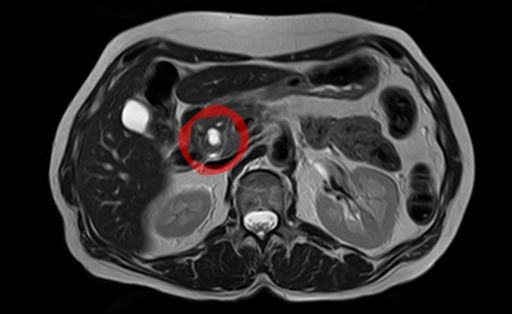
EUS guided therapy has been performed successfully, though more data is necessary, particularly prospective study. [3] [4] An EUS-guided approach appears more effective with smaller sized MCNs. [5] Epidemiology [ edit ] MCNs are much more common in women. [6] A study in 2012 found that amongst individuals undergoing surgical resection of a pancreatic cyst, about 23 percent were mucinous cystic neoplasms. [7] The rate of malignancy present in MCN is about 10 percent. [1] Malignancy is more often present in older individuals. [8] See also [ edit ] Intraductal papillary mucinous neoplasm Pancreatic serous cystadenoma References [ edit ] ^ a b c d e f g h Elta, GH; Enestvedt, BK; Sauer, BG; Lennon, AM (April 2018). "ACG Clinical Guideline: Diagnosis and Management of Pancreatic Cysts".
This article needs additional citations for verification . Please help improve this article by adding citations to reliable sources . Unsourced material may be challenged and removed. Find sources: "Pancreatic mucinous cystadenoma" – news · newspapers · books · scholar · JSTOR ( January 2017 ) ( Learn how and when to remove this template message ) Pancreatic mucinous cystadenoma , also known as " mucinous cystadenoma of the pancreas ", is a benign tumour of pancreas . It is one of the cystic lesions of the pancreas . [1] Contents 1 Pathology 1.1 Microscopy 2 See also 3 References Pathology [ edit ] Microscopy [ edit ] Mucinous cystadenoma of the pancreas Mucinous cystadenoma of the pancreas Mucinous cystadenoma of the pancreas Mucinous cystadenoma of the pancreas See also [ edit ] Mucinous cystadenoma Pancreatic cysts Pancreatic serous cystadenoma References [ edit ] ^ Parra-Herran, C. E.; Garcia, M. T.; Herrera, L; Bejarano, P. A. (2010). "Cystic lesions of the pancreas: Clinical and pathologic review of cases in a five year period". Journal of the Pancreas . 11 (4): 358–64. PMID 20601810 . v t e Digestive system neoplasia GI tract Upper Esophagus Squamous cell carcinoma Adenocarcinoma Stomach Gastric carcinoma Signet ring cell carcinoma Gastric lymphoma MALT lymphoma Linitis plastica Lower Small intestine Duodenal cancer Adenocarcinoma Appendix Carcinoid Pseudomyxoma peritonei Colon/rectum Colorectal polyp : adenoma , hyperplastic , juvenile , sessile serrated adenoma , traditional serrated adenoma , Peutz–Jeghers Cronkhite–Canada Polyposis syndromes: Juvenile MUTYH-associated Familial adenomatous / Gardner's Polymerase proofreading-associated Serrated polyposis Neoplasm: Adenocarcinoma Familial adenomatous polyposis Hereditary nonpolyposis colorectal cancer Anus Squamous cell carcinoma Upper and/or lower Gastrointestinal stromal tumor Krukenberg tumor (metastatic) Accessory Liver malignant : Hepatocellular carcinoma Fibrolamellar Hepatoblastoma benign : Hepatocellular adenoma Cavernous hemangioma hyperplasia : Focal nodular hyperplasia Nodular regenerative hyperplasia Biliary tract bile duct : Cholangiocarcinoma Klatskin tumor gallbladder : Gallbladder cancer Pancreas exocrine pancreas : Adenocarcinoma Pancreatic ductal carcinoma cystic neoplasms : Serous microcystic adenoma Intraductal papillary mucinous neoplasm Mucinous cystic neoplasm Solid pseudopapillary neoplasm Pancreatoblastoma Peritoneum Primary peritoneal carcinoma Peritoneal mesothelioma Desmoplastic small round cell tumor
. ^ a b Sechi E, Morris PP, McKeon A, Pittock SJ, Hinson SR, Weinshenker BG, Aksamit AJ, Krecke KN, Kaufmann TJ, Jolliffe EA, Zalewski NL, Zekeridou A, Wingerchuk DM, Jitprapaikulsan J, Flanagan EP (July 2018). ... Retrieved 21 March 2018 . ^ Scott TF, Frohman EM, De Seze J, Gronseth GS, Weinshenker BG (December 2011). "Evidence-based guideline: clinical evaluation and treatment of transverse myelitis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology" .
