFS 33% Immediate nephrectomy + lymph node sampling followed by abdominal radiotherapy, whole-lung radiotherapy, and regimen I Diffuse anaplastic (preoperative treatment) 31% EFS 44% Preoperative treatment with regimen I followed by nephrectomy + lymph node sampling followed by abdominal radiotherapy, whole-lung radiotherapy Stage V [23] Overall 61% EFS80% Favorable histology 65% 87% Preoperative treatment with regimen DD-4A , followed by nephron sparing surgery or nephrecomy, staging of tumors, and chemotherapy and/or radiotherapy based on pathology and staging Focal anaplastic 76% 88% Preoperative treatment with regimen DD-4A , followed by nephron sparing surgery or nephrecomy, staging of tumors, and chemotherapy and/or radiotherapy based on pathology and staging Diffuse anaplastic 25% 42% Preoperative treatment with regimen DD-4A , followed by nephron sparing surgery or nephrecomy, staging of tumors, and chemotherapy and/or radiotherapy based on pathology and staging In case of relapse of Wilms' tumor, the 4-year survival rate for children with a standard-risk has been estimated to be 80%. [24] Epidemiology [ edit ] Wilms tumor is the most common malignant renal tumor in children. [25] There are a number of rare genetic syndromes that have been linked to an increased risk of developing Wilms Tumor. [26] Screening guidelines vary between countries; however health care professionals are recommending regular ultrasound screening for people with associated genetic syndromes. [26] Wilms' tumor affects approximately one person per 10,000 worldwide before the age of 15 years. [27] People of African descent may have slightly higher rates of Wilms' tumor. [27] The peak age of Wilms' tumor is 3 to 4 years and most cases occur before the age of 10 years. [28] A genetic predisposition to Wilms' tumor in individuals with aniridia has been established, due to deletions in the p13 band on chromosome 11. [29] History [ edit ] Dr. ... Cell . 60 (3): 509–20. doi : 10.1016/0092-8674(90)90601-A . PMID 2154335 . S2CID 29092372 . ^ Huff V (October 1998). ... "Frequent association of beta-catenin and WT1 mutations in Wilms tumors" . Cancer Research . 60 (22): 6288–92. PMID 11103785 . ^ Ruteshouser EC, Robinson SM, Huff V (June 2008). ... Medical and Pediatric Oncology . 21 (3): 172–81. doi : 10.1002/mpo.2950210305 . PMID 7680412 . ^ Breslow NE, Beckwith JB, Perlman EJ, Reeve AE (September 2006).
A number sign (#) is used with this entry because of evidence that susceptibility to Wilms tumor can be caused by mutation in the REST (600571) gene on 4q12. For a general phenotypic description and discussion of genetic heterogeneity of Wilms tumor, see WT1 (194070). Molecular Genetics To identify predisposition genes for Wilms tumor, Mahamdallie et al. (2015) performed exome sequencing in 24 individuals with Wilms tumor from 12 families and identified 2 different frameshift mutations (600571.0001, 600571.0002) that segregated with the disease in 2 unrelated families. Neither was present in the ICR1000 and ExAC browsers. The mutations were confirmed by Sanger sequencing. Subsequently, Mahamdallie et al. (2015) performed Sanger sequencing of the full coding sequence and intron-exon boundaries of the REST gene in 38 individuals with familial Wilms tumor from 27 families.
Vernon et al. (2003) found that the B1 gene (607968) was interrupted by a translocation t(1;7)(q42;p15) associated with WT5 in a child with Wilms tumor and skeletal abnormalities. The breakpoint bisected intron 1 of the obscurin gene (608616) on chromosome 1 and intron 22 of the B1 gene on chromosome 7. The translocation altered expression of 2 B1 isoforms. Vernon et al. (2003) also identified additional B1 splice isoforms and aberrant isoform expression in 2 of 8 additional WT5 tumors that showed 7p loss of heterozygosity.
A number sign (#) is used with this entry because of evidence that Wilms tumor-2 (WT2) is caused by mutation of the H19/IGF2-imprinting control region (ICR1; 616186) on chromosome 11p15. ICR1 controls imprinted expression of H19 (103280) and IGF2 (147470). ICR1 and a neighboring imprinted gene cluster are implicated in Beckwith-Wiedemann syndrome (BWS; 130650), of which Wilms tumor is a common feature. For a general phenotypic description and discussion of genetic heterogeneity of Wilms tumor, see WT1 (194070). Mapping Using a range of probes for chromosome 11, Mannens et al. (1988) demonstrated that loss of heterozygosity in Wilms tumors may involve chromosome 11p15.5 in addition to 11p13.
