Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
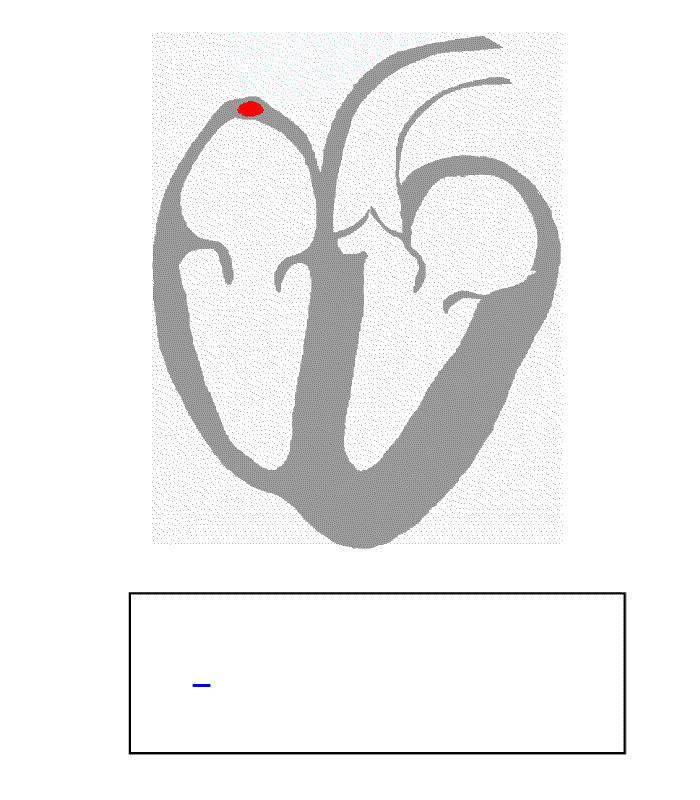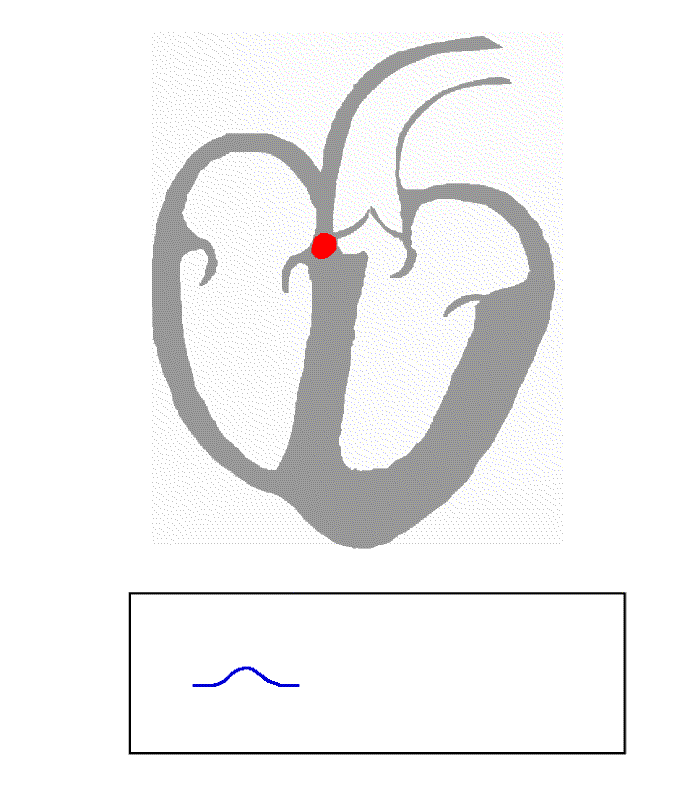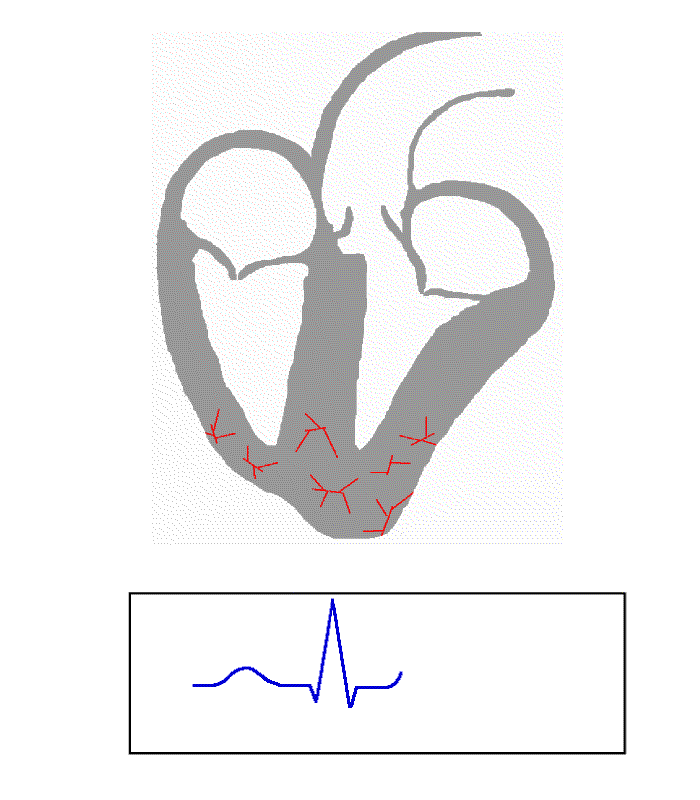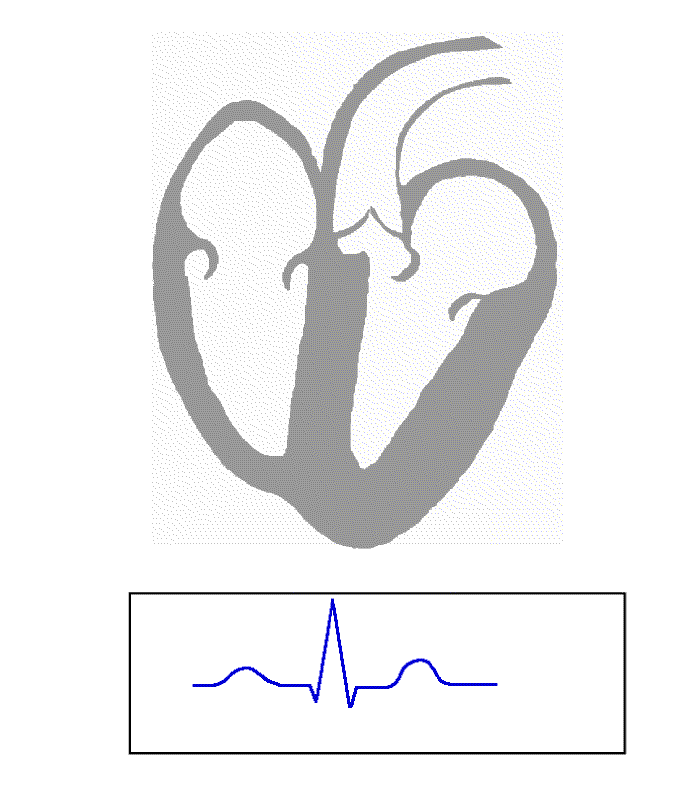
Atrioventricular Block
Wikipedia
The signal travels from the SA node to the ventricles through the atrioventricular node (AV node). In an AV block, this electrical signal is either delayed or completely blocked. ... The electrical signal then travels to the AV node located on the lower portion of the interatrial septum . ... An ECG is used to differentiate between the different types of AV block. However, one important consideration when diagnosing AV blocks from ECGs is the possibility of pseudo- AV blocks which are due to concealed junctional extrasystoles. ... In a second-degree AV block, the impairment results in a failure to conduct an impulse, which causes a skipped beat. [9] Mobitz I [ edit ] Mobitz I is characterized by a progressive, yet, reversible block of the AV node. ... On ECG, there is no relationship between P waves and QRS complexes, meaning the P waves and QRS complexes are not in a 1:1 ratio. Third-degree AV block is the most severe of the AV blocks.LMNA, GNAI2, SCN5A, KCNH2, DES, KCNQ1, PRKAG2, TRPM4, TTN, ACADVL, MTM1, KCNJ5, MMP2, AGXT, SCN4B, MYBPC3, TTR, BVES, SYNE2, SYNE1, TMEM43, KCNJ2, MMP14, CACNA2D1, GLA, GPX4, FHL1, EMD, SLC25A20, NKX2-5, CALR, TDP2, ARSD, TBX5, TRIM21, ARFGEF1, GJC1, TOPORS, ITGAM, SDS, PKD2L1, CACNA1C, SMUG1, RHOA, APRT, ARID2, HCN4, TBX6, CALCR, TBX3, SSB, SLC6A8, SCN10A, CAV1, PKD2, PITX2, CUX1, FLT4, MFAP1, GATA4, KRT5, GNB2, MIR19B1
-
Heart Block
Wikipedia