Seckel syndrome is a genetic disorder characterized by growth retardation, very small head (microcephaly( with intellectual disability , and unique facial features such as large eyes, beak-like nose, narrow face, and receding lower jaw. About less than 25% of the patients also have blood abnormalities. Seckel syndrome is inherited in an autosomal recessive fashion. The condition may be divided in 8 different subtypes , according to the specific gene alteration (mutation ). Treatment is supportive.
A number sign (#) is used with this entry because of evidence that autosomal recessive primary microcephaly-13 (MCPH13) is caused by compound heterozygous mutation in the CENPE gene (117143) on chromosome 4q24. One such family has been reported. For a phenotypic description and a discussion of genetic heterogeneity of primary microcephaly, see MCPH1 (251200). Clinical Features Mirzaa et al. (2014) reported a brother and sister, born of unrelated parents of European descent, with microcephaly, poor overall growth, and developmental delay. Both had intrauterine growth retardation and microcephaly apparent on prenatal ultrasound, as well as similar dysmorphic facial features, including sloping forehead, prominent nose, and mild micrognathia. At age 5 years, the older sib, a boy, had microcephaly (-9 SD), short stature (-7 SD), small hands and feet, mild spasticity, and severely delayed psychomotor development with absent speech and poor gross and fine motor skills.
Seckel syndrome is a type of microcephalic primordial dwarfism that is characterized by a proportionate dwarfism of prenatal onset, a severe microcephaly, a typical dysmorphic face (bird-like), and mild to severe intellectual disability.
A number sign (#) is used with this entry because Seckel syndrome-1 (SCKL1) is caused by homozygous or compound heterozygous mutation in the ATR gene (601215) on chromosome 3q23. Description Seckel syndrome is a rare autosomal recessive disorder characterized by intrauterine growth retardation, dwarfism, microcephaly with mental retardation, and a characteristic 'bird-headed' facial appearance (Shanske et al., 1997). Genetic Heterogeneity of Seckel Syndrome Other forms of Seckel syndrome include SCKL2 (606744), caused by mutation in the RBBP8 gene (604124) on chromosome 18q11; SCKL4 (613676), caused by mutation in the CENPJ gene (609279) on chromosome 13q12; SCKL5 (613823), caused by mutation in the CEP152 gene (613529) on chromosome 15q21; SCKL6 (614728), caused by mutation in the CEP63 gene (614724) on chromosome 3q22; SCKL7 (614851), caused by mutation in the NIN gene (608684) on chromosome 14q22; SCKL8 (615807), caused by mutation in the DNA2 gene (601810) on chromosome 10q21; SCKL9 (616777), caused by mutation in the TRAIP gene (605958) on chromosome 3p21; and SCKL10 (617253), caused by mutation in the NSMCE2 gene (617246) on chromosome 8q24. The report of a Seckel syndrome locus on chromosome 14q, designated SCKL3, by Kilinc et al. (2003) was found to be in error; see History section. Clinical Features This condition was given the 2 names bird-headed dwarfism and nanocephaly by Virchow.