Among 121 cases of Beckwith-Wiedemann syndrome (BWS; 130650), 96% were diagnosed by age 8 years; the oldest BWS patient had WT detected at 10 years, 2 months. Among 203 patients with hemihyperplasia, 94% were detected by age 8; the oldest HH patient had WT detected at 12 years, 4 months. ... Among 52 patients with Denys-Drash syndrome (DDS; 194080), WT was detected in 96% by age 5 years; the oldest DDS patient had WT detected at 6 years of age. ... Thus, PTH must be proximal to 11p13, the cytologically determined site of the Wilms tumor 'gene.' Scoggin et al. (1985) showed that E7-associated cell-surface antigen encoded by chromosome 11 and defined by a monoclonal antibody is deleted in cases of WAGR. This antigen is probably the same as that previously called 'a1' (151250). The studies in cases of WAGR with small deletions of 11p permitted regionalization of the assignment of antigen a1 to 11p13.
For a general phenotypic description and a discussion of genetic heterogeneity of Wilms tumor, see WT1 (194070). Mapping With loss of heterozygosity studies, Maw et al. (1992) concluded that a third Wilms tumor locus (WT3) is on 16q. In addition to loss on chromosome 11p (11 of 25 informative Wilms tumors), there was significant loss on 16q (9 of 45 informative tumors), while the total frequency of allele loss excluding these loci was low (9 of 426 total informative loci). They screened loci on 33 autosomal arms. The parental origin of the lost chromosome 16q allele was paternal in 4 and maternal in 4 sporadic tumors tested. Thus, unlike chromosome 11p, alleles of either parental origin are lost on 16q.
Etiology Nephroblastoma is sporadic in 99% of cases and, among these cases, 10% are associated with congenital anomalies (aniridia, hemihypertrophy, genitourinary defects) or form part of specific syndromes (Beckwith-Wiedemann, Denys-Drash, WAGR or Perlman syndromes; see these terms). ... Prognosis In the majority of cases, the prognosis is favorable with a survival rate of over 90%. Adult forms have the same prognosis and should be treated following the same methods, even when adult patients tolerate chemotherapy less well than children (which may lead to a reduction in treatment and as a result a worse prognosis).
For a general phenotypic description and discussion of genetic heterogeneity of Wilms tumor, see WT1 (194070). Clinical Features Rahman et al. (1996) described a large Canadian family with 7 confirmed cases of Wilms tumor in 3 generations. No congenital abnormalities or other cancers had been observed in the family. The average age of presentation was 5 years (age range of 2 to 12 years), which is older than the average age of diagnosis of sporadic WT (3 to 4 years). Typical triphasic histology with stromal, blastemal, and epithelial elements was found in 5 tumors, while the sixth tumor was predominantly myogenic.
Overview Wilms tumor is a rare kidney cancer that mainly affects children. Also known as nephroblastoma, it's the most common cancer of the kidneys in children. Wilms tumor most often affects children ages 3 to 4. It becomes much less common after age 5, but it can affect older children and even adults. Wilms tumor mostly occurs in just one kidney. But it can sometimes be in both kidneys at the same time. Over the years, progress in the diagnosis and treatment of Wilms tumor has greatly improved the prognosis for children with this disease.
With proper treatment, children with Wilms tumor have a 90 percent survival rate. However, the risk that the cancer will come back (recur) is between 15 and 50 percent, depending on traits of the original tumor. ... Learn more about the genes associated with Wilms tumor AMER1 CTNNB1 H19 IGF2 TP53 WT1 Additional Information from NCBI Gene: DGCR8 DROSHA POU6F2 REST Inheritance Pattern Most cases of Wilms tumor are not caused by inherited genetic factors and do not cluster in families. Approximately 90 percent of these cancers are due to somatic mutations, which means that the mutations are acquired during a person's lifetime and are present only in the tumor cells.
"Regulatory Roles of MAPK Phosphatases in Cancer" . Immune Network . 16 (2): 85–98. doi : 10.4110/in.2016.16.2.85 . ... "SOCS1 in cancer: An oncogene and a tumor suppressor". Cytokine . 82 : 87–94. doi : 10.1016/j.cyto.2016.01.005 . ... "Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment" . American Journal of Hematology . 94 (5): 604–616. doi : 10.1002/ajh.25460 . ... PMID 29988902 . ^ a b c Silva RNF, Mendonça EF, Batista AC, Alencar RCG, Mesquita RA, Costa NL (December 2019). ... PMID 28706431 . ^ Barut F, Kandemir NO, Gun BD, Ozdamar SO (July 2016). "T-cell/histiocyte-rich large B-cell lymphoma of stomach".