Blockages are therefore classified based on where the blockage occurs - namely the SA node (" Sinoatrial block "), AV node (" AV block " or AVB ), and at or below the bundle of His ("Intra-Hisian" or "Infra-Hisian block" respectively). ... Therefore, most of the important heart blocks are AV nodal blocks and infra-Hisian blocks. ... Sinus rhythm (rate = 100/min) with 3:2 and 2:1 Type II AV block; right bundle branch block Sinus tachycardia with complete AV block and resulting junctional escape Following the path of the electrical signals, the places where conduction can be blocked give rise to different kinds of heart blocks: Location Name Within the sinoatrial node (SA node or Sinus node), where the heart's signals originate Sinoatrial nodal blocks (often abbreviated "SA nodal block" or "SA block", sometimes written "Sinuatrial block") Within the atrioventricular node (AV node) Atrioventricular block (often abbreviated "AV nodal block", "AV block" or AVB). ... In an SA block, the electrical impulse is delayed or blocked on the way to the atria, thus delaying atrial depolarization. By contrast, an AV block occurs in the AV node and delays ventricular depolarization. ... In SA node suppression, on the other hand, the SA node doesn't generate an electrical impulse because it is reset by the electrical impulse that enters the SA node. AV nodal blocks [ edit ] There are three basic types of AV nodal block : First-degree AV block Second-degree AV block Type I second-degree AV block (Mobitz I), also known as a Wenckebach block [5] Type 2 second-degree AV block (Mobitz II) - due to a block in or below the bundle of His [5] Third-degree AV block (complete heart block) Infra-Hisian block [ edit ] Infra-Hisian block is that of the distal conduction system.