A number sign (#) is used with this entry because of evidence that Seckel syndrome-9 (SCKL9) is caused by homozygous mutation in the TRAIP gene (605958) on chromosome 3p21. For a general phenotypic description and a discussion of genetic heterogeneity of Seckel syndrome, see SCKL1 (210600). Clinical Features Silengo et al. (2001) described 2 Italian sibs with a Seckel-like malformation syndrome characterized by intrauterine growth retardation (IUGR), microcephaly, beaked nose, failure to thrive, and early death. The first sib was a 46,XX girl who at birth underwent surgery for right diaphragmatic hernia. External genitalia appeared normal, with slight clitoral hypertrophy.
A number sign (#) is used with this entry because of evidence that autosomal recessive microcephaly and chorioretinopathy-2 (MCCRP2) is caused by homozygous mutation in the PLK4 gene (605031) on chromosome 4q28. Description Microcephaly and chorioretinopathy-2 is an autosomal recessive developmental disorder characterized by delayed psychomotor development, visual impairment, and short stature (summary by Martin et al., 2014). For a discussion of genetic heterogeneity of microcephaly and chorioretinopathy, see MCCRP1 (251270). Clinical Features Martin et al. (2014) reported 7 members of a highly consanguineous Pakistani family with a severe developmental disorder characterized by primary microcephaly (up to -15 SD), profoundly delayed psychomotor development, and growth retardation with dwarfism (up to -8 SD). Many patients had ocular abnormalities, including microphthalmia, microcornea, and cataracts.
A number sign (#) is used with this entry because of evidence that Seckel syndrome-2 (SCKL2) is caused by homozygous mutation in the RBBP8 gene (604124) on chromosome 18q11. Jawad syndrome (251255), a microcephaly syndrome involving mental retardation and digital anomalies, is also caused by mutation in the RBBP8 gene. Description Seckel syndrome is a rare autosomal recessive disorder characterized by growth retardation, microcephaly with mental retardation, and a characteristic facial appearance (Borglum et al., 2001). For a general phenotypic description and a discussion of genetic heterogeneity of Seckel syndrome, see SCKL1 (210600). Clinical Features Borglum et al. (2001) studied a consanguineous family of Iraqi descent with Seckel syndrome.
Woods et al. (1995) reported the case of an infant with pre- and postnatal microcephaly and growth retardation, a distinctive face, and developmental delay. Seckel syndrome was the initial diagnosis. The infant became pancytopenic at 16 months of age and died soon thereafter. His bone marrow was of normal cellularity but had an infiltration of small lymphocytes. Increased spontaneous chromosome breakage was seen in blood and fibroblasts. Mitomycin C-induced chromosome damage was increased and comparable to that seen in Fanconi anemia.
A number sign (#) is used with this entry because of evidence that Seckel syndrome-8 (SCKL8) is caused by homozygous mutation in the DNA2 gene (601810) on chromosome 10q21. One such family has been reported. Description Seckel syndrome is a rare autosomal recessive disorder characterized by intrauterine growth retardation, dwarfism, microcephaly with mental retardation, and a characteristic 'bird-headed' facial appearance (summary by Shanske et al., 1997). For a discussion of genetic heterogeneity of Seckel syndrome, see SCKL1 (210600). Clinical Features Shaheen et al. (2014) studied an uncle and niece, both born of consanguineous marriages, with short stature and strikingly similar facial features consistent with Seckel syndrome. Both uncle and niece had decreased length at birth (-4.9 and -5.1 SD, respectively) and continued to have short stature at ages 18 years and 9 years (-9.5 and -6 SD) as well as severe microcephaly (-7.5 and -11.8 SD).