T-cell/histiocyte rich large B cell lymphoma (THRLBCL) is a rare variant of diffuse large B-cell lymphoma (DLBCL; see this term), mainly affecting middle-aged men and often not being discovered until an advanced disease stage, with involvement of the spleen, liver and bone marrow occurring at a greater frequency than in DLBCL. It is often difficult to diagnose due to its similarity with other lymphoid diseases such as classic Hodgkin lymphoma and nodular lymphocyte-predominant Hodgkin lymphoma (see these terms) and has an aggressive clinical course.
The role of HPV in the remaining 25-30% is not yet clear. [43] Oral sex is not risk free and results in a significant proportion of HPV-related head and neck cancer. [44] Positive HPV16 status is associated with improved prognosis over HPV-negative OSCC. [45] HPV can induce tumor by several mechanisms: [46] E6 and E7 oncogenic proteins. Disruption of tumor suppressor genes . ... A total of 11,170 people died of their disease in 2006. [80] The worldwide incidence exceeds half a million cases annually. ... Journal of the National Cancer Institute . 91 (8): 726–8. doi : 10.1093/jnci/91.8.726a . hdl : 2434/520105 . ... The New England Journal of Medicine . 340 (23): 1773–80. CiteSeerX 10.1.1.460.1056 . doi : 10.1056/NEJM199906103402301 . ... Journal of the National Cancer Institute . 97 (7): 481–8. doi : 10.1093/jnci/dji095 .
A rare, acquired peripheral neuropathy disease characterized by chronic neuropathic pain involving the sensory territory of the pudendal nerve (from clitoris to anus or from penis to anus), aggravated by sitting and for which no organic cause can be found by imaging studies or laboratory tests. It is often associated with pelvic dysfunction. Epidemiology The prevalence of Pudendal neuralgia (PN) is unknown. A female predominance is reported, with a female/male ratio of 6:4. Clinical description PN usually presents between the ages of 50-70 years. and manifests with neuropathic pain of varying intensity in the perineal region. The pain is described as an intense, sharp, burning sensation, and sometimes as numbness. Rectal or vaginal foreign body sensations (sympathalgia) are commonly reported.
Pudendal neuralgia occurs when the pudendal nerve is injured, irritated, or compressed. Symptoms include burning pain (often unilateral), tingling, or numbness in any of the following areas: buttocks, genitals, or perineum (area between the buttocks and genitals). Symptoms are typically present when a person is sitting but often go away when the person is standing or lying down. The pain tends to increase as the day progresses. Additional symptoms include pain during sex and needing to urinate frequently and/or urgently. Damage to the pudendal nerve can result from surgical procedures, childbirth, trauma, spasms of the pelvic floor muscles, or tumors.
However, the precise mechanism is not well characterized. [3] There is limited evidence implicating the large tumor antigen as responsible for inducing cellular proliferation through pathways involving phosphorylated retinoblastoma protein (pRB). [9] Diagnosis [ edit ] Overview of histological findings in trichodysplasia spinulosa. Top row (A1-A3) shows healthy control; bottom row (B1-B3) shows TS skin. Low-magnification-power overview (A1, B1); high-power representative examples of epidermis showing thickening of the skin ( acanthosis ) in TS (A2, B2); high-power representative examples of hair follicles showing enlarged and dysmorphic appearance in TS (A3, B3). Inset in B3 shows characteristic eosinophilic protein granules, probably trichohyalin (arrowheads). ... There is compelling evidence that TSPyV is the direct causative agent of TS. [2] [3] References [ edit ] ^ a b van der Meijden E, Janssens RW, Lauber C, Bouwes Bavinck JN, Gorbalenya AE, Feltkamp MC (July 2010). "Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient" . ... PMID 21801610 . ^ Gossai, A; Waterboer, T; Nelson, HH; Michel, A; Willhauck-Fleckenstein, M; Farzan, SF; Hoen, AG; Christensen, BC; Kelsey, KT; Marsit, CJ; Pawlita, M; Karagas, MR (1 January 2016).
Virus-associated trichodysplasia spinulosa is a rare infectious skin disease characterized by the development of follicular papules with keratin spicules in various parts of the body, predominantly in the face (e.g. nose, eyebrows, auricles), that is due to polyomavirus infection in immunocompromized patients.
., comparisons of unrelated patients and control subjects, had demonstrated an increased frequency of the extended HLA haplotype A1-B8-DR3 (8.1) in Caucasian MG patients with thymus hyperplasia, in women, and in patients with an early onset of disease (Fritze et al., 1974; Vieira et al., 1993; Machens et al., 1999). ... The 8.1 haplotype of the major histocompatibility complex (MHC) associates not only with the HLA DR3, B8, and A1 antigens but also alleles of many other HLA loci and extends over 3 Mb stably across generations in Caucasians.
