-
Bronchiectasis With Or Without Elevated Sweat Chloride 2
OMIM
He had a plasma renin of 1 ng/ml/h, plasma aldosterone of 101 pg/ml, and he had been hypertensive for several years (160/90). ... Mutesa et al. (2009) analyzed the CFTR gene in 60 unrelated Rwandan children who had CF-like symptoms and identified heterozygosity for a CFTR mutation in 5 patients (none were homozygous). Sequencing of the genes encoding the 3 subunits of the epithelial sodium channel (ENaC) revealed heterozygous mutations in SCNN1A and SCNN1B (600760) in 4 patients, respectively, whereas the remaining patient was heterozygous for a mutation in both SCNN1B and SCNN1G (600761).
-
Gastrinoma
Wikipedia
One fourth of gastrinomas are related to Multiple endocrine neoplasia type 1 , Zollinger- Ellison Syndrome , Peptic Ulcer disease . [4] Contents 1 Signs and symptoms 2 Pathophysiology 3 Diagnosis 4 Treatment 5 Prognosis 6 Epidemiology 7 Research 8 See Also 8.1 References 8.2 External links Signs and symptoms [ edit ] Gastrinoma in the early stages will have signs and symptoms of indigestion or similar to Irritable Bowl disease (IBD) such as: Hypergastrinemia Ulcers of the duodenum , stomach, and small intestine. ... Pain and Bleeding in stool. Obstruction of intestine. [5] Weight loss/ poor appetite Anemia (Due to Vitamin B12 malabsorption, and bleeding) Hematemesis Acid Reflux Esophageal complications ( Barrett esophagus , esophagitis , stricture formation) [6] Vomiting Steatorrhea [4] Depression Pathophysiology [ edit ] Gastrin is secreted by the G cells . ... The normal levels of gastrin are 150 pg/mL ( > 72.15 pmol/L); therefore elevated levels of > 1000 pg/mL (> 480 pmol/L) would establish the diagnosis of gastrinoma. [12] Another test that can be conducted is the secretin stimulated test, [4] which is useful in patents who have the sign and symptoms of gastrinoma but the gastrin levels are below < 1000 pg/mL. ... If patients present with hepatic metastases they might have remaining life span of 1 year with a 5 year survival rate of 20-30%. In patients with localized tumor or localized lymph spread the survival rate of 5 years id 90%.MEN1, GAST, CHGA, SST, SCT, CDKN2A, ACTB, SCTR, GRPR, GRP, ERBB2, POTEF, CASR, MET, NMBR, POMC, PYGM, S100B, BBS2, ATP4A, ATP12A, SSTR5, TCF3, TNFRSF1B, TP53, VIP, KHSRP, PSIP1, SIGLEC7, NMB, CUX1, EGF, SMAD4, EGFR, CDKN2D, CDKN2B, GFAP, GH1, FFAR1, CDH1, CD44, HCLS1, HGF, APC, IGF1, IGF1R, IGFBP1, IL2RB, HRH2

-
Acute Motor Axonal Neuropathy
Wikipedia
Contents 1 Causes 2 Diagnosis 3 Treatment 4 History 5 References 6 External links Causes [ edit ] A link to Campylobacter jejuni was suspected when a young girl was admitted to Second Teaching Hospital. ... These cases showed deposition of antibody and complement along the motor axolemma and associated macrophage infiltration. [5] References [ edit ] ^ Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, Asbury AK, Blaser MJ, McKhann GM (1995). ... Lippincott Williams and Wilkins; 2002:Pg 361 ^ McKhann GM, Cornblath DR, Ho T, Li CY, Bai AY, Wu HS, Yei QF, Zhang WC, Zhaori Z, Jiang Z, et al. (1991).
-
Anosodiaphoria
Wikipedia
Contents 1 Causes 2 Neurology 3 Treatment 4 Research 5 See also 6 Footnotes Causes [ edit ] A few possible explanations for anosodiaphoria exist: The patient is aware of the deficit but does not fully comprehend it or its significance for functioning May be related to an affective communication disorder and defective arousal. These emotional disorders cannot account for the verbal explicit denial of illness of anosognosia . [5] Other explanations include reduced emotional experience, impaired emotional communication, alexithymia , behavioral abnormalities , dysexecutive syndrome , and the frontal lobes . [6] Neurology [ edit ] Anosodiaphoria occurs after stroke of the brain. 27% of patients suffering from an acute hemispheric stroke suffer the stroke in the right hemisphere, while 2% suffer it in their left. [7] Anosodiaphoria is thought to be related to unilateral neglect , a condition often found after damage to the non-dominant (usually the right) hemisphere of the cerebral cortex in which sufferers seem unable to attend to, or sometimes comprehend, anything on a certain side of their body (usually the left). ... New York, New York: Oxford University Press https://books.google.com/books?id=d4S-T0NboMQC&pg=PA89&lpg=PA89&dq=treatment+for+anosodiaphoria&source=bl&ots=Pe3e3wmEDI&sig=Xh5nwCPHgBvxrJqWfYuicyOMJ4E&hl=en&sa=X&ei=DiipT5yANcmJ6AHT7JHKBA&ved=0CEsQ6AEwAQ#v=onepage&q&f=false . ^ Prigatano, G. (1991).
-
Mucolipidosis Iv
GeneReviews
Neurodegeneration is thought to occur in no more than 15% of individuals. About 5% of individuals have atypical mucolipidosis IV, often manifest as less severe psychomotor retardation and/or eye findings. ... Plasma gastrin concentration is elevated in virtually all individuals with mucolipidosis IV (mean 1507 pg/mL; range 400-4100 pg/mL) (normal 0-200 pg/mL) [Schiffmann et al 1998, Altarescu et al 2002]. ... In individuals of Ashkenazi Jewish ancestry targeted analysis for the two common pathogenic variants – c.406-2A>G and a 6.4-kb deletion beginning in the 5’UTR and extending into exon 6 – can be performed first, as they account for 95% of pathogenic variants in this population. ... For issues to consider in interpretation of sequence analysis results, click here. 5. Cannot detect 6.4 kb del, one the two pathogenic variants common in persons of Ashkenazi Jewish heritage 6. ... The phenotype in affected individuals can be either typical (~95% of individuals) or atypical (~5% of individuals) [Altarescu et al 2002].
