-
Pancreatic Neuroendocrine Tumor
Wikipedia
PanNETs are a type of neuroendocrine tumor , representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign , while some are malignant . ... However, morphological imaging alone is not sufficient for a definite diagnosis [14] [16] On biopsy , immunohistochemistry is generally positive for chromogranin and synaptophysin . [17] Genetic testing thereof typically shows altered MEN1 and DAXX / ATRX . [17] Staging [ edit ] The 2010 WHO classification of tumors of the digestive system grades all the neuroendocrine tumors into three categories, based on their degree of cellular differentiation (from well-differentiated "NET G1" through to poorly-differentiated "NET G3"). ... Combinations of several medicines have been used, such as doxorubicin with streptozocin and fluorouracil (5-FU) [12] and capecitabine with temozolomide. [ citation needed ] Although marginally effective in well-differentiated PETs, cisplatin with etoposide has some activity in poorly differentiated neuroendocrine cancers (PDNECs), [12] particularly if the PDNEC has an extremely high Ki-67 score of over 50%. [8] : 30 Several targeted therapy agents have been approved in PanNETs by the FDA based on improved progression-free survival (PFS): everolimus (Afinitor) is labeled for treatment of progressive neuroendocrine tumors of pancreatic origin in patients with unresectable, locally advanced or metastatic disease. [20] [21] The safety and effectiveness of everolimus in carcinoid tumors have not been established. [20] [21] sunitinib (Sutent) is labeled for treatment of progressive, well-differentiated pancreatic neuroendocrine tumors in patients with unresectable locally advanced or metastatic disease. [22] [23] Sutent also has approval from the European Commission for the treatment of 'unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors with disease progression in adults'. [24] A phase III study of sunitinib treatment in well differentiated pNET that had worsened within the past 12 months (either advanced or metastatic disease) showed that sunitinib treatment improved progression-free survival (11.4 months vs. 5.5 months), overall survival , and the objective response rate (9.3% vs. 0.0%) when compared with placebo. [25] Genetics [ edit ] Pancreatic neuroendocrine tumors may arise in the context of multiple endocrine neoplasia type 1 , Von Hippel–Lindau disease , neurofibromatosis type 1 (NF-1) or tuberose sclerosis (TSC) [26] [27] Analysis of somatic DNA mutations in well-differentiated pancreatic neuroendocrine tumors identified four important findings: [28] [6] as expected, the genes mutated in NETs, MEN1 , ATRX , DAXX , TSC2 , PTEN and PIK3CA , [28] are different from the mutated genes previously found in pancreatic adenocarcinoma . [29] [30] one in six well-differentiated pancreatic NETs have mutations in mTOR pathway genes, such as TSC2 , PTEN and PIK3CA . [28] The sequencing discovery might allow selection of which NETs would benefit from mTOR inhibition such as with everolimus , but this awaits validation in a clinical trial . mutations affecting a new cancer pathway involving ATRX and DAXX genes were found in about 40% of pancreatic NETs. [28] The proteins encoded by ATRX and DAXX participate in chromatin remodeling of telomeres ; [31] these mutations are associated with a telomerase -independent maintenance mechanism termed ALT (alternative lengthening of telomeres) that results in abnormally long telomeric ends of chromosomes . [31] ATRX / DAXX and MEN1 mutations were associated with a better prognosis . [28] References [ edit ] ^ Burns WR, Edil BH (March 2012).MEN1, ATRX, DAXX, ELK3, TP53, EPHB1, SLC6A2, CEACAM5, CEACAM7, UQCRFS1, DHDDS, CHPT1, RALBP1, CIB1, SEMA4D, RIPK1, CXCR4, VEGFA, TTR, GNA12, TSC2, TFE3, CDKN1B, PSG2, POMC, MYCN, CEACAM3, GRN, MUC16

-
Estrogen And Neurodegenerative Diseases
Wikipedia