Myasthenia gravis is a disorder that causes weakness of the skeletal muscles, which are muscles that the body uses for movement. The weakness most often starts in the muscles around the eyes, causing drooping of the eyelids (ptosis) and difficulty coordinating eye movements, which results in blurred or double vision. In a form of the disorder called ocular myasthenia, the weakness remains confined to the eye muscles. In most people with myasthenia gravis, however, additional muscles in the face and neck are affected. Affected individuals may have unusual facial expressions, difficulty holding up the head, speech impairment (dysarthria), and chewing and swallowing problems (dysphagia) that may lead to choking, gagging, or drooling.
Tola et al. (1994) examined HLA antigens in 47 Italian patients with sporadic myasthenia gravis. The frequency of B8 and DR3 in patients was 19.1 and 27.3%, respectively, compared to 9.7 and 14.1% in controls. There was also an association between the B8 allele and early onset of generalized myasthenia gravis sustained by thymic hyperplasia. ... Namba et al. (1971) pointed out, on the basis of 85 families with multiple cases (excluding transient neonatal myasthenia in offspring of myasthenic mothers), that the familial cases most often involved sibs. ... The authors also noted that the younger sister had recently had 3 children and, unlike her sister, was homozygous for the HLA-DR3-B8-A1 phenotype, which is known to associate with autoimmune myasthenia gravis. ... Lin et al. (2002) demonstrated enhanced susceptibility to experimental autoimmune myasthenia gravis in mice lacking decay-accelerating factor (DAF; 125240), an intrinsic complement regulator. Following anti-AChR Ab injection, Daf1 -/- mice (devoid of neuromuscular DAF protein) showed dramatically greater muscle weakness than their Daf1 +/+ littermates.
Myasthenia gravis (MG) is a chronic autoimmune neuromuscular disease characterized by weakness of the skeletal muscles. Common symptoms include weakness of the muscles that control the eye and eyelid, facial expressions, chewing, talking, and swallowing. Weakness tends to increase during periods of activity and improve after periods of rest. The condition results from a defect in the transmission of nerve impulses to muscles, which is due to the presence of antibodies against acetylcholine . The exact reason this occurs is not known. Some cases have been linked to tumors in the thymus gland .
Overview Myasthenia gravis (my-us-THEE-nee-uh GRAY-vis) causes muscles under your voluntary control to feel weak and get tired quickly. This happens when the communication between nerves and muscles breaks down. There's no cure for myasthenia gravis. Treatment can help with symptoms. These symptoms can include weakness of arm or leg muscles, double vision, drooping eyelids, and problems with speaking, chewing, swallowing and breathing. This disease can affect people of any age, but it's more common in women younger than 40 and in men older than 60.
Clinical Features Among 314 patients with classic myasthenia gravis (MG; 254200), Oh and Kuruoglu (1992) found 12 (3.8%) who presented with chronic limb-girdle myasthenia. None of the 12 patients had oculobulbar involvement, which had initially excluded the diagnosis of myasthenia gravis. Disease onset occurred between the ages of 28 and 69 years and was characterized by lower proximal muscle weakness, usually followed by upper proximal muscle weakness. All patients showed a decremental response to repetitive nerve stimulation, and all patients had normal serum creatine kinase. Three patients had thymic hyperplasia, 1 patient had systemic lupus erythematosus (SLE; 152700), and 1 patient had hyperthyroidism.
Myasthenia gravis (MG) is a rare, clinically heterogeneous, autoimmune disorder of the neuromuscular junction characterized by fatigable weakness of voluntary muscles. Epidemiology The prevalence is estimated to be 1/5,000 and the incidence to be 1/250,000 to 1/33,000 in Europe. MG affects both males and females: mainly females before the age of 40 years and males and females equally after the age of 50 years. Clinical description Myasthenia gravis can develop at any age but there is a bimodal peak in the age of onset in the adult-onset form, with primarily female patients before 40 years of age, and primarily males after 50 years of age (adult-onset myasthenia gravis; see this term). Patients have fluctuating weakness, worsening with repetitive activities, heat and stress while improving with rest and with involvement of skeletal muscle groups of ocular, bulbar, extremities and neck.
American Journal of Obstetrics and Gynecology . 199 (5): 514.e1–514.e8. doi : 10.1016/j.ajog.2008.03.050 . ... American Journal of Obstetrics and Gynecology . 201 (4): 417.e1–417.e7. doi : 10.1016/j.ajog.2009.07.046 . ... American Journal of Obstetrics and Gynecology . 198 (2): e4–e7. doi : 10.1016/j.ajog.2007.08.073 .