-
Blue Toe Syndrome
Wikipedia
"Blue toe syndrome: treatment with intra-arterial stents and review of therapies". J Vasc Interv Radiol . 11 (5): 585–92. doi : 10.1016/s1051-0443(07)61610-8 . ... "Evaluation and management of cholesterol embolization and the blue toe syndrome". Curr Opin Cardiol . 11 (5): 533–42. doi : 10.1097/00001573-199609000-00013 . ... "Making the diagnosis when the patient has 'blue toes ' ". Geriatrics . 49 (12): 37–9, 43–5. PMID 7982584 . ^ Kopani K, Liao S, Shaffer K (2009). ... "Making the diagnosis when the patient has 'blue toes ' ". Geriatrics . 49 (12): 37–9, 43–5. PMID 7982584 . ^ Blackshear JL, Oldenburg WA, Cohen MD (Dec 1994). "Making the diagnosis when the patient has 'blue toes ' ". Geriatrics . 49 (12): 37–9, 43–5. PMID 7982584 . ^ Blackshear JL, Oldenburg WA, Cohen MD (Dec 1994).
-
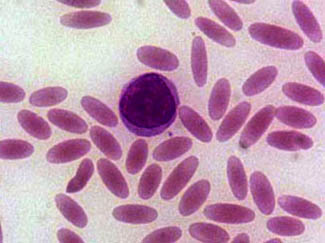
Cigar Cell
Wikipedia
Churchill Livingstone Publishing, ISBN 0-443-06556-X , pg. 276-78 External links [ edit ] Classification D ICD - 10 : D58.1 ICD - 9-CM : 282.1 MeSH : D004612 DiseasesDB : 4172 External resources eMedicine : ped/987 med/648 v t e Myeloid blood cells and plasma Hematopoiesis Myelopoiesis ( CFU-GEMM ) CFU-GM Granulopoiesis Myeloblast Promyelocyte Myelocyte Metamyelocyte Band cell Monocytopoiesis Monoblast Promonocyte MEP Thrombopoiesis Megakaryoblast Promegakaryocyte Erythropoiesis Proerythroblast Normoblast Reticulocyte General Extramedullary hematopoiesis Myeloid tissue Granulocytes Myeloblast Band cell Neutrophil Basophil CFU-Baso Eosinophil CFU-Eos Mast cell CFU-Mast Monocytes Macrophages Histiocytes Kupffer cells Alveolar macrophage Microglia Osteoclasts Epithelioid cells giant cells Langhans giant cells Foreign-body giant cell Touton giant cells Other Antigen-presenting cells Dendritic cells Langerhans cell CFU-DL Monoblast MPS Platelets CFU-Meg Megakaryoblast Promegakaryocyte Megakaryocyte Red blood cells Reticulocyte Nucleated red blood cell CFU-E Immune response Leukocyte extravasation Phagocytosis Intrinsic immunity Other Precursor cells CFU-GM Megakaryocyte–erythroid progenitor cell CFU-GEMM Myelomonocyte Other Phagocyte Plasma Hematopoietic system Hematopoietic stem cell This article related to pathology is a stub .
-
Canalicular Adenoma
Wikipedia
Although seldom necessary, a pathologist can do immunohistochemistry studies to confirm the diagnosis, with the cells strongly reactive with pancytokeratin, S100 protein and SOX10, with a delicate GFAP reaction around the periphery. [5] [1] [6] [7] Even though it is a benign tumor, it must be separated from a basal cell adenoma, pleomorphic adenoma, adenoid cystic carcinoma, and polymorphous adenocarcinoma. ... PMID 4709966 . ^ Suarez P, Hammond HL, Luna MA, Stimson PG (Aug 1998). "Palatal canalicular adenoma: report of 12 cases and review of the literature".

-
Corticobasal Syndrome
Wikipedia
Corticobasal syndrome (CBS) is a rare, progressive atypical Parkinsonism syndrome and is a tauopathy related to frontotemporal dementia . [1] [2] CBS is typically caused by the deposit of tau proteins forming in different areas of the brain. [1] [3] Contents 1 Classification 2 Symptoms 3 Pathophysiology 4 Diagnosis 4.1 Differential 5 Prognosis 6 References Classification [ edit ] CBS is the most common type of corticobasal degeneration (CBD) although the terms CBD and CBS have been used interchangeably in the past. [2] The other three phenotypes of CBD are: frontal-behavioral dysexecutive-spatial syndrome (FBS) nonfluent/agrammatic variant of primary progressive aphasia (naPPA), and progressive supranuclear palsy syndrome (PSPS). [1] [4] Symptoms [ edit ] Symptoms of CBS include apraxia , alien limb phenomenon, frontal deficits, and extrapyramidal motor symptoms such as myoclonus or rigidity. [5] Movement deficits often begin on one side and progress to the other. [1] Pathophysiology [ edit ] CBD is the pathology underlying approximately 50% of CBS cases. [6] Diagnosis [ edit ] The Armstrong criteria were proposed in 2013; the accuracy of these is limited and further research is needed. [7] Symptoms may be symmetric or asymmetric, with one or more of the following: limb rigidity or akinesia limb dystonia limb myoclonus, plus one of: orobuccal or limb apraxia cortical sensory deficit alien limb phenomena (more than simple levitation) The onset is insidious with gradual progression, lasting one year or more, with no exclusion criteria present. ... PMID 29213468 . ^ a b c Constantinides VC, Paraskevas GP, Paraskevas PG, Stefanis L, Kapaki E (August 2019). ... "Corticobasal syndrome: neuroimaging and neurophysiological advances". Eur. J. Neurol . 26 (5): 701–e52. doi : 10.1111/ene.13928 . hdl : 11573/1225619 . ... Neurology (Multicenter study). 80 (5): 496–503. doi : 10.1212/WNL.0b013e31827f0fd1 .
-
Pancoast Tumor
Wikipedia
Pancoast tumors are named for Henry Pancoast , an American radiologist , who described them in 1924 and 1932. [1] Contents 1 Signs and symptoms 2 Diagnosis 3 Treatment 4 References 5 External links Signs and symptoms [ edit ] Aside from cancer general symptoms such as malaise, fever, weight loss and fatigue, Pancoast tumor can include a complete Horner's syndrome in severe cases: miosis (constriction of the pupils), anhidrosis (lack of sweating), ptosis (drooping of the eyelid), and pseudoenophathalmos (because of the ptosis). ... Surgical access may be via thoracotomy from the back [2] or the front of the chest [3] and modifications. [4] Nonsurgical treatment may consist of radiation therapy alone or clinical trials of new combinations of treatment. [5] CT scan showing a Pancoast tumor (labeled as P, non-small cell lung carcinoma, right lung), from a 47-year-old female smoker References [ edit ] ^ synd/2953 at Who Named It? ... "Results of bronchoplastic procedures for bronchogenic carcinoma" . Ann. Surg . 151 (5): 729–40. doi : 10.1097/00000658-196005000-00013 . PMC 1613696 . PMID 14431029 . ^ Dartevelle PG, Chapelier AR, Macchiarini P, et al. ... Surg . 63 (2): 563–6. doi : 10.1016/S0003-4975(96)01023-5 . PMID 9033349 . Anterior Access for radical resection of Pancoast tumors on YouTube ^ "Non-Small Cell Lung Cancer Treatment (PDQ®)–Patient Version - National Cancer Institute" . www.cancer.gov . 2020-05-22 .