-
Ataxia With Vitamin E Deficiency
GARD
Ataxia with vitamin E deficiency (AVED) is a progressive disease affecting motor control and movement. Symptoms of AVED include slurred speech (dysarthria), difficulty coordinating movements ( ataxia ), numbness in the hands and feet (peripheral neuropathy), and progressive leg weakness. ... Symptoms typically begin during childhood or adolescence and worsen with age, resulting in the need for a wheelchair by early adulthood. AVED is caused by a mutation to the TTPA gene. ... Vitamin E is important because it protects the cells of the neurological system (neurons) from dangerous molecules called free radicals. AVED is inherited in an autosomal recessive manner. Treatment for AVED includes vitamin E supplements, which will prevent AVED from developing if given before symptoms begin and may reverse some neurological symptoms if begun after AVED develops.TTPA, APOB, APOA1, FXN, SH3BP4, ZFP36, SETX, APTX, COQ8A, COPRS, RRS1, AFP, TNF, NOS3, MTHFR, IL6, GNB3, SRR

-
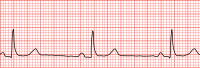
First-Degree Atrioventricular Block
Wikipedia
First-degree AV block Other names First degree heart block, PR prolongation An ECG showing a first degree AV block of greater than 300 ms Specialty Cardiology Symptoms Asymptomatic Complications Progression to second or third degree AV block Causes Fibrosis in AV node, medication, vagal tone , electrolyte disturbances Diagnostic method Electrocardiogram Treatment Avoidance of AV-nodal-blocking medication First-degree atrioventricular block (AV block) is a disease of the electrical conduction system of the heart in which electrical impulses conduct from the cardiac atria to the ventricles through the atrioventricular node (AV node) more slowly than normal. First degree AV block does not generally cause any symptoms, but may progress to more severe forms of heart block such as second - and third-degree atrioventricular block . ... The medications that most commonly cause first-degree heart block are those that increase the refractory time of the AV node, thereby slowing AV conduction. ... Diagnosis [ edit ] In normal individuals, the AV node slows the conduction of electrical impulse through the heart. ... In first-degree heart block, the diseased AV node conducts the electrical activity more slowly.CACNA1C, CACNA2D1, TRPM4, RANGRF, KCNE5, GPD1L, AKAP9, ABCC9, HCN4, KCNE3, SLMAP, TBX5, SCN10A, SCN5A, SCN2B, SCN1B, PKP2, KCNJ8, KCND3, GJA5, GJA1, DMPK, CALM2, CACNB2, SCN3B, EHD1