With each subsequent cycle of chemotherapy, the reaction will appear more quickly, be more severe and will take longer to heal. [24] History [ edit ] Hand-foot syndrome was first reported in association with chemotherapy by Zuehlke in 1974. [25] Synonyms for acral erythema (AE) include: hand-foot syndrome, palmar-plantar erythrodysesthesia, peculiar AE, chemotherapy-induced AE, toxic erythema of the palms and soles, palmar-plantar erythema, and Burgdorf's reaction. ... Palmar-plantar rash with cytarabine therapy". N. Engl. J. Med . 364 (3): e5. doi : 10.1056/NEJMicm1006530 . PMID 21247311 . ^ a b c Baack BR, Burgdorf WH (1991). ... Dermatol . 24 (3): 457–61. doi : 10.1016/0190-9622(91)70073-b . PMID 2061446 . ^ Demirçay Z, Gürbüz O, Alpdoğan TB, Yücelten D, Alpdoğan O, Kurtkaya O, Bayik M (1997). ... "Pyridoxine therapy for palmar-plantar erythrodysesthesia associated with taxotere" . J. Natl. Cancer Inst . 85 (17): 1432–3. doi : 10.1093/jnci/85.17.1432 . ... "Erythematous eruption of the palms and soles associated with mitotane therapy". Dermatologica . 148 (2): 90–2. doi : 10.1159/000251603 . PMID 4276191 .
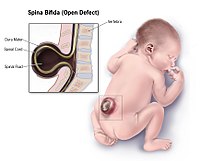
A group of rare neural tube defect disorders characterized by improper closure of the spinal column during embryonal development, not associated with other major congenital malformations nor ventriculomegaly. The extent of the closure defect may vary, ranging from spina bifida occulta, in which the site of the lesion is not exposed (e.g. an isolated posterior vertebral arch defect), to spina bifida aperta, in which the lesion may be conformed of proturding spinal cord and meninges (myelomeningocele) or meninges exposure only (meningocele), with or without a proturding sac at the site of the lesion, to the most severe defect which includes total exposure of the spinal cord along its full length (rachischisis). Depending on the type, size and site of the defect, severe morbidity, typically inlcuding motor, sensory and sphincter dysfunction, and mortality may be associated. Spina bifida occulta may be asymptomatic.
Christensen et al. (1999) assessed genotypes and folate status in 56 patients with spina bifida, 62 mothers of patients, 97 children without NTDs (controls), and 90 mothers of controls to determine the impact of these factors on NTD risk.
For a general phenotypic description and a discussion of genetic heterogeneity of neural tube defects, see 182940 and 601634. Inheritance Toriello et al. (1980) observed either anencephaly or spina bifida in 5 males in 5 different sibships spanning 4 generations genealogically connected through females. Baraitser and Burn (1984) and Toriello (1984) reported additional kindreds with pedigree patterns strongly supporting X-linked recessive inheritance. Baraitser and Burn (1984) reported a nonconsanguineous Pakistani Muslim family in which a woman had 3 brothers and 3 sons with neural tube defects, including posterior encephalocele and spina bifida cystica. Jensson et al. (1988) reported an Icelandic family in which 5 males had either anencephaly or spina bifida: 2 had spina bifida, 2 sibs had anencephaly, and 1 had both high and low spinal lesions.
Overview Spina bifida is a birth defect that occurs when the spine and spinal cord don't form properly. It's a type of neural tube defect. The neural tube is the structure in a developing embryo that eventually becomes the baby's brain, spinal cord and the tissues that enclose them. Typically, the neural tube forms early in pregnancy and it closes by the 28th day after conception. In babies with spina bifida, a portion of the neural tube doesn't close or develop properly, causing problems in the spinal cord and in the bones of the spine. Spina bifida can range from mild to severe, depending on the type of defect, size, location and complications.
Spina bifida is a type of neural tube defect in which the neural tube (the structure in an embryo that becomes the brain and spinal cord) does not completely close during development in the womb. This may result in part of the spinal cord sticking out through an opening in the spine, leading to permanent nerve damage. Babies born with spina bifida often have a fluid-filled sac, covered by skin, on their back. This is called a meningocele . If the sac contains part of the spinal cord and its protective covering, it is known as a myelomeningocele . The signs and symptoms of spina bifida can range from mild to severe, depending on the location and extent of spinal cord involvement.
A number sign (#) is used with this entry because of evidence that susceptibility to the development of neural tube defects (NTDs) is conferred by variation in the VANGL1 (610132), VANGL2 (600533), CELSR1 (604523), or FUZ (610622) genes. An association has been reported with variants in the T locus (601397) on chromosome 6q. Description Neural tube defects are the second most common type of birth defect after congenital heart defects. The 2 most common NTDs are open spina bifida, also known as spina bifida cystica (SBC) or myelomeningocele, and anencephaly (206500) (Detrait et al., 2005). Spina bifida occulta (SBO), a bony defect of the spine covered by normal skin, is a mild form of spina bifida that is often asymptomatic.