-
Cutis Verticis Gyrata
Wikipedia
The condition was first reported by Jean-Louis-Marc Alibert in 1837, [3] who called it cutis sulcata . [4] A clinical description of the condition was provided by Robert [ who? ] in 1843 [5] and it was named by Paul Gerson Unna in 1907. [6] It has also been called Robert-Unna syndrome , bulldog scalp , corrugated skin , cutis verticis plicata , and pachydermia verticis gyrata . [7] Contents 1 Cause 2 Diagnosis 2.1 Classifications 3 Treatment 4 See also 5 References 6 External links Cause [ edit ] At this time, causes are unknown, but it is believed to not be congenital. ... This occurs mainly in men, with a male:female ratio of 5:1 or 6:1, and develops during or soon after puberty. ... "Primary essential cutis verticis gyrata in an adult female patient: a case report". J. Dermatol . 33 (7): 492–5. doi : 10.1111/j.1346-8138.2006.00116.x . ... Cite journal requires |journal= ( help ) ^ Unna PG. Cutis verticis gyrata. Monatschr Prakt Derm. 1907. 45:227-33. ^ Levine, Norman (2004).

-
Hellp Syndrome
Wikipedia
"Long-term maternal and subsequent pregnancy outcomes 5 years after hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome". Am J Obstet Gynecol . 201 (4): 385 e1–5. doi : 10.1016/j.ajog.2009.06.033 . ... PMID 16631593 . ^ Martin JN Jr, Blake PG, Perry KG, Jr, McCaul JF, Hess LW, Martin RW (June 1992). ... "Preeclampsia: an endothelial cell disorder". Am J Obstet Gynecol . 161 (5): 1200–4. doi : 10.1016/0002-9378(89)90665-0 . ... "Preeclampsia/eclampsia with hemolysis, elevated liver enzymes, and thrombocytopenia". Obstet Gynecol . 66 (5): 657–60. PMID 4058824 . ^ Martin JN Jr, Blake PG, Lowry SL, Perry KG Jr, Files JC, Morrison JC (November 1990).CD46, CFH, CFI, HELLPAR, FASLG, FAS, PGF, F5, LEP, HADHA, F2, TNF, HPGDS, LGALS13, FLT1, VEGFA, MTHFR, MAPK14, TLR4, AIMP2, TLR2, MAPK3, TPBG, VEGFC, TGFB3, VWF, MAPK1, ABCG2, TFPI2, IL18R1, GRAP2, EBI3, AHSA1, ADAMTS13, SIRT4, RNF19A, POLDIP2, SLC17A5, ERVW-1, MBL3P, AHSP, NOD2, POTEF, SERPINE2, ACTB, SERPINE1, PAH, APC, CFB, CA9, CD40LG, CD59, CDKN1C, COX8A, CP, CRK, ENG, EPHX1, GAPDH, GNB3, GPT, NR3C1, GSTM1, GSTT1, HSPA4, HSPG2, IFNG, IL1B, IL1RN, CXCL8, IL10, LEPR, LNPEP, ADM, NOS3, PAEP, MBL2
-
Somatostatinoma
Wikipedia
Contents 1 Pathophysiology 2 Diagnosis 3 Treatment 4 References 5 External links Pathophysiology [ edit ] Main article: Somatostatin In a normal subject actions of somatostatin include: [ citation needed ] In the anterior pituitary gland , the effects of somatostatin are: Inhibit the release of growth hormone thus opposing the effects of growth hormone-releasing hormone (GHRH) Inhibit the release of thyroid-stimulating hormone (TSH) Somatostatin suppresses the release of gastrointestinal hormones Gastrin Cholecystokinin (CCK) Secretin Motilin Vasoactive intestinal peptide (VIP) Gastric inhibitory polypeptide (GIP) Enteroglucagon Lowers the rate of gastric emptying, and reduces smooth muscle contractions and blood flow within the intestine Suppresses the release of pancreatic hormones Inhibits the release of insulin Inhibits the release of glucagon Suppresses the exocrine secretory action of pancreas . This explains how abnormally elevated somatostatin can cause diabetes mellitus , by inhibiting insulin secretion, steatorrhoea by inhibiting cholecystokinin and secretin , gall stones by inhibiting cholecystokinin which normally induce gallbladder myocytes to contract, and hypochlorhydria caused by inhibiting gastrin , which normally stimulate acid secretion. [ citation needed ] Somatostatinomas are associated with calcium deposits called psammoma bodies . [ citation needed ] Diagnosis [ edit ] Fasting plasma somatostatin greater than 30 pg/mL. [ citation needed ] SRS(Somatostatin Receptor Scintigraphy)- It is a radio-nucleotide scan by giving Octreotide tagged with Indium111 isotope, which shows an increase in uptake by the tumour cells. [ citation needed ] Treatment [ edit ] Treatment is by chemotherapy with streptozocin , dacarbazine , doxorubicin or by 'watchful waiting' and surgical debulking via Whipple procedure and other resections of the gastrointestinal organs affected. [1] References [ edit ] ^ Ellison TA, Edil BH (2012).
-
Unexplained Infertility
Wikipedia
Unexplained infertility is infertility that is idiopathic in the sense that its cause remains unknown even after an infertility work-up, usually including semen analysis in the man and assessment of ovulation and fallopian tubes in the woman. [1] Contents 1 Possible causes 2 Prevalence 3 Management 4 Prognosis 5 See also 6 References Possible causes [ edit ] In unexplained infertility abnormalities are likely to be present but not detected by current methods. ... However, a growing body of evidence suggests that epigenetic modifications in sperm may be partially responsible. [3] [4] Prevalence [ edit ] Data from UK, 2009. [5] Globally, about 10% of infertile couples have unexplained infertility. [6] Management [ edit ] Potential methods in unexplained infertility include oral ovarian stimulation agents (such as clomifene citrate , anastrozole or letrozole ) as well as intrauterine insemination (IUI), intracervical insemination (ICI) and in vitro fertilization (IVF). ... PMID 22290605 . ^ Regulated fertility services: a commissioning aid - June 2009 , from the Department of Health UK ^ [1] Merck Manuals ^ a b c "IVF for unexplained infertility" . Human Reproduction Update . 19 (5): 431. 2013. doi : 10.1093/humupd/dmt005 . ^ a b c van den Boogaard NM, Bensdorp AJ, Oude Rengerink K, Barnhart K, Bhattacharya S, Custers IM, Coutifaris C, Goverde AJ, Guzick DS, Hughes EC, Factor-Litvak P, Steures P, Hompes PG, van der Veen F, Mol BW, Bossuyt P (2013).