A number sign (#) is used with this entry because Seckel syndrome-5 is caused by homozygous or compound heterozygous mutation in the CEP152 gene (613529) on chromosome 15q21. Description Seckel syndrome is an autosomal recessive disorder characterized by proportionate short stature, severe microcephaly, mental retardation, and a typical 'bird-head' facial appearance (summary by Kalay et al., 2011). For a general phenotypic description and a discussion of genetic heterogeneity of Seckel syndrome, see 210600. Clinical Features Kalay et al. (2011) clinically evaluated 5 consanguineous families with Seckel syndrome originating from an isolated rural area in Turkey. The patients presented with microcephaly, sloping forehead, high nasal bridge, beaked nose, and retrognathia.
A number sign (#) is used with this entry because of evidence that Seckel syndrome-4 (SCKL4) is caused by homozygous mutation in the CENPJ gene (609279) on chromosome 13q12. One such family has been reported. Homozygous mutations in the CENPJ gene can also cause primary microcephaly-6 (MCPH6; 608393). Description Seckel syndrome is a rare autosomal recessive disorder characterized by severe pre- and postnatal growth retardation, severe microcephaly with mental retardation, and specific dysmorphic features (Faivre et al., 2002). For a general description and a discussion of genetic heterogeneity of Seckel syndrome, see 210600. Clinical Features Al-Dosari et al. (2010) described a consanguineous Saudi family in which several members had clinical features of Seckel syndrome.
A number sign (#) is used with this entry because of evidence that erythrokeratodermia variabilis et progressiva-4 (EKVP4) is caused by compound heterozygous mutation in the KDSR gene (136440) on chromosome 18q21. Description Erythrokeratodermia variabilis et progressiva-4 is characterized by severe lesions of thick scaly skin on the face and genitals, as well as thickened, red, and scaly skin on the hands and feet (summary by Boyden et al., 2017). For a discussion of genetic heterogeneity of EKVP, see EKVP1 (133200). Clinical Features Boyden et al. (2017) reported 4 unrelated patients with a similar skin phenotype, presenting either at birth or in the perinatal period with thickened red skin with vernix, thickened skin in the diaper area, or tight red skin with deep fissures and collodion membrane. Erythema faded in infancy, and by 4 months of age all 4 probands developed well-demarcated, thickened, scaly lesions on the cheeks and periocular areas, with erythema and thickening of the palms and soles.
A number sign (#) is used with this entry because of evidence that erythrokeratodermia variabilis et progressiva-3 (EKVP3) is caused by heterozygous mutation in the gene encoding connexin-43 (GJA1; 121014) on chromosome 6q22. Description Erythrokeratodermia variabilis et progressiva is a rare skin disease. Patients with EKVP3 have normal skin at birth but develop hyperpigmentation and scaling at sites of friction in childhood, with progression to near-confluent corrugated hyperkeratosis, palmoplantar keratoderma, and transient figurate erythema (summary by Boyden et al., 2015). For a discussion of genetic heterogeneity of EKVP, see EKVP1 (133200). Clinical Features Boyden et al. (2015) reported 3 unrelated patients with erythrokeratodermia variabilis.
A number sign (#) is used with this entry because of evidence that erythrokeratodermia variabilis et progressiva-1 (EKVP1) is caused by heterozygous mutation in the gene encoding connexin-31 (GJB3; 603324) on chromosome 1p34. One family with EKVP1 has been reported with a homozygous mutation in the GJB3 gene. Description The erythrokeratodermias are a clinically variable and genetically heterogeneous group of inherited disorders characterized by widespread erythematous plaques, stationary or migratory, associated with nonmigratory hyperkeratoses (summary by Ishida-Yamamoto et al., 1997). The condition is usually present at birth or occurs during the first year but may begin later in childhood or even in early adulthood. Lesions preferentially affect the face, buttocks, and extensor surfaces of the limbs.