The risks of maternal chorioamnionitis or fetal death as a result of the fetoscopic procedure run below 5%. [92] [93] [94] Women are discharged home from hospital one week after the procedure. ... Part A, Clinical and Molecular Teratology . 97 (7): 437–43. doi : 10.1002/bdra.23153 . ... PMID 9055372 . S2CID 41462354 . ^ Iwamoto J, Abe H, Tsukimura Y, Wakano K (2005). ... American Journal of Sports Medicine . 32 (3): 781–86. doi : 10.1177/0363546503261721 . ... Archives of Physical Medicine and Rehabilitation . 88 (8): 1064–73. doi : 10.1016/j.apmr.2007.04.018 .
The American Journal of Medicine . 19 (1): 78–86. doi : 10.1016/0002-9343(55)90276-X . ... "Prophylactic diet: A treatment for night eating syndrome". Hypothesis . 10 (1): e5. ^ a b c d e Young, SN (2007), "How to increase serotonin in the human brain without drugs", J Psychiatry Neurosci , 32 (6): 394–399, PMC 2077351 , PMID 18043762 . ^ O'Reardon J.P.; Stunkard A.J.; Allison K.C. (2004).
Signs and symptoms of foreign body aspiration vary based on the site of obstruction, the size of the foreign body, and the severity of obstruction. [2] 20% of foreign bodies become lodged in the upper airway, while 80% become lodged in a bronchus . [6] Signs of foreign body aspiration are usually abrupt in onset and can involve coughing, choking, and/or wheezing ; however, symptoms can be slower in onset if the foreign body does not cause a large degree of obstruction of the airway. [2] With this said, aspiration can also be asymptomatic on rare occasions. [1] Classically, patients present with acute onset of choking. [2] In these cases, the obstruction is classified as a partial or complete obstruction. [2] Signs of partial obstruction include choking with drooling, stridor , and the patient maintains the ability to speak. [2] Signs of complete obstruction include choking with inability to speak or absence of bilateral breath sounds among other signs of respiratory distress such as cyanosis . [2] A fever may be present. ... Retrieved 2008-12-16 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa abac ad ae Federico, Monica (2018). ... Current Diagnosis & Treatment: Emergency Medicine, 8e, "Respiratory Distress" . New York, NY: McGraw-Hill. ... "Pediatric Foreign Body Aspiration" . Pediatrics in Review . 21 (3): 86–90. doi : 10.1542/pir.21-3-86 . ISSN 0191-9601 . ... Annals of Emergency Medicine . 66 (6): 570–582.e5. doi : 10.1016/j.annemergmed.2015.07.499 .
Mapping By HLA typing of 62 unrelated IgA-deficient blood donors, Oen et al. (1982) showed a significant increase in the prevalence of HLA-B8. In the family of a 57-year-old woman with IgA deficiency and Still disease, Lakhanpal et al. (1988) found a suggestion of linkage to the HLA haplotype A1-B8. The maternal HLA-A1-B8 haplotype was associated with IgA deficiency in all 3 of her children, whereas all 5 family members with exclusively paternally derived A1-B8 haplotype had normal IgA levels. In a third generation, of 3 family members whose A1-B8 haplotype was of indeterminate origin--that is, potentially either maternally or paternally derived--2 had IgA deficiency and 1 did not. ... Both the recipient and the donor were homozygous HLA-A1, B8, DR3, a haplotype associated with selective IgA deficiency.
Caister Academic Press . ISBN 978-1-904455-99-8 . ^ a b Poulin DL, DeCaprio JA (September 2006). ... PMID 16963733 . ^ Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX (February 2002). "Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control" . ... New Science Press. ISBN 978-1-904455-99-8 . ^ a b c Levine AJ (February 2009). ... PMID 19150725 . ^ Scheffner M, Huibregtse JM, Vierstra RD, Howley PM (November 1993). "The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53". ... PMID 18202256 . ^ Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (December 1990).