-
Narcolepsy Type 1
Orphanet
The presence of low hypocretin-1 levels (<110 pg/ml) in the cerebrospinal fluid can confirm the diagnosis with an excellent sensibility and specificity.HLA-DQB1, HCRT, HLA-DRB1, MOG, HCRTR2, P2RY11, CTSH, ZNF365, CPT1B, CHKB, TRA, EIF3G, TNFSF4, TAC1, TRH, PENK, SOCS2, HLA-DQA1, DNMT1, PPAN, PPAN-P2RY11, CHKB-CPT1B, MAP3K7, NOTCH4, HLA-DQA2, NRXN1, ANO3, TMEM108, TBL1XR1, COLGALT1, ZYG11B, GALNT14, MMP26, MRPL24, JPH1, NUP37, FSTL5, SLC28A3, CA10, LYRM4, ADAMTSL3, TENM2, LRRC7, WDR48, DPP10, LRRN1, VAT1L, LRRC4C, ASIC2, RBFOX1, BTNL2, OXR1, AGTR1, CACNG2, TSBP1, NPFFR2, TCERG1, POLI, KLF12, MRAS, DENND3, ATF6, PALLD, SWAP70, TMEM131L, MTUS2, SRGAP2, DDAH1, KCNE4, TANC2, AK5, PDE7B, BBS9, RBMS3, REM1, NXPH1, NOX4, PHF20, DYNC2LI1, FHOD3, CDKAL1, ZNF385D, SERAC1, CCDC68, LINC00687, ZEB1-AS1, FAM171A1, NT5DC1, SDK1, LINC01619, HS6ST3, SUMF1, IQCM, HLA-F-AS1, NKAIN3, FREM2, RPS10P7, CATSPER4, SHC4, DCDC2C, GLIS3, MAPT-AS1, C9orf92, LINC00861, ATP5MC1P6, HOXD-AS2, SPRY4-AS1, LINC01847, LINC01687, LINC01182, TSBP1-AS1, LINC01500, LINC02336, LINC01839, LINC02511, CEP112, TOGARAM2, CYB5B, ANTXR2, ITCH, ACSS1, PYROXD2, ELMO1, ERI1, ALKBH8, SNX29, SHKBP1, FOXP2, PIK3IP1, TRIM9, CSMD3, TMEM132B, C8orf34, PIK3AP1, MDGA2, AGBL1, HSD3BP4, RBM45, EGFLAM, HINT3, UBXN2B, UNC5D, FAUP1, SUN5, FRMPD2, PLB1, LINC00691, NKAIN2, LINC00964, RAD50, FRY, LINC02248, SFRP1, PSMA5, PSMB9, EPHB1, EDNRA, RIT2, RYR2, DPYD, DPP6, PRCP, DNAH8, SLC18A2, STIM1, TAP1, TAP2, TF, TIAM1, FGF10, PPARA, MCC, GRM5, HLA-F, HLA-DRB9, NFRKB, HLA-DRA, HLA-DQB2, HLA-DOB, GRM8, GRID1, GPC5, GNAO1, GABRG3, FXN, FOLR1, PDE4B, FHIT, SERPINB5, TJP1, ITGA3, DHODH, CTNND2, RNGTT, KALRN, BLMH, BCR, MAGI1, DLGAP2, DLGAP1, NDST3, AKAP6, ATG5, ATP2B2, ASTN1, MICAL2, HDAC9, JAKMIP2, NOL4, DCHS1, BARX2, CTNNA2, SERPING1, H2AC6, SLC14A2, EPM2A, CDK8, CENPC, DAP3, VDR, CILP, VAV2, COL14A1, VAV1, TNF, COMT, HLA-DRB5, HRH3, CCR3, P2RX5-TAX1BP3, ATXN3, P2RY2, P2RY1, P2RX5, P2RX4, TNFRSF1B, P2RX3, P2RX1, TRIB2, P2RX7, P2RX2, P2RX6, LEP, HCRTR1, PSG5, HLA-A, PICSAR, HLA-DPB1, MX2, NFATC2, TAAR1, TNFRSF1A, MIR30C2, SLC6A2, LINC00163, SLC6A3, MIR320A, SKOR1, ZGLP1, MIR130A, MIR30C1, TRAC, CSF3, CRP, TRAJ60, TRAV29DV5, MIR4455, GDNF-AS1, CCR1, SLC17A5, FGF21, CD40LG, SMUG1, CLOCK, ABCG1, SALL4, CIITA, RANGAP1, NAA50, ABCC9, CXCL8, IFNG, IFNA17, IFNA13, IFNA2, IFNA1, IFN1@, HTR2A, MS, FAM3D, CD200R1, HLA-DRB4, HDC, PTPRD, EHMT1, GLP1R, FBXO15, GHRH, GH1, GDNF, GCG, GAD2, MAOA, PRKN, PDYN, POLE, QRFP, CCS

-
Side Effects Of Bicalutamide
Wikipedia
"The role of antiandrogen monotherapy in the treatment of prostate cancer". BJU Int . 91 (5): 455–61. doi : 10.1046/j.1464-410X.2003.04026.x . ... "The role of antiandrogen monotherapy in the treatment of prostate cancer" . BJU International . 91 (5): 455–61. doi : 10.1046/j.1464-410X.2003.04026.x . ... Expert Opinion on Drug Safety . 11 (5): 779–95. doi : 10.1517/14740338.2012.712109 . ... "Managing Male Mammary Maladies" . Eur J Breast Health . 14 (1): 5–9. doi : 10.5152/ejbh.2017.3841 . ... Journal of Clinical Oncology . 30 (30): 3720–5. doi : 10.1200/JCO.2012.41.8509 .
-
Eosinophilic Bronchitis
Wikipedia
If the cause of the eosinophilic bronchitis is unknown, the first line treatment is inhaled corticosteroids. [3] [9] [11] Patients respond well to inhaled corticosteroids and their eosinophil counts in their sputum usually decrease after treatment. [1] [5] There has not been a study to determine the ideal dosage of inhaled corticosteroids for patients with eosinophilic bronchitis, and there is no consensus on whether the treatment should be discontinued once the patient's symptoms resolve or to continue long-term. [1] The use of oral corticosteroids for eosinophilic bronchitis is rare, but it may be considered when inhaled corticosteroids are ineffective in managing the symptoms. [1] [5] Epidemiology [ edit ] Approximately 10–30% of people who present with a chronic cough are suspected to be symptomatic due to eosinophilic bronchitis. [4] [11] References [ edit ] ^ a b c d e f g h i j k Gonlugur U, Gonlugur TE (2008). ... International Archives of Allergy and Immunology . 147 (1): 1–5. doi : 10.1159/000128580 . PMID 18446047 . ^ a b c d e Gibson PG, Fujimura M, Niimi A (February 2002). ... Jornal Brasileiro de Pneumologia . 40 (5): 552–63. doi : 10.1590/S1806-37132014000500012 . ... The Medical Clinics of North America . 100 (5): 1033–45. doi : 10.1016/j.mcna.2016.04.008 . ... American Journal of Roentgenology . 208 (5): 1002–1010. doi : 10.2214/ajr.16.17315 .