A number sign (#) is used with this entry because of evidence that erythrokeratodermia variabilis et progressiva-2 (EKVP2) is caused by heterozygous mutation in the gene encoding connexin-30.3 (GJB4; 605425) on chromosome 1p34. Description Erythrokeratodermia variabilis et progressiva-2 is a genodermatosis characterized by persistent plaque-like or generalized hyperkeratosis and transient red patches of variable size, shape, and location. The severity and dominating features of the disease vary strikingly within families and also during an individual's course of disease. The erythematous component usually prevails in young children, whereas hyperkeratosis is the dominant or sole feature in adults. Some patients with EKVP2 display lesions resembling erythema gyratum repens (summary by Richard et al., 2003).
"Mendes da Costa" redirects here. For people with this name, see Mendes (name) . "Mendes da Costa syndrome" redirects here. It is not to be confused with Da Costa's syndrome . Erythrokeratodermia variabilis Other names Progressive symmetric erythrokeratodermia, Gottron type Erythrokeratodermia variabilis is inherited in an autosomal dominant manner of inheritance Specialty Dermatology , medical genetics Erythrokeratodermia variabilis (also known as " erythrokeratodermia figurata variabilis ", " keratosis extremitatum progrediens ", " keratosis palmoplantaris transgrediens et progrediens ", [2] : 509 " Mendes da Costa syndrome ", [3] " Mendes da Costa type erythrokeratodermia ", and " progressive symmetric erythrokeratoderma ") is a rare autosomal dominant disorder that usually presents at birth or during the first year of life. [4] To date, it is thought to be caused by mutations in genes encoding for connexin channels proteins in the epidermis, leading to the misregulation of homeostasis in keratinocytes. [5] One type is characterized by generalized, persistent, brown hyperkeratosis with accentuated skin markings, while a second type is localized, with involvement that is limited in extent and characterized by sharply demarcated, hyperkeratotic plaques. [2] [6] : 565 It can be associated with GJB3 [7] and GJB4 . [8] It was characterized in 1925. [9] See also [ edit ] List of cutaneous conditions References [ edit ] ^ "OMIM Entry - # 133200 - ERYTHROKERATODERMIA VARIABILIS ET PROGRESSIVA 1; EKVP1" . omim.org . Retrieved 3 September 2017 . ^ a b Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine . (6th ed.). McGraw-Hill. ISBN 0-07-138076-0 . ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
Tetrasomy X is a chromosome disorder that only affects females and is caused by having four copies of the X chromosome instead of two. Females with tetrasomy X have a total of 48 chromosomes in their cells, so this condition is sometimes written as 48, XXXX. The signs and symptoms of tetrasomy X vary, but can include mild to moderate speech and learning difficulties; developmental delay; distinctive facial features; dental abnormalities; hypotonia and joint laxity; radioulnar synostosis ; heart defects; hip dysplasia ; and problems with ovarian function. An increased risk of childhood infections has also been reported. Tetrasomy X is caused by a random error that occurs during the development of an egg cell and is not caused by anything a mother does during her pregnancy.
Tetrasomy X is a sex chromosome anomaly caused by the presence of two extra X chromosomes in females (48,XXXX instead of 46,XX). Epidemiology Prevalence is unknown but only around 40 cases have been reported in the literature so far. Clinical description Tetrasomy X is associated with delayed speech, learning difficulties, developmental delay and facial dysmorphism. Although disease severity is variable, the learning difficulties and developmental delay are generally mild to moderate. Commonly associated facial features include hypertelorism, upslanting palpebral fissures, epicanthal folds and a flat nasal bridge.
Summary Purpose. Many human disorders of glycosylation pathways have now been identified; they include defects in synthetic pathways for N-linked oligosaccharides, O-linked oligosaccharides, shared substrates, glycophosphatidylinositol (GPI) anchors, and dolichols. This overview will focus on disorders of the N-linked glycan synthetic pathway and some disorders that overlap this metabolic network (multiple-pathway disorders). The goals of this overview on congenital disorders of glycosylation are the following: Goal 1. To describe the clinical characteristics of congenital disorders of N-linked glycosylation Goal 2. To review the causes of congenital disorders of N-linked glycosylation Goal 3.