Most deletions were of the AZFc-b2/b4 subtype and were associated with a variable spermatogenic phenotype, with sperm present in 72% of cases. ... The authors identified 18 cases of partial AZFc deletions in the infertile group (5.3%) and 1 case in the control group (0.4%); 17 deletions had the so-called gr/gr pattern, 1 had the b2/b3 pattern, and 1 represented a novel deletion with breakpoints in the b3 and b4 amplicons. ... Haplogroup Q1 was found to be gr/gr-deleted, and fixation of the b2/b3 deletion was confirmed in haplogroup N. ... Thirty-one men (3.7%) had deletion of 1 or more STS markers: 22 had gr/gr deletions, 4 had b2/b4 deletions, 4 had b2/b3 deletions, and 1 had a b1/b3 deletion. ... Rozen et al. (2012) screened 20,884 anonymized DNA samples from men of 5 populations (India, Poland, Tunisia, United States, and Vietnam) for 6 recurrent interstitial deletions in the AZFc region, and found that 1 of every 27 men carried 1 of 4 deletions: gr/gr, b2/b3, b1/b3, and b2/b4, in descending order of prevalence.
In this region, the structure of the Y-chromosome is rich in repeated palindromes and recombination between two flanking sequences sharing a high degree of homology leads to various deletions: 1) AZFa deletions (recombination between the sequences HERV15yq1 and HERV15yq2 ); the rarest - ii) AZFb or P5/proximal-P1 deletions, iii) AZFb+c deletions, of which two types are distinguished: P5/distal-P1 or P4/distal-P1 and iv) AZFc deletions caused by recombination between palindromes b2 and b4. Diagnostic methods Diagnosis is made on the basis of azoospermia or oligozoospermia in otherwise healthy males after exclusion of other causes of infertility.
Y chromosome microdeletion (YCM) is a family of genetic disorders caused by missing gene(s) in the Y chromosome . Many men with YCM exhibit no symptoms and lead normal lives. However, YCM is also known to be present in a significant number of men with reduced fertility . [1] Men with reduced sperm production (in up to 20% of men with reduced sperm count , some form of YCM has been detected [2] ) varies from oligozoospermia , significant lack of sperm, or azoospermia , complete lack of sperm. Contents 1 Cause 2 Diagnosis 3 Infertility 4 See also 5 References 6 Further reading Cause [ edit ] The mechanism of mutation is not different for Y-chromosome microdeletion. However, the ability to repair it differs from other chromosomes. The human Y chromosome is passed directly from father to son, and is not protected against accumulating copying errors, whereas other chromosomes are error corrected by recombining genetic information from mother and father. This may leave natural selection as the primary repair mechanism for the Y chromosome.
A number sign (#) is used with this entry because Sertoli cell-only (SCO) syndrome has been found to be associated with interstitial deletions in the 'azoospermia factor' (AZF) region on the long arm of the Y chromosome, particularly deletions of the AZFa region, which includes the ubiquitin-specific protease 9 gene (USP9Y; 400005), the DEAD/H box 3 gene (DBY; 400010), and the ubiquitously transcribed tetratricopeptide repeat gene (UTY; 400009). Description In the evaluation of male infertility, the Sertoli cell-only (SCO) syndrome is diagnosed on testicular biopsy when either no germ cells are visible in any seminiferous tubules (SCO type I) or germ cells are present in a minority of tubules (SCO type II). It is believed that the latter variant arises from a failure to complete differentiation and maturation of spermatocytes and spermatids, leading to degeneration of germ cells within most tubules (Sargent et al., 1999). Another, possibly X-linked, form of Sertoli cell-only syndrome has also been reported (305700). Heterogeneity of Spermatogenic Failure See 415000 for a general discussion of the AZF region of the Y chromosome and Y-linked nonobstructive spermatogenic failure.
If ACE-I is not well tolerated by the patient, it can be replaced by angiotensin receptor blockers (ARB). Hydralazine with nitrates may replace ACE-I in breastfeeding mothers or before delivery; however, evidence suggests that this course of treatment may not be as effective as ACE-I but beneficial when necessary. [2] [4] [5] [6] [8] [10] [15] If EF is less than 35%, anticoagulation is indicated, as there is a greater risk of developing left ventricular thrombi (blood clots). ... PMID 15496265 . ^ Ansari AA, Fett JD, Carraway RE, Mayne AE, Onlamoon N, Sundstrom JB (December 2002). ... "Myocarditis and long-term survival in peripartum cardiomyopathy". Am. Heart J . 140 (5): 785–91. doi : 10.1067/mhj.2000.110091 . PMID 11054626 . ^ a b Palmer BA, Janosko KM, McTiernan C, Sherman F, McNamara DM (2007). ... "Frequency of peripartum cardiomyopathy". Am. J. Cardiol . 97 (12): 1765–8. doi : 10.1016/j.amjcard.2006.01.039 .