-
Estrogen Insensitivity Syndrome
Wikipedia
Specialty Endocrinology Estrogen insensitivity syndrome ( EIS ), or estrogen resistance , is a form of congenital estrogen deficiency or hypoestrogenism [2] which is caused by a defective estrogen receptor (ER) – specifically, the estrogen receptor alpha (ERα) – that results in an inability of estrogen to mediate its biological effects in the body. [3] Congenital estrogen deficiency can alternatively be caused by a defect in aromatase , the enzyme responsible for the biosynthesis of estrogens, a condition which is referred to as aromatase deficiency and is similar in symptomatology to EIS. [4] EIS is an extremely rare occurrence. [5] [6] As of 2016, there have been three published reports of EIS, involving a total of five individuals. [6] The reports include a male case published in 1994, [7] [8] a female case published in 2013, [5] [9] and a familial case involving two sisters and a brother which was published in 2016. [6] EIS is analogous to androgen insensitivity syndrome (AIS), a condition in which the androgen receptor (AR) is defective and insensitive to androgens , such as testosterone and dihydrotestosterone (DHT). ... Contents 1 History 1.1 Male case 1.2 Female case 1.3 Familial case 2 Research 2.1 αERKO mice 2.1.1 Females 2.1.2 Males 2.2 βERKO mice 2.2.1 Females 2.2.2 Males 2.3 GPERKO mice 3 Androgen insensitivity syndrome 4 References 5 Further reading 6 External links History [ edit ] Male case [ edit ] In 1994, a 28-year-old man with EIS was reported. [7] [8] He was fully masculinized. [10] At 204 cm, he had tall stature . [7] His epiphyses were unfused, and there was evidence of still-occurring slow linear growth (for comparison, his height at 16 years of age was 178 cm). [7] He also had markedly delayed skeletal maturation ( bone age 15 years), a severely undermineralized skeleton, evidence of increased bone resorption , and very early-onset osteoporosis . [7] The genitalia , testes , and prostate of the patient were all normal and of normal size/volume. [7] The sperm count of the patient was normal (25 million/mL; normal, >20 million/mL), but his sperm viability was low (18%; normal, >50%), indicating some degree of infertility . [7] The patient also had early-onset temporal hair loss . [7] He reported no history of gender identity disorder , considered himself to have strong heterosexual interests, and had normal sexual function , including morning erections and nocturnal emissions . [7] Follicle-stimulating hormone and luteinizing hormone levels were considerably elevated (30–33 mIU/mL and 34–37 mIU/mL, respectively) and estradiol and estrone levels were markedly elevated (145 pg/mL and 119–272 pg/mL, respectively), while testosterone levels were normal (445 ng/dL). [7] Sex hormone-binding globulin levels were mildly elevated (6.0–10.0 nmol/L), while thyroxine-binding globulin , corticosteroid-binding globulin , and prolactin levels were all normal. [7] Osteocalcin and bone-specific alkaline phosphatase levels were both substantially elevated (18.7–21.6 ng/mL and 33.3–35.9 ng/mL, respectively). [7] Treatment with up to very high doses of estradiol (fourteen 100-μg Estraderm patches per week) had no effect on any of his symptoms of hypoestrogenism , did not produce any estrogenic effects such as gynecomastia , and had no effect on any of his physiological parameters (e.g., hormone levels or bone parameters), suggesting a profile of complete estrogen insensitivity syndrome. [7] Female case [ edit ] In 2013, an 18-year-old woman with EIS was reported. [5] [9] DNA sequencing revealed a homozygous mutation in ESR1, the gene that encodes the ERα. [9] Within the ligand-binding domain , the neutral polar glutamine 375 was changed to a basic, polar histidine . [9] An in vitro assay of ERα-dependent gene transcription found that the EC 50 for transactivation had been reduced by 240-fold relative to normal, non-mutated ERα, indicating an extreme reduction in the activity of the receptor. [9] Clinical signs suggested a profile of complete estrogen insensitivity syndrome with a resemblance to ERα knockout mice . [9] The patient presented with delayed puberty , including an absence of breast development ( Tanner stage I) and primary amenorrhea , as well as intermittent pelvic pain . [9] Examination revealed markedly enlarged ovaries with multiple hemorrhagic cysts as the cause of the lower abdominal pain. [9] Estrogen levels were dramatically and persistently elevated ( estradiol levels were 2,340 pg/mL, regarded as being about 10 times the normal level, and ranged from 750–3,500 pg/mL), gonadotropin levels were mildly elevated ( follicle-stimulating hormone and luteinizing hormone levels were 6.7–19.1 mIU/mL and 5.8–13.2 mIU/mL, respectively), and testosterone levels were slightly elevated (33–88 ng/dL). [9] Inhibin A levels were also markedly elevated. [9] Sex hormone-binding globulin , corticosteroid-binding globulin , thyroxine-binding globulin , prolactin , and triglycerides , which are known to be elevated by estrogen, were all within normal ranges in spite of the extremely high levels of estrogen, and inhibin B levels were also normal. [9] Her relatively mildly elevated levels of gonadotropins were attributed to retained negative feedback by progesterone as well as by her elevated levels of testosterone and inhibin A, although it was acknowledged that possible effects of estrogen mediated by other receptors such as ERβ could not be excluded. [9] The patient had a small uterus , with an endometrial stripe that could not be clearly identified. [9] At the age of 15 years, 5 months, her bone age was 11 or 12 years, and at the age of 17 years, 8 months, her bone age was 13.5 years. [9] Her bone mass was lower than expected for her age, and levels of osteocalcin and C-terminal telopeptide were both elevated, suggesting an increased rate of bone turnover. [9] She was 162.6 cm tall, and her growth velocity indicated a lack of estrogen-induced growth spurt at puberty. [9] The patient had normal pubic hair development (Tanner stage IV) and severe facial acne , which could both be attributed to testosterone. [9] Her ovarian pathology was attributed to the elevated levels of gonadotropins. [9] In addition to her absence of breast development and areolar enlargement, the patient also appeared to show minimal widening of the hips and a lack of subcutaneous fat deposition, which is in accordance with the established role of estrogen and ERα in the development of female secondary sexual characteristics . [9] [11] Treatment of