In addition, estrogen deprivation is likely to initiate or enhance degenerative changes caused by oxidative stress , and to reduce the brain's ability to maintain synaptic connectivity and cholinergic integrity leading to the cognitive decline seen in aged and disease-afflicted individuals. [5] There is sufficient evidence that estradiol is a powerful neuroprotectant which might have use against AD, stroke and Parkinson's disease both in women and men. [5] Estrogen and Alzheimer's disease [ edit ] This figure shows how APP cleavage produces toxic Abeta in Alzheimer's disease. Amyloid plaques formed by amyloid-β (Aβ) deposition and neurofibrillary tangles formed by tau protein phosphorylation are dominant physiological features of Alzheimer's disease. Amyloid precursor protein (APP) proteolysis is fundamental for production of Aβ peptides implicated in AD pathology. [6] By using a cell line that contains high levels of estrogen receptors, scientists found that treatment with physiological concentrations of 17 beta-estradiol is associated with accumulation in the conditioned medium of an amino-terminal cleavage product of APP (soluble APP or protease nexin-2), indicative of non-amyloidogenic processing. [7] Estrogen and Parkinson's disease [ edit ] Recommendations on the use of postmenopausal hormonal replacement therapy in women with Parkinson's disease or those genetically at risk. [8] But another group of scientists found a positive association between estrogen use and lower symptom severity in women with early PD not yet taking L-dopa . [9] Estrogen and Huntington's disease [ edit ] Huntington's disease (HD) is a polyglutamine disorder based on an expanded CAG triplet repeat [10] leading to cerebral and striatal neurodegeneration. [11] Potential sex differences concerning the age of onset and the course of the disease are poorly defined, as the difficulties of matching female and male HD patients regarding their CAG repeat lengths limit comparability. [12] Estrogen and Amyotrophic lateral sclerosis [ edit ] ALS occurs more commonly in men than in women, and women get the disease later in life compared to men. [13] This suggested the possible protective role of estrogen in ALS. ... "Octyl Gallate Markedly Promotes Anti-amyloidogenic Processing of APP through Estrogen Receptor-Mediated ADAM10 Activation" .
-
Photoinhibition
Wikipedia
In 1966, Jones and Kok measured the action spectrum of photoinhibition and found that ultraviolet light is highly photoinhibitory. [2] The visible-light part of the action spectrum was found to have a peak in the red-light region, suggesting that chlorophylls act as photoreceptors of photoinhibition. ... Thus, the redox state of Quinone A is no longer active and there is, again, no change in the concentration of carbon dioxide in the intracellular airspaces of the leaf. All these factors work to have a net decrease of stomatal conductance. ... Photoinhibition damages PSII at the same rate whether the leaf stalk is in water or lincomycin, but, in the “leaf stalk in water” sample, repair is so rapid that no net decrease in (F V /F M ) occurs Photoinhibition can be measured from isolated thylakoid membranes or their subfractions, or from intact cyanobacterial cells by measuring the light-saturated rate of oxygen evolution in the presence of an artificial electron acceptor ( quinones and dichlorophenol-indophenol have been used). ... Ecology of photoinhibition [ edit ] Photoinhibition may cause coral bleaching . [27] See also [ edit ] Anthocyanin Chlorophyll Kautsky effect Light reaction Photosynthetic reaction centre Photosynthesis References [ edit ] ^ Kok B (1956). "On the inhibition of photosynthesis by intense light". Biochimica et Biophysica Acta . 21 (2): 234–244. doi : 10.1016/0006-3002(56)90003-8 . PMID 13363902 . ^ Jones LW, Kok B (1966). "Photoinhibition of Chloroplast Reactions.

- Dowling-Degos Disease GARD
-
Azotemia, Familial
OMIM
Furthermore, urea is reabsorbed actively by the tubule; this process is apparently brought into play particularly in states of low protein intake. Net reabsorption might be due to exaggerated active reabsorption or to deficient secretion.
-
Microvasculature Remodeling
Wikipedia
What makes vessels grow with exercise training? J App Physiol 97: 1119–28, 2004. This cardiovascular system article is a stub .