Characterization of lymphocyte responses to Ca2+ in Scott syndrome. Thromb Haemost 2004; 91:412-415 ^ Sims PJ, Wiedmer T. Unraveling the mysteries of phospholipid scrambling. Thromb Haemost 2001; 86:266-275 ^ Zhou Q, Sims PJ, Wiedmer T. ... Platelet activation and blood coagulation, Thromb Haem 2002; 88:186-194 Martinez MC, Martin S, Toti F, Fressinaud E, Dachary-Prigent J, Meyer D, et al. ... Store-mediated Ca2+ entry in the regulation of phoshatidylserine exposure in blood cells from Scott patients. Thromb Haemost 2003; 89:687-695 Weiss HJ, Vicic WJ, Lages BA, Rogers J. ... Production and characterization of transformed B-lymphocytes expressing the membrane defect of Scott Syndrome. J Clin Invest 1994; 94:2237-2244 Stout JG, Basse F, Luhm RA, Weiss HJ, Wiedmer T, Sims PJ.
A number sign (#) is used with this entry because of evidence that Scott syndrome (SCTS) is caused by homozygous mutation in the TMEM16F gene (608663) on chromosome 12q12. Description Scott syndrome is a mild platelet-type bleeding disorder characterized by impaired surface exposure of procoagulant phosphatidylserine (PS) on platelets and other blood cells, following activation with Ca(2+)-elevating agents (Munnix et al., 2003). Clinical Features In 4 generations of a family (with 1 instance of male-to-male transmission), Robinson et al. (1967) described a mild bleeding disorder with no spontaneous hemorrhage. Analysis of blood coagulation in 2 generations revealed normal values for all clotting factors and other hemostatic systems except prothrombin consumption and thromboplastin inactivation. Robinson et al. (1967) demonstrated the presence of what they termed an 'inactivator' of active factor X (F10; 613872) in the plasma of these patients which accelerated the decay of blood thromboplastin.
Causes [ edit ] The cause is not known but is often associated with some: fetal chromosomal anomalies like triploidy intra uterine infections premature rupture of membrane drugs; COX inhibitors like indomethacin, ACE inhibitors renal agenesis or obstruction of the urinary tract of the fetus preventing micturition such as posterior urethral valves in males intrauterine growth restriction (IUGR) associated with placental insufficiency amnion nodosum ; failure of secretion by the cells of the amnion covering the placenta postmaturity (dysmaturity) Diagnosis [ edit ] uterine size is much smaller than the period of amenorrhoea fewer fetal movements, the uterus "full of fetus" because of scanty liquid, malpresentation ( breech ) evidences of IUGR of the fetus, sonographic diagnosis is made when largest liquid pool is less than 2 cm, visualization of normal filling and emptying of fetal bladder essentially rule out urinary tract abnormality, Oligohydramnios with fetal symmetric growth retardation is associated with increased chromosomal abnormality. ... CS1 maint: DOI inactive as of December 2020 ( link ) ^ Johnson JM, Chauhan SP, Ennen CS, Niederhauser A, Magann EF (2007). "A comparison of 3 criteria of oligohydramnios in identifying peripartum complications: a secondary analysis". ... Gynecol . 197 (2): 207.e1–7, discussion 207.e7–8. doi : 10.1016/j.ajog.2007.04.048 .
Train-of-four ratio (TOFR): [ edit ] A TOF ratio (TOFR) is calculated by dividing the amplitude of the fourth response by the amplitude of the first response (requires an quantitative measure of the response to stimulation). [13] Train-of-four count (TOFC) [ edit ] The TOF count (TOFC) is defined as the "number of detectable evoked responses, and it correlates with the degree of neuromuscular block, as follows: TOFC = 1 : >95 percent of nicotinic acetylcholine receptors (nAChRs) blocked TOFC = 2 : 85 to 90 percent of nAChRs blocked TOFC = 3 : 80 to 85 percent of nAChRs blocked TOFC = 4 : 70 to 75 percent of nAChRs blocked [13] Train-of-four ratio <0.9 [ edit ] Data suggests that a TOF ratio measured qualitatively with EMG, MMG, or AMG must reach the threshold value of >0.9 to assure recovery of neuromuscular function. ... New England Journal of Medicine . 378 (4): e6. doi : 10.1056/nejmvcm1603741 . ISSN 0028-4793 . ... "Advances in Neurobiology of the Neuromuscular Junction: Implications for the Anesthesiologist". Anesthesiology . 96 (1): 202–231. doi : 10.1097/00000542-200201000-00035 . ... S2CID 9670756 . ^ Bhananker SM, Treggiari MM, Sellers BA, Cain KC, Ramaiah R, Thilen SR (October 2015). ... "Efficacy of Tactile-guided Reversal from Cisatracurium-induced Neuromuscular Block". Anesthesiology . 96 (1): 45–50. doi : 10.1097/00000542-200201000-00013 .