the patient with conjugated estrogens and high doses of estradiol had no effect. [9] Although the authors of the paper considered her ERα to be essentially unresponsive to estrogen, they stated that they "[could not] exclude the possibility that some residual estrogen sensitivity could be present in some tissues", which is in accordance with the fact that the EC 50 of her ERα had been reduced 240-fold but had not been abolished. [9] Treatment with a progestin , norethisterone , reduced her estradiol concentrations to normal levels and decreased the size of her ovaries and the number of ovarian cysts, alleviating her hypothalamic-pituitary-gonadal axis hyperactivity and ovarian pathology. [9] Familial case [ edit ] In 2016, a familial instance of EIS involving three siblings was reported. [6] The afflicted individuals were a 25-year-old female, a 21-year-old female, and an 18-year-old male. [6] The family was consanguineous , with the parents of the siblings being first cousins. [6] The parents were both heterozygous for the causative mutation and were healthy and normal, while the three affected siblings were homozygous for the mutation, and a fourth sibling, an unaffected sister, was heterozygous. [6] The fact that the heterozygous parents and heterozygous sister were unaffected indicates that the disorder is transmitted in an autosomal recessive manner and that a single normal allele is sufficient to achieve normal puberty and fertility , which is consistent with what has been observed in ERα knockout mice . [6] All three siblings presented with pubertal failure . [6] Both of the sisters had no breast development (i.e., Tanner stage I), illustrating how the ERα is absolutely required for normal mammary gland development. [6] The older sister was overweight ( BMI 26.3) and had mild incidental adipomastia , [6] or adipose tissue deposition in the breasts without true glandular tissue , a trait that is not indicative of pubertal development. [12] [13] The sisters had complete pubic hair maturation (i.e., Tanner stage V), while the brother had Tanner stage II pubic hair development and Tanner stage I gonadal maturation. [6] The right testis of the brother was cryptorchid , while the left testis was severely hypoplastic , with a volume of less than 1 mL. [6] Both of the sisters presented with primary amenorrhea and enlarged, multicystic ovaries , and the older sister had a small uterus and a thin endometrium . [6] The older sister had chest acne , which could be attributed to hyperandrogenism (see below). [6] All three siblings showed markedly delayed bone maturation for their chronological ages. [6] Surprisingly, the older sister was of normal height, while the younger sister was tall. [6] In all three siblings, estradiol levels were markedly elevated and gonadotropin levels were elevated. [6] In the sisters, estradiol levels were extremely high, more than 50-fold greater than normal levels, while gonadotropin levels were elevated 3-fold above the normal range. [6] Levels of progesterone , 17α-hydroxyprogesterone , androstenedione , testosterone , and dihydrotestosterone (DHT) were elevated in the sisters, while concentrations of adrenal steroids including cortisol , dehydroepiandrosterone (DHEA), 11β-hydroxyandrostenedione , 11-deoxycortisol , and 21-deoxycortisol were within normal ranges. [6] Levels of sex hormone-binding globulin (SHBG) were very low, which can be attributed to the absence of hepatic actions of estrogen. [6] In the older sister, anti-Müllerian hormone (AMH) levels were normal, while levels of inhibin A and inhibin B were significantly increased. [6] In the brother, levels of AMH and inhibin B were low, in conjunction with the patient's low concentrations of testosterone. [6] The low testosterone levels of the brother were probably related to his cryptorchidism, this symptom having not been previously reported in the earlier male case report of EIS. [6] Consistent with the brother's phenotype , cryptorchidism has been described in ERα knockout mice. [6] Because of the brother's low inhibin B levels, it was stated by the researchers that it was very likely that spermatogenesis would not occur in him. [6] Impaired negative feedback by estrogen on the hypothalamic-pituitary-gonadal (HPG) axis would account for the elevated estradiol and gonadotropin levels in the siblings and for the ovarian enlargement and cyst formation in the sisters. [6] All three siblings were homozygous for a missense mutation in the fifth coding exon of the ESR1 gene . [6] The mutation caused a change from guanine to adenine at complementary DNA nucleotide 1181 (c.1181G>A) in the gene, which resulted in the substitution of a histidine for an arginine at residue 394 (p.Arg394His) in the helix H5 of the ligand-binding domain (LBD) of the ERα protein . [6] This is a critical residue that is completely conserved among species and in the androgen receptor (AR) and mineralocorticoid receptor (MR). [6] Mutations involving the corresponding residue in the AR and MR have previously been associated with androgen insensitivity syndrome (AIS) and diminished sensitivity to mineralocorticoids , respectively. [6] Assays revealed that the mutated ERα showed strongly reduced transcriptional activity in response to stimulation by estradiol, with an ED 50 that was approximately 65-fold greater than that of normal/wild-type ERα. [6] In the normal ERα, estradiol is anchored in the binding pocket of the receptor by three hydrogen bonds ; the C3 and C17 hydroxyl groups of estradiol are anchored by the Glu353 and Arg394, and His524 residues of the ERα protein, respectively. [6] In the mutated ERα, the His394 residue is unable to properly anchor estradiol, which results in the dramatically reduced sensitivity and response of the receptor to estradiol relative to the normal ERα. [6] A group of other ERα agonists that included ethinylestradiol , diethylstilbestrol , tamoxifen , clomifene , and raloxifene were tested in their ability to promote transcriptional activity of the mutated ERα, but none of them were found to be more efficacious than estradiol in activating the mutated receptor and hence in overcoming the estrogen insensitivity of the siblings. [6] As the sisters had very high, supraphysiological levels of circulating estradiol, the authors cautioned that it could not be ruled out that estradiol may have exerted some functional influence on their phenotypes via signaling through the ERβ and GPER (i.e., that not all of the observed phenotypes may have simply been due to loss of ERα