Congenital disorders of glycosylation (CDG) are a group of inherited metabolic disorders that affect a process called glycosylation. Glycosylation is the complex process by which all human cells build long sugar chains that are attached to proteins, which are called glycoproteins. There are many steps involved in this process, and each step is triggered by a type of protein called an enzyme. Individuals with a CDG are missing one of the enzymes that is required for glycosylation. The type of CDG that a person has depends on which enzyme is missing.
A fast growing group of inborn errors of metabolism characterized by defective activity of enzymes that participate in glycosylation (modification of proteins and other macromolecules by adding and processing of oligosaccharide side chains). This group is comprised of phenotypically diverse disorders affecting multiple systems including the central nervous system, muscle function, immunity, endocrine system, and coagulation. The numerous entities in this group are subdivided, based on the synthetic pathway affected, into disorder of protein N-glycosylation, disorder of protein O-glycosylation, disorder of multiple glycosylation, and disorder of glycosphingolipid and glycosylphosphatidylinositol anchor glycosylation.
Defazio et al. (1998) described 10 new patients with 'so-called apraxia of eyelid opening.' They concluded that the term 'apraxia' may not be the correct descriptive term even when the eyelid disturbance occurs without any other central nervous system disease. Familial clustering of the isolated finding in 1 patient was consistent with a genetic contribution: 2 brothers, their father, and 2 paternal aunts were thought to be affected. Combining their 10 patients with 11 previously reported cases of isolated so-called apraxia of eyelid opening, Defazio et al. (1998) found that the peak age at onset was the sixth decade and that there was a female preponderance of 2 to 1. The characteristic inability to initiate lid elevation was frequently associated with failure to sustain lid elevation, thus suggesting that eyelid motor control may be abnormal.
Benign childhood occipital epilepsy, Gastaut type is a rare, genetic neurological disorder characterized by childhood to mid-adolescence onset of frequent, brief, diurnal simple partial seizures which usually begin with visual hallucinations (e.g. phosphenes) and/or ictal blindness and may associate non visual seizures (such as deviation of the eyes, oculoclonic seizures), forced eyelid closure and blinking and sensory hallucinations. Post-ictal headache is common while impairment of consciousness is rare.
. ^ Voermans NC, van Alfen N, Pillen S, Lammens M, Schalkwijk J, Zwarts MJ, van Rooij IA, Hamel BC, van Engelen BG (June 2009). "Neuromuscular involvement in various types of Ehlers-Danlos syndrome".
. - "This article is distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/ )" ^ a b c d Elta, GH; Enestvedt, BK; Sauer, BG; Lennon, AM (April 2018). "ACG Clinical Guideline: Diagnosis and Management of Pancreatic Cysts".
A rare, benign, epidermal disease characterized by a solitary, asymptomatic, verrucous, skin-coloured to red-brown papule or nodule, which contains a central pore and keratotic plug, occuring most frequently on the scalp, face and neck (rarely, in the mouth, under the nail plate or on the mons pubis). Occasionally, lesions may be multiple and/or pruritic. Histologically, a well-circumscribed, cup-shaped, keratin-filled invagination, with prominent acantholytic dyskeratosis, suprabasilar clefts and villi projecting into the clefts, is observed.
The American Surgeon . 76 (3): 292–5. PMID 20349659 . ^ Verghese BG, Ravikanth R (May 2012). "Breast abscess, an early indicator for diabetes mellitus in non-lactating women: a retrospective study from rural India".
Current Opinion in Genetics & Development . 10 (3): 262–9. doi : 10.1016/s0959-437x(00)00084-8 . PMID 10826992 . ^ Rash BG, Grove EA (October 2007). "Patterning the dorsal telencephalon: a role for sonic hedgehog?"