-
Isolated Atrial Amyloidosis
Wikipedia
Retrieved 17 November 2010 . v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2
-
Al Amyloidosis
Wikipedia
British Journal of Haematology . 140 (4): 365–377. doi : 10.1111/j.1365-2141.2007.06936.x . ... External links [ edit ] Classification D ICD - 10 : E85 ICD - 9-CM : 277.3 OMIM : 254500 MeSH : C531616 DiseasesDB : 315 External resources MedlinePlus : 000533 eMedicine : med/3363 v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2 v t e Immunoproliferative immunoglobulin disorders PCDs / PP Plasmacytoma Multiple myeloma ( Plasma cell leukemia ) MGUS IgM ( Macroglobulinemia / Waldenström's macroglobulinemia ) heavy chain ( Heavy chain disease ) light chain ( Primary amyloidosis ) Other hypergammaglobulinemia CryoglobulinemiaCCND1, LINC02343, CBX7, LINC00457, SMARCD3, TTR, DNAH11, CD38, SCT, GDF15, NPPB, SPP1, IGKV2-29, MIR34A, SMN1, SMN2, ST2, TNF, BMS1P20, KRT20, IGKV1-8, BABAM2, GLIPR1, WDHD1, MAGEC2, SLC1A4, PLXNB2, IGKV3D-15, IGKV1D-8, IGKV3-20, PRAME, ALB, SDC1, FGA, BCR, PRDM1, CAT, MS4A1, CDA, CDH1, CRP, CSF3, CYP1B1, DDT, ETFA, GSN, FAS, IGKV@, LGALS3, LGALS4, MDK, MMP1, MMP2, MUC1, MYD88, NCAM1, PFDN5, RPS27A, SAA1
-
Amyloidosis
Orphanet
A vast group of diseases defined by the presence of insoluble protein deposits in tissues. Amyloidoses are classified according to clinical signs and biochemical type of amyloid protein involved. Clinical description Most amyloidoses are multisystemic, 'generalize' or 'diffuse'. Mainly affected are the kidneys, heart, GI tract, liver, skin, peripheral nerves and eyes, but any organ can be affected. Progression is usually severe, as affected organs are destroyed. There are a few forms of localized amylosis.TTR, PSEN1, GSN, APP, ACHE, APOE, HMOX1, APOC3, ZDHHC13, APOC2, TNFRSF1A, CCND1, POLA1, B2M, BCHE, BDNF, NLRP3, PRNP, OSMR, CASP3, MME, CSF2, CST3, SAA2, MEFV, MAPT, LYZ, LIG4, LAMC2, IL6, IL1B, TREM2, IDE, BACE1, IAPP, SAA1, GFAP, APCS, TNF, AGER, SNCA, APOA1, NEFL, TGFBI, CLU, ADAM10, IL1A, SUCLA2, DPYD, IL4, NGF, NFE2L2, APOA2, LPAL2, ABCB1, BECN1, PPARG, NRGN, TARDBP, BIN1, BCL2, TLR4, MMP9, ABCA7, CTSD, DCLRE1B, LRP1, TYROBP, SIRT1, TGFB1, LECT2, CHI3L1, MOK, RELN, PSEN2, S100A9, TSPO, LEP, CTSB, SOD1, SYP, ALOX5, KHDRBS1, DLG4, MCIDAS, CAT, CRP, IGF1, FGA, SMUG1, NUP62, DCTN4, HP, S100B, GTF2H1, GSK3B, SOD2, AQP4, CHRNA4, VEGFA, PIK3CD, ABCB6, CDK5, PLG, SQSTM1, PIK3CG, PIK3CB, MAPK3, ITM2B, PIK3CA, SORL1, IL10, LPA, VSNL1, ROS1, RXRA, PTPRC, EGR1, PARP1, ALB, CD40, STS, TLR2, PLD3, CD36, GABPA, HTT, SDC1, PYCARD, SH2D1A, LINC01672, ACE, MAPK8, CNR2, MAP3K5, CYP46A1, TSHZ1, CD55, PINK1, SERPINA3, ADIPOQ, GRM5, HTRA1, MAPK1, TRPC6, DYSF, TYRP1, RNR2, UBQLN1, QPCT, NCAM1, NFIB, MMP2, HLA-DRB1, NFIA, HCRT, PPARA, HDAC3, UCHL1, ABCG1, MAP2, PTGS2, PPARGC1A, PDB1, LDLR, NFIC, LBP, TXN, DKK1, INSR, SYNM, NCSTN, IL17A, IL13, GH1, P2RX7, RBP4, IL1RN, MFAP1, PDE5A, NFIX, A2M, CD40LG, FYN, NGB, DDIT3, CRYAB, CD38, APLP2, C4BPA, MIR200A, ARG1, ESCO1, EPO, DDR1, CYLD, CALCA, FCGRT, FAP, DECR1, LRRK2, APRT, CR1, GCG, DYRK1A, CHRM3, GDNF, ECE1, EDN1, ACTB, DRD1, IGAN1, TNMD, CHAT, DPYSL2, BAX, MFT2, PCSK9, GCSAM, LRRTM3, STOX1, CDK5R1, SUMO4, GOLGA6A, OSTN, ANO5, LOC390714, ARHGEF7, DUOX2, CD163, UBR1, FSIP1, SV2A, SNAP91, MAGI2, ZEB2, KEAP1, UCN3, TOMM20, PCLAF, CLSTN3, HDAC9, CD109, ACTRT1, CARTPT, MAD2L1BP, BCAR1, GDF15, TRIM69, TMC4, GRAP2, TICAM1, RMDN2, DAB2IP, CRADD, MSC, SLC2A12, RAB11A, MIR137, MIR107, RIPK1, VWF, DEFB103A, VTN, DNM1P33, LOC643387, SCFV, VIP, SFTPA1, VDR, VCAM1, UTRN, NR1H2, C20orf181, BACE1-AS, OCLN, TYRO3, MICA, TMX2-CTNND1, LOC102723407, LOC102724971, LINC02210-CRHR1, UPK3B, HSP90B2P, TRAF6, LOC105379528, WNT1, XBP1, XK, AGPS, MIR132, URI1, WDR1, EIF3A, MIR15B, MIR181C, UNC5C, DENR, DEGS1, MIR29A, MIR29B1, CST7, LRP8, ULK1, AXIN1, PICALM, MIR29C, BAS, FXR1, PSCA, AIMP2, TFEB, PLA2G7, NPHS2, DBA2, PPIF, DGCR2, RAB21, TP53, HSPA14, DCDC2, GDE1, DUOX1, GSAP, DAPK2, TLR9, TREM1, QPCTL, DDAH1, ODAM, SLC25A38, RMDN3, RCBTB1, SYBU, SIRT3, USE1, DEFB103B, CRTC1, CTNNBL1, ARC, SLC17A7, ARL6IP1, TBC1D9, PRDX5, BACE2, RNF19A, ADIPOR1, SNX12, EEF2K, SNX8, FLVCR1, TMEM176B, F11R, IGKV2-29, IGKV3-20, ASCC1, PRLH, GAL, IGKV1D-8, SH2B1, IGKV3D-15, IGHV3-69-1, IGHV3OR16-7, APH1A, HTRA2, VPS4A, RMDN1, HSPB8, POLDIP2, SDF4, CHMP2B, KIF1B, SHANK2, HDAC5, ATF6, GPNMB, OLFM1, PRMT5, PHF6, RTN3, COL25A1, LILRB2, CALCOCO2, MFSD2A, ABI2, NAV3, CHRFAM7A, BMF, NLRP12, BMS1P20, OPTN, PRRT2, MBL3P, NR1H3, DNM1L, CDCA5, SH2D3C, TOM1, BCL2L11, PDCD6IP, CIB1, MAP1LC3B, KAT5, FERMT2, NLGN1, NLRP1, HPS5, CENPK, KLK8, WDHD1, TPPP, ABCG4, RIPK3, HHIP, SDS, STIP1, SLC8B1, CLEC7A, UBE2Z, EDAR, CARD14, MMEL1, CLDN16, PAGR1, TXNIP, EHMT1, AHSA1, ATG7, TPSG1, PPP3R1, CLDN5, EREG, NR5A1, MTOR, FPR1, FN1, FOXO1, FGF2, FCN2, EFEMP1, FAT1, FANCD2, F10, F3, ESR2, ESR1, EPHB2, TLR5, EPHA3, EPHA1, ELN, SERPINB1, EHHADH, E2F1, DUSP6, DPP4, DPEP1, DNMT3A, DNMT1, DNM1, DLX4, DLG2, GAST, FUS, G6PD, GALNS, HSPA9, HSPA4, HSF1, HSD11B1, FOXA2, NR4A1, HMGB1, HMBS, HLA-DMA, NRG1, HFE, HDAC2, GSK3A, CXCL2, GRM3, GRM2, GRIN2B, GRIN1, GRN, GRIA1, GPX1, GNA12, GEM, GDF2, GCHFR, GC, GATA6, GAS6, GAPDH, DES, DCX, DCN, C1QB, C1QBP, BSG, BRCA2, BGN, BCR, TNFRSF17, AVP, ATF4, SERPINC1, ARSA, ABCC6, FAS, APOA4, APLP1, BIRC3, APBB2, APAF1, ANXA5, ANXA1, ALOX15, ALOX12, AKT1, AIF1, AGTR1, AGT, ACAN, ACAT1, ABCA4, ABCA1, C1QA, C1QC, CYBB, C3, CTNND1, CSF3, CSE1L, MAPK14, CRMP1, CRK, CRHR1, CRH, CPB2, COL11