signaling). [6] Moreover, the authors noted that this might partially explain the variability in the phenotypes. [6] Research [ edit ] EIS can be experimentally induced in animals via knockout of the ER. [14] In these so-called ERKO mice , different ERs can be disabled allowing to study the role of these receptors. [14] ERKO mice show development of the respective female or male reproductive systems , and male and female αERKO mice are infertile, βERKO males are fertile while females are subfertile, male and female double αERKO and βERKO mice are infertile. [14] The uterus and mammary glands are hypoplastic and do not respond to exogenous stimulation by estrogens. [14] Males are infertile with atrophy in the testes . [14] Bone age is delayed and bones are more brittle . [ citation needed ] Variations in these patterns can be achieved by selectively disabling the ERα or ERβ. [14] The following sections are an extensive though partial/incomplete list of deficits observed in ERKO mice. [14] αERKO mice [ edit ] Females [ edit ] Estradiol and LH levels are dramatically elevated due to loss of negative feedback by estradiol on the HPG axis. [14] FSH levels, in contrast, are normal. [14] Testosterone levels are also substantially elevated. [14] Prolactin levels are decreased by 5-fold, which is due to a loss of its estradiol-induced secretion from the anterior pituitary . [14] The uterus and endometrium show hypoplasia and hypotrophy , respectively, and the vagina is atrophic . [14] The oviduct is normal. [14] The ovary is normal until sexual maturity , at which point there is complete anovulation and the ovaries become enlarged, hemorrhagic , and cystic . [14] Because there is complete anovulation, female αERKO mice are infertile . [14] The ovarian phenotype closely resembles that of polycystic ovary syndrome (PCOS) in humans. [14] It is caused by chronic exposure to abnormally high levels of LH. [14] By 18 months of age, there is a 30 to 40% incidence of ovarian tumors . [14] The mammary gland is normal until puberty , at which point there is a complete absence of pubertal development and the gland remains in a prepubertal state. [14] Body weight and body fat are increased. [14] There are signs of insulin resistance , as in PCOS in humans. [14] Due to the substantially elevated testosterone levels, there is hyperandrogenism , including masculinization of the preputial glands . [14] In addition, female αERKO mice exhibit behavior that is similar to that of males in terms of parental , aggressive , and sexual activities . [14] There is a complete lack of sexual receptivity , measured as lordosis behavior . [14] There are significant deficits in parental behavior, including a tendency toward infanticide , and aggressive behavior is increased. [14] Males [ edit ] LH and testosterone levels are both increased 2-fold due to loss of negative feedback by estradiol on the HPG axis. [14] The testes develop relatively normally initially, but are slightly smaller than normal and possess various defects. [14] By 20 weeks, the weights of the testes, epididymis , and vas deferens are significantly decreased relative to those of normal mice. [14] However, there is a severe testicular phenotype with age, such that the testes are completely atrophied by 150 days of age. [14] Also, the testes show Leydig cell hyperplasia , which is due to the increased levels of LH and intratesticular testosterone. [14] Further, there is a greater incidence of cryptorchidism (undescended/retracted testes). [14] There is complete infertility, which is due both to testicular defects and to severely compromised normal sexual behavior (see below). [14] Males can produce viable sperm , but there are severe deficits in both spermatogenesis and sperm function, the latter rendering produced sperm ineffective. [14] Sperm counts are significantly reduced, at 55% of those of normal mice, and further diminish with age, at 13% of those of normal mice by 16 weeks of age. [14] There are deficits in sperm motility , an increased incidence of sperm defects (specifically, sperm heads separated from the flagellum (tail)), and a complete inability of sperm to fertilize oocytes (assessed in vitro ). [14] There are no obvious abnormalities in the male accessory glands , including the prostate gland , bulbourethral glands , coagulating gland , and seminal vesicles . [14] However, there is a significant increase in weight of the seminal vesicles/coagulating gland that becomes more apparent with age, which is likely due to elevated testosterone levels. [14] Aggressive behavior is dramatically reduced, whereas parental behavior, in terms of infanticide, is relatively normal. [14] There is little effect on sexual behavior in terms of mounting and sexual attraction to females. [14] However, there is an almost complete lack of intromission and ejaculation , in spite of the relatively normal mounting rate. [14] This contributes to infertility. [14] βERKO mice [ edit ] Females [ edit ] The uterus, vagina, and oviducts are normal. [14] The ovary is normal prior to puberty, and there is still no gross aberrant phenotype during adulthood. [14] However, there is partial anovulation and subfertility, which is due to ovarian defects, namely compromised follicular maturation via loss of estradiol signaling in ovarian granulosa cells . [14] The mammary gland appears to be normal. [14] Body weight and fat distribution appear to be normal. [14] Increased anxiety -like behavior is seen. [15] In addition, the antidepressant -like effects of exogenous estradiol in the forced swim test are lost. [15] Males [ edit ] Fertility is full and normal, with a lack of relevant phenotypes observed. [14] The male accessory glands, including the prostate gland, bulbourethral glands, coagulating gland, and seminal vesicles, all seem to be normal. [14] However, there is an increased incidence of prostate hyperplasia with age. [16] Body weight and fat distribution appear to be normal. [14] There is a lack of grossly apparent behavioral phenotypes, including in regards to sexual behavior. [14] However, increased aggressive behavior is observed. [15] GPERKO mice [ edit ] GPER knockout mice have also been generated, and exhibit obesity , cardiovascular dysfunction , insulin resistance , glucose intolerance , differences in mammary carcinogenesis and metastasis , and differences in central nervous system function. [17] [18] Androgen insensitivity syndrome [ edit ] Main article: Androgen insensitivity syndrome In contrast to EIS, androgen insensitivity syndrome (AIS), a condition in