A number sign (#) is used with this entry because it represents a contiguous gene deletion syndrome. A form of holoprosencephaly (HPE10) has been mapped within the deleted region of chromosome 1q41-q42. For a general phenotypic description and a discussion of genetic heterogeneity of holoprosencephaly, see HPE1 (236100). See also congenital diaphragmatic hernia (DIH; 142340), which has been associated with deletion of chromosome 1q41-q42. See also Skraban-Deardorff syndrome (SKDEAS; 617616), caused by mutation in the WDR26 gene (617424) on chromosome 1q42, which shows overlapping features with chromosome 1q41-q42 deletion syndrome.
A number sign (#) is used with this entry because of evidence that holoprosencephaly-4 (HPE4) is caused by heterozygous mutation in the TGIF gene (602630) on chromosome 18p11. For phenotypic information and a general discussion of genetic heterogeneity in holoprosencephaly, see HPE1 (236100). Cytogenetics Johnson and Bachman (1976) described a normal female who appeared to have a nonreciprocal translocation from the short arm of one chromosome 18 to the long arm of a chromosome 12. She gave birth to a cebocephalic child whose karyotype included an 18p- chromosome. The association of loss of 18p with holoprosencephaly was suggested by the patient reported by Munke et al. (1988); cytogenetic and molecular studies indicated a Y/18 translocation with loss of 18p and distal Yq material in a holoprosencephalic fetus.
A number sign (#) is used with this entry because holoprosencephaly-11 (HPE11) is caused by heterozygous mutation in the CDON gene (608707) on chromosome 11q24. For a general phenotypic description and a discussion of genetic heterogeneity of holoprosencephaly, see HPE1 (236100). Clinical Features Bae et al. (2011) reported 4 unrelated patients with HPE11. One patient had agenesis of the corpus callosum, hypotelorism, growth hormone deficiency, global developmental delay, and thick eyebrows with synophrys. Another had agenesis of the corpus callosum, alobar HPE, hypotelorism, cleft lip/palate, and absent columella; absent pituitary and polysplenia were noted in this patient at autopsy.
For a phenotypic description and a discussion of genetic heterogeneity of holoprosencephaly, see HPE1 (236100). Clinical Features Levin and Surana (1991) described holoprosencephaly in association with an interstitial deletion of chromosome 14q11.1-q13. Parental karyotypes were normal. The white female, born to nonconsanguineous young parents after an uncomplicated pregnancy, showed hypotelorism, lack of nasal bridge, flattened nasal tip with no visible septum, wide midline cleft of lip and hard palate, and ptosis of the left upper eyelid. Axial CT scan of the head was interpreted as showing semilobar holoprosencephaly. The infant died at 8 days of age. Kamnasaran et al. (2005) reported 6 patients with HPE and interstitial deletions on proximal chromosome 14q: 1 had alobar HPE and 5 had lobar HPE.
For phenotypic information and a general discussion of genetic heterogeneity in holoprosencephaly (HPE), see HPE1 (236100). Clinical Features Lehman et al. (2001) described a female infant who survived for 5.5 hours after delivery at 33 weeks' gestation. Autopsy showed a lobar variant of holoprosencephaly. Cytogenetics By cytogenetic analysis in an infant with a lobar variant of holoprosencephaly, Lehman et al. (2001) identified a 2q37.1-q37.3 deletion. This case represented the fourth reported case of HPE associated with partial monosomy 2q37 and the first with an apparently isolated 2q37 deletion. Lehman et al. (2001) suggested that the deleted segment may contain yet another locus, here designated HPE6, which, when disrupted, can lead to brain malformations within the HPE spectrum.
Nonsyndromic holoprosencephaly is an abnormality of brain development that also affects the head and face. Normally, the brain divides into two halves (hemispheres ) during early development. Holoprosencephaly occurs when the brain fails to divide properly into the right and left hemispheres. This condition is called nonsyndromic to distinguish it from other types of holoprosencephaly caused by genetic syndromes, chromosome abnormalities, or substances that cause birth defects (teratogens). The severity of nonsyndromic holoprosencephaly varies widely among affected individuals, even within the same family.