A2, CLCN3, CHIT1, AKR1C4, CETN1, CEBPA, CDR1, CD74, CD68, CD33, CD19, CD14, RUNX1T1, CAV1, CASR, CASP1, CAMK2G, CAMK4, CACNA1C, C4BPB, HSP90AA1, HSPD1, HSPG2, MAPK12, S100A8, S100A6, SORT1, S100A1, RPL29, RHD, RET, RARB, PVALB, PTX3, PTGS1, MAP2K6, MAP2K1, PRKCB, PRKCA, PPY, PTPA, PPID, PPARD, POR, POLB, PMP22, PLK1, PLA2G4A, PITX3, PIN1, SERPINB9, SERPINB6, SLC25A3, SAA4, ATXN7, PAWR, SCD, TIMP1, TGFBR2, PRDX2, TAT, SYT1, SYN1, VAMP1, STK11, ST13, SST, TRIM21, SPRR2A, SPP1, SOAT1, SNCG, SMN2, SMN1, SLC6A3, SLC5A5, SLC1A3, PMEL, SH3GL2, SGCG, SRSF2, SEMG1, SELPLG, CXCL12, CCL5, SCT, SERPINF1, REG3A, HTR1B, MAZ, MAOA, MAG, TACSTD2, LTB, LNPEP, LMNB1, LIMK1, LCN2, LAIR1, KRAS, KLC1, KNG1, KIR3DL1, ITIH4, ITGAM, IRS1, PDX1, INS, CXCL10, ING1, IL18, IL12A, CXCR2, CXCL8, IL2, IFNG, IFNB1, ID2, HTR2A, MATN1, MBL2, SERPINE1, MBP, PAEP, P2RY2, P2RX4, OCA2, NTRK2, YBX1, SLC11A2, NPTX1, NPPA, NPC1, NPY, NOTCH1, NOS3, NFKB1, NEFH, NCF2, NAGLU, MYOC, COX1, MT3, ABCC1, MPZ, MPV17, MNAT1, MMP3, AFF1, MFGE8, DNAJB9, CHST6, H3P40

-
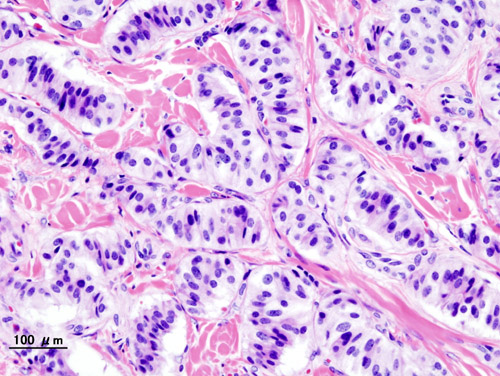
Insulinoma
GARD
Insulinoma is a type of pancreatic neuroendocrine tumor (pancreatic NET), which refers to a group of rare tumors that form in the hormone-making cells of the pancreas.MEN1, RPS15, CDKN2B, CDKN2C, IAPP, GCG, CDKN1B, CDKN1A, SST, FOXM1, GLP1R, PDX1, INS, IL1B, RIT2, PTPRN2, GAD1, EHMT1, IGF2, ZGLP1, CDKN2A, SLC30A8, SLC30A10, GCK, SSTR2, FFAR1, YY1, LEP, DPP4, INSM1, MNX1, HSPD1, GAD2, SLC2A2, CASR, RALBP1, RIPK1, PDHX, BTC, UQCRFS1, TP53, TGM2, SSTR5, CDKN1C, INSR, ABCC8, SLC6A2, SSTR4, SSTR3, WFS1, NIT1, SERPINA1, PTPRN, GIP, GCKR, CORO1A, H3P47, PRL, H3P10, ERBB2, GAST, EGR1, ELK3, CALCA, CASP3, EPHB1, G6PC, DLK1, CCN5, SQSTM1, PTTG1, GCM2, LHX2, KL, MAPK8IP1, INSL5, IRS2, ZNRD2, KHDRBS1, DCTN6, LILRB1, FASTK, CCND1, PDIA5, FAS, ATF6, KDM1A, PDZD2, BCL2, BRCA1, TNKS, PLA2G6, HNF1A, TCF19, TGFA, TGFB1, CASP8, THBD, TKT, TSPAN7, TPD52, TRP-AGG2-5, TRPC1, EIPR1, TXN, TYRP1, UCP2, VDR, CACNA1D, BRAF, STAB1, ERP44, NUP62, KCNH4, CAT, KCNH8, GPR119, STOML3, AKT1, HCAR2, GOLGA6A, TICAM2, HES3, MIR107, MIR144, MIR155, MIR204, MIR21, MIR375, INS-IGF2, ADSS2, TMED7-TICAM2, ECT, LINC02210-CRHR1, H3P23, ADM, SLC22A12, TXNDC5, TRABD, RCBTB1, FGF21, MCAT, MCTS1, TMED7, ADIPOR1, DCTN4, CDKAL1, SLC25A38, BANK1, MEG3, ZC3H12A, APOC2, SOX6, SELENOS, IGSF9, SEMA6A, HAMP, G6PC2, PDIA2, ANGPT2, SYP, STAT5A, STC1, STAT5B, KCNJ1, KCNJ6, KRT8, KRT16, KRT19, DECR1, LEPR, LGALS3, LMO2, EPCAM, SMAD2, SMAD3, SMAD4, MAPT, MC2R, MDK, RAB8A, CUX1, MET, CIITA, MLH1, EGF, EGFR, INPPL1, HK1, MTOR, FGF13, GNA12, GPD2, FBN1, GRN, GSK3B, GSR, GTF2H1, ESR2, ELK1, HLA-DQB1, HMGN2, HNF4A, EPHB2, IFI27, IGFBP1, IGFBP2, IL4, IL10, MRC1, NCAM1, NEDD4, SLC2A1, RAP1A, REG1A, CPE, CMA1, S100A8, SCT, CCL2, CXCL12, SDHD, CHGA, RAB3A, CDKN2D, SLC16A1, SNX1, CDC42, CDK1, CCND3, CCNC, CCK, STAT1, RANBP2, CR2, NF1, PIK3CG, NFE2L1, CTSB, NME1, OPA1, PAX4, PAX6, PCSK1, ENPP1, CTNNB1, PKD1, CRHR1, POLD1, MAPK1, MAPK3, MAPK8, ADCYAP1, PRSS1, PSEN2, PSMD9, PTEN, ACO2