which the androgen receptor (AR) is defective, is relatively common. ... External links [ edit ] Classification D ICD - 10 : E34.5 OMIM : 300068 External resources Orphanet : 99429 v t e Gonadal disorder Ovarian Polycystic ovary syndrome Premature ovarian failure Estrogen insensitivity syndrome Hyperthecosis Testicular Enzymatic 5α-reductase deficiency 17β-hydroxysteroid dehydrogenase deficiency aromatase excess syndrome Androgen receptor Androgen insensitivity syndrome Familial male-limited precocious puberty Partial androgen insensitivity syndrome Other Sertoli cell-only syndrome General Hypogonadism Delayed puberty Hypergonadism Precocious puberty Hypoandrogenism Hypoestrogenism Hyperandrogenism Hyperestrogenism Postorgasmic illness syndrome Cytochrome P450 oxidoreductase deficiency Cytochrome b5 deficiency Androgen-dependent condition Aromatase deficiency Complete androgen insensitivity syndrome Mild androgen insensitivity syndrome Hypergonadotropic hypogonadism Hypogonadotropic hypogonadism Fertile eunuch syndrome Estrogen-dependent condition Premature thelarche Gonadotropin insensitivity Hypergonadotropic hypergonadism v t e Genetic disorders relating to deficiencies of transcription factor or coregulators (1) Basic domains 1.2 Feingold syndrome Saethre–Chotzen syndrome 1.3 Tietz syndrome (2) Zinc finger DNA-binding domains 2.1 ( Intracellular receptor ): Thyroid hormone resistance Androgen insensitivity syndrome PAIS MAIS CAIS Kennedy's disease PHA1AD pseudohypoaldosteronism Estrogen insensitivity syndrome X-linked adrenal hypoplasia congenita MODY 1 Familial partial lipodystrophy 3 SF1 XY gonadal dysgenesis 2.2 Barakat syndrome Tricho–rhino–phalangeal syndrome 2.3 Greig cephalopolysyndactyly syndrome / Pallister–Hall syndrome Denys–Drash syndrome Duane-radial ray syndrome MODY 7 MRX 89 Townes–Brocks syndrome Acrocallosal syndrome Myotonic dystrophy 2 2.5 Autoimmune polyendocrine syndrome type 1 (3) Helix-turn-helix domains 3.1 ARX Ohtahara syndrome Lissencephaly X2 MNX1 Currarino syndrome HOXD13 SPD1 synpolydactyly PDX1 MODY 4 LMX1B Nail–patella syndrome MSX1 Tooth and nail syndrome OFC5 PITX2 Axenfeld syndrome 1 POU4F3 DFNA15 POU3F4 DFNX2 ZEB1 Posterior polymorphous corneal dystrophy Fuchs' dystrophy 3 ZEB2 Mowat–Wilson syndrome 3.2 PAX2 Papillorenal syndrome PAX3 Waardenburg syndrome 1&3 PAX4 MODY 9 PAX6 Gillespie syndrome Coloboma of optic nerve PAX8 Congenital hypothyroidism 2 PAX9 STHAG3 3.3 FOXC1 Axenfeld syndrome 3 Iridogoniodysgenesis, dominant type FOXC2 Lymphedema–distichiasis syndrome FOXE1 Bamforth–Lazarus syndrome FOXE3 Anterior segment mesenchymal dysgenesis FOXF1 ACD/MPV FOXI1 Enlarged vestibular aqueduct FOXL2 Premature ovarian failure 3 FOXP3 IPEX 3.5 IRF6 Van der Woude syndrome Popliteal pterygium syndrome (4) β-Scaffold factors with minor groove contacts 4.2 Hyperimmunoglobulin E syndrome 4.3 Holt–Oram syndrome Li–Fraumeni syndrome Ulnar–mammary syndrome 4.7 Campomelic dysplasia MODY 3 MODY 5 SF1 SRY XY gonadal dysgenesis Premature ovarian failure 7 SOX10 Waardenburg syndrome 4c Yemenite deaf-blind hypopigmentation syndrome 4.11 Cleidocranial dysostosis (0) Other transcription factors 0.6 Kabuki syndrome Ungrouped TCF4 Pitt–Hopkins syndrome ZFP57 TNDM1 TP63 Rapp–Hodgkin syndrome / Hay–Wells syndrome / Ectrodactyly–ectodermal dysplasia–cleft syndrome 3 / Limb–mammary syndrome / OFC8 Transcription coregulators Coactivator: CREBBP Rubinstein–Taybi syndrome Corepressor: HR ( Atrichia with papular lesions )ESR1, CCDC170, TMEM59, EGFR, BCAR1, BCAR3, AR, PRL, RTN4, SLC39A6, ZFYVE9, TFF3, SHOX, S100B, S100A1, RTN1, PIK3CG, BCL2, PIK3CD, PIK3CB, PIK3CA, NEDD9, INSR, CCN1, GPER1, FOXM1, ERBB3, ERBB2, CDC42, SPECC1
-
Pancreatic Serous Cystadenoma
Wikipedia
Contents 1 Signs and symptoms 2 Classification 3 Pathology 4 Treatment 5 Epidemiology 6 See also 7 References 8 External links Signs and symptoms [ edit ] In most cases, serous cystadenomas of the pancreas are asymptomatic. [5] However, large cysts may cause symptoms related to their size. [5] Classification [ edit ] Pathologists classify serous cystic neoplasms into two broad groups. ... Since these lesions do not have malignant potential, long-term observation with imaging surveillance is unnecessary. [5] Surgery can include the removal of the head of the pancreas (a pancreaticoduodenectomy ), removal of the body and tail of the pancreas (a distal pancreatectomy ), or rarely removal of the entire pancreas (a total pancreatectomy). [7] In selected cases the surgery can be performed using minimally invasive techniques such as laparoscopy . [8] Epidemiology [ edit ] Serous cystadenomas of the pancreas are more common in women. [5] SCAs are usually diagnosed in people 50-60 years of age. [5] See also [ edit ] Ovarian serous cystadenoma Pancreatic mucinous cystadenoma Solid pseudopapillary neoplasm References [ edit ] ^ Jais, B; Rebours, V; Malleo, G; Salvia, R; Fontana, M; Maggino, L; Bassi, C; Manfredi, R; Moran, R; Lennon, AM; Zaheer, A; Wolfgang, C; Hruban, R; Marchegiani, G; Fernández Del Castillo, C; Brugge, W; Ha, Y; Kim, MH; Oh, D; Hirai, I; Kimura, W; Jang, JY; Kim, SW; Jung, W; Kang, H; Song, SY; Kang, CM; Lee, WJ; Crippa, S; Falconi, M; Gomatos, I; Neoptolemos, J; Milanetto, AC; Sperti, C; Ricci, C; Casadei, R; Bissolati, M; Balzano, G; Frigerio, I; Girelli, R; Delhaye, M; Bernier, B; Wang, H; Jang, KT; Song, DH; Huggett, MT; Oppong, KW; Pererva, L; Kopchak, KV; Del Chiaro, M; Segersvard, R; Lee, LS; Conwell, D; Osvaldt, A; Campos, V; Aguero Garcete, G; Napoleon, B; Matsumoto, I; Shinzeki, M; Bolado, F; Fernandez, JM; Keane, MG; Pereira, SP; Acuna, IA; Vaquero, EC; Angiolini, MR; Zerbi, A; Tang, J; Leong, RW; Faccinetto, A; Morana, G; Petrone, MC; Arcidiacono, PG; Moon, JH; Choi, HJ; Gill, RS; Pavey, D; Ouaïssi, M; Sastre, B; Spandre, M; De Angelis, CG; Rios-Vives, MA; Concepcion-Martin, M; Ikeura, T; Okazaki, K; Frulloni, L; Messina, O; Lévy, P (February 2016).

-
Proteus Syndrome
Wikipedia
"Clinical experience of the Klippel-Trenaunay syndrome" . Arch Plast Surg . 42 (5): 552–8. doi : 10.5999/aps.2015.42.5.552 . ... PMID 26430625 . ^ Nguyen, TA; Krakowski, AC; Naheedy, JH; Kruk, PG; Friedlander, SF (2015). "Imaging Pediatric Vascular Lesions" . ... "Proteus syndrome: differential diagnosis, and patient evaluation". Am J Med Genet . 84 (5): 389–95. doi : 10.1002/(SICI)1096-8628(19990611)84:5<389::AID-AJMG1>3.0.CO;2-O . ... Nature Clinical Practice Oncology . 5 (6): 357–361. doi : 10.1038/ncponc1112 . ... Br Med J (Clin Res Ed) . 293 (6548): 683–5. doi : 10.1136/bmj.293.6548.683 .