Holoprosencephaly is an abnormality of brain development in which the brain doesn't properly divide into the right and left hemispheres. The condition can also affect development of the head and face. There are 4 types of holoprosencephaly, distinguished by severity. From most to least severe, the 4 types are alobar, semi-lobar, lobar, and middle interhemispheric variant (MIHV). In general, the severity of any facial defects corresponds to the severity of the brain defect. The most severely affected people have one central eye (cyclopia) and a tubular nasal structure (proboscis) located above the eye.
A number sign (#) is used with this entry because of evidence that holoprosencephaly-3 (HPE3) is caused by heterozygous mutation in the SHH gene (600725), which encodes the human Sonic hedgehog homolog, on chromosome 7q36. For a phenotypic description and a discussion of genetic heterogeneity of holoprosencephaly, see HPE1 (236100). Clinical Features Berry et al. (1984) and Johnson (1989) provided information on a family (family 2 in Johnson, 1989) in which holoprosencephaly occurred in 2 sibs and their first cousin, who were offspring of parents with a single central maxillary incisor. Johnson (1989) reported a second patient (family 1) with full-blown holoprosencephaly whose mother and sister had only a single central maxillary incisor. Johnson (1989) suggested that holoprosencephaly is a developmental field defect of which the mild forms can be single median incisor, hypotelorism, bifid uvula, or pituitary deficiency.
A number sign (#) is used with this entry because of evidence that holoprosencephaly-7 (HPE7) is caused by heterozygous mutation in the PTCH1 gene (601309) on chromosome 9q22. For phenotypic information and a general discussion of genetic heterogeneity in holoprosencephaly, see HPE1 (236100). Description Holoprosencephaly (HPE) is the most commonly occurring congenital structural forebrain anomaly in humans. HPE is associated with mental retardation and craniofacial malformations. Considerable heterogeneity in the genetic causes of HPE has been demonstrated (Ming et al., 2002).
A number sign (#) is used with this entry because of evidence that solitary median maxillary central incisor (SMMCI) and SMMCI syndrome are caused by heterozygous mutation in the Sonic hedgehog gene (SHH; 600725) on chromosome 7q36. Clinical Features Rappaport et al. (1976, 1977) reported 7 unrelated patients with single (unpaired) deciduous and permanent maxillary central incisors and short stature. Five of them had isolated growth hormone deficiency. The other 2 had normal growth hormone responses but were short of stature. No similar or possibly related abnormalities were present in the 7 families. Rappaport et al. (1976) used the term monosuperoincisivodontic dwarfism to describe the association of short stature and solitary incisor.
Description Holoprosencephaly (HPE) is the most common structural malformation of the human forebrain and occurs after failed or abbreviated midline cleavage of the developing brain during the third and fourth weeks of gestation. HPE occurs in up to 1 in 250 gestations, but only 1 in 8,000 live births (Lacbawan et al., 2009). Classically, 3 degrees of severity defined by the extent of brain malformation have been described. In the most severe form, 'alobar HPE,' there is a single ventricle and no interhemispheric fissure. The olfactory bulbs and tracts and the corpus callosum are typically absent.
A rare complex brain malformation characterized by incomplete cleavage of the prosencephalon, and affecting both the forebrain and face and resulting in neurological manifestations and facial anomalies of variable severity. Epidemiology Prevalence is estimated to be 1/10,000 live and still births and 1/250 conceptuses, with worldwide distribution. Clinical description Three classical forms of holoprosencephaly (HPE) of increasing severity are described based on the degree of anatomical separation: lobar, semi-lobar and alobar HPE. Milder subtypes include midline interhemispheric variant and septopreoptic HPE. There is, however, a continuous spectrum of abnormal separation of the hemispheres that extends from aprosencephaly/atelencephaly, the most severe end of the spectrum, to microform HPE, a less severe midline defect without the typical HPE brain characteristics.