- Sneddon Syndrome GARD
-
Heredofamilial Amyloidosis
Wikipedia
ISBN 978-1-4160-2999-1 . v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2 This cutaneous condition article is a stub .
-
Secondary Systemic Amyloidosis
Wikipedia
ISBN 978-1-4160-2999-1 . v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2 This cutaneous condition article is a stub .

-
Secondary Cutaneous Amyloidosis
Wikipedia
ISBN 978-1-4160-2999-1 . v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2 This cutaneous condition article is a stub .
-
Leukoencephalopathy
Wikipedia
Leukoencephalopathy ( leukodystrophy -like diseases) is all of the brain white matter diseases, whether their molecular cause is known or not. [1] It can refer specifically to any of these diseases: Progressive multifocal leukoencephalopathy Toxic leukoencephalopathy Leukoencephalopathy with vanishing white matter Leukoencephalopathy with neuroaxonal spheroids Reversible posterior leukoencephalopathy syndrome Megalencephalic leukoencephalopathy with subcortical cysts . It can also refer to gene MLC1 or Megalencephalic leukoencephalopathy with subcortical cysts 1 , a human gene related to the former disease. Hypertensive leukoencephalopathy The classification of leukoencephalopathies is a matter of debate. Some authors divide leukoencephalopathies into hereditary disorders and acquired disorders. The hereditary demyelinating disorders are then classified according to the localization of the underlying metabolic defect, and they include the leukodystrophies when myelin growth is the underlying problem.CSF1R, COL4A1, TYMP, SCP2, SPP1, APP, TCN2, RHOA, MTHFR, MTR, DPYD, DARS2, BDNF, CDKN1B, NOTCH3, EIF2B2, EIF2B1, EIF2B4, CLCN2, DARS1, POLG, EIF2B5, SNORD118, RPIA, NDUFS1, NDUFAF3, HTRA1, L2HGDH, VPS11, GJC2, CLN6, TYROBP, COA8, NUBPL, EARS2, NAXE, NDUFV1, PLAA, NFE2L2, SAMHD1, NDUFS8, NDUFS6, NDUFS4, MRPS22, NDUFS3, NDUFS2, NDUFV2, TACO1, SCN8A, SCO1, YME1L1, PMPCB, AIFM1, NDUFAF1, RRM2B, TREX1, EIF2B3, NDUFB10, NDUFB11, TMEM70, FASTKD2, FOXRED1, TIMMDC1, KMT2E, NDUFAF4, TMEM126B, NDUFB9, MARS2, CTC1, PET100, AUH, NDUFS7, COX6B1, COX8A, COX10, B3GALNT2, NDUFA11, COX20, NDUFAF2, HMGCL, COX14, LAMB1, HIBCH, COLGALT1, ND2, TRNS1, ND3, NDUFAF5, NDUFA1, NDUFA6, TRNN, ND1, NDUFB3, AARS2, MLC1, EIF2S2, HEPACAM, SDHAF1, RNASET2, TUBB4A, ABCB6, LGALS14, GFAP, APOE, WARS2, CSF2, DEAF1, NFU1, PSEN1, LAMA2, KARS1, ISCA2, COA7, KCNT1, POLR3B, NOTCH2NLC, DDX59, SLC2A4RG, SHANK3, SLC26A5, GGCT, ACBD5, LYRM7, SLC5A8, RMND1, GLRX5, TREM2, MTFMT, IBA57, HIKESHI, SLC13A3, AARS1, SZT2, MS, MOG, MBP, MANBA, KIF5A, IL6, IFNG, IFNB1, HMGB1, HMBS, GJA1, GFPT1, FMR1, EIF2S3, EIF2S1, EEF1A1, EDN1, SARDH, CST3, COL4A2, CLCN1, C1QBP, ALDH3A2, ABCD1, JAG1, ADORA2A, MPV17, COX2, POLR3A, MYL2, SDS, MYL9, SCO2, FIG4, ARHGEF2, DEGS1, CUL4B, VEGFA, TUFM, TLR2, SURF1, ACTA2, SORD, SLC16A2, SLC2A1, SDHB, CCL5, PTGS2, PRPS1, PLP1, PDGFB, PAFAH1B1, NEFL, NDUFB8, NDUFA2, MTCO2P12
-
Mosquito Bites
Mayo Clinic
Avoid and exclude mosquitoes Limit exposure to mosquitoes by: Repairing any tears in the screens on windows, doors and camping gear Using mosquito netting over strollers and cribs Using mosquito netting when sleeping outdoors Selecting self-care products that don't have scents Use insect repellent Use insect repellent when mosquitoes are active. ... Some sporting goods stores sell clothing pretreated with permethrin. Don't wash bed nets or set them in sunlight, as this breaks down permethrin.
-
Clanging
Wikipedia
. ^ Spitzer, Manfred (1999). The mind within the net: Models of learning, thinking, and acting .
-
Occupational Hearing Loss
Wikipedia
Relationship between noise exposure levels and duration of allowable exposure at that level for NIOSH and OSHA The NIOSH Sound Level Meter app Sound level meters and dosimeters are two types of devices that are used to measure sound levels in the workplace. ... Some recent studies suggest that some smartphone applications may be able to measure noise as precisely as a Type 2 SLM. [15] [16] Although most smartphone sound measurement apps are not accurate enough to be used for legally required measurements, the NIOSH Sound Level Meter app met the requirements of IEC 61672/ANSI S1.4 Sound Level Meter Standards (Electroacoustics - Sound Level Meters - Part 3: Periodic Tests). [17] Ototoxic chemical exposure [ edit ] Chemically-induced hearing loss (CIHL) is a potential result of occupational exposures. ... "Evaluation of smartphone sound measurement applications (apps) using external microphones-A follow-up study" . ... "Smartphone-based sound level measurement apps: Evaluation of compliance with international sound level meter standards".

-
Organ-Limited Amyloidosis
Wikipedia
External links [ edit ] Classification D ICD - 10 : E85.4 ICD - 9-CM : 277.3 v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2
-
Chronic Hallucinatory Psychosis
Wikipedia
Others, again, might be swept into the widespread net of dementia praecox . This state of affairs cannot be regarded as satisfactory, for they are not truly cases of melancholia, paranoia, dementia praecox or any other described affection.