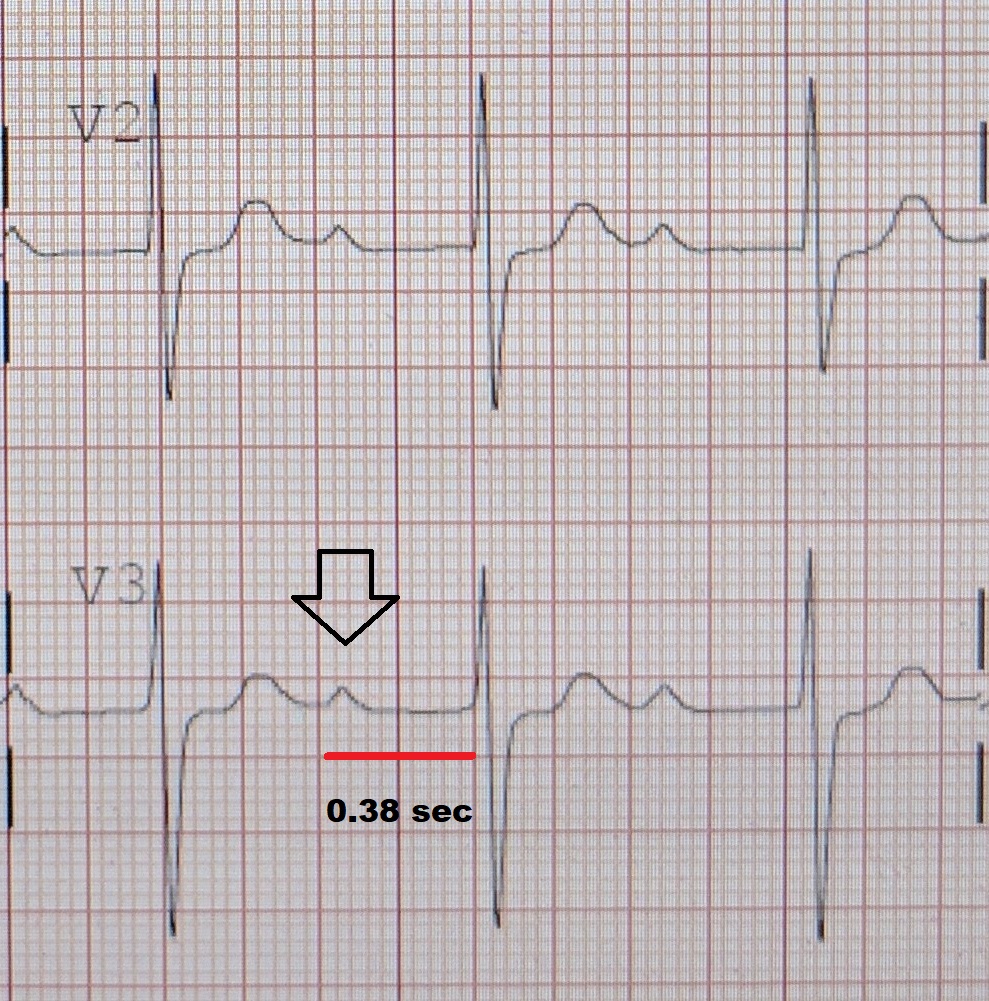
-
Av Nodal Reentrant Tachycardia
Wikipedia
Not to be confused with Atrioventricular reentrant tachycardia . AV-nodal reentrant tachycardia Other names Atrioventricular-nodal reentrant tachycardia An example of an ECG tracing typical of uncommon AV nodal reentrant tachycardia. ... The fast pathway is usually located just superior and posterior to the AV node. These pathways are formed from tissue that behaves very much like the AV node, and some authors regard them as part of the AV node. ... In AVNRT, the fast and slow pathways are located within the right atrium close to or within the AV node and exhibit electrophysiologic properties similar to AV nodal tissue. ... "Typical", "common", or "slow-fast" AVNRT uses the slow AV nodal pathway to conduct towards the ventricle (the anterograde limb of the circuit) and the fast AV nodal pathway to conduct to the atria (the retrograde limb). The re-entrant circuit can be reversed such that the fast AV nodal pathway is the anterograde limb and the slow AV nodal pathway is the retrograde limb, referred to as "atypical", "uncommon", or "fast-slow" AVNRT.

-
Second-Degree Atrioventricular Block
Wikipedia
Second-degree atrioventricular block Other names Second-degree heart block ECGs demonstrating forms of second-degree AV block Specialty Cardiology Symptoms Dizziness , Fainting , Shortness of breath Types Type 1 (Wenckebach), Type 2 Causes Fibrosis in AV node, medication, vagal tone , electrolyte disturbances Diagnostic method Electrocardiogram Treatment Avoidance of AV-nodal-blocking medication, pacemaker Second-degree atrioventricular block (AV block) is a disease of the electrical conduction system of the heart . ... It is classified as a block of the AV node and is categorized in between first-degree (slowed conduction) and third degree blocks (complete block). ... Both types are named after Woldemar Mobitz . [3] [4] Type I is also named for Karel Frederik Wenckebach , [5] and type II is also named for John Hay . [6] [7] Type 1 (Mobitz I/Wenckebach) [ edit ] Type 1 Second-degree AV block , also known as Mobitz I or Wenckebach periodicity , is almost always a disease of the AV node . ... The definitive treatment for this form of AV Block is an implanted pacemaker . ... Thus one may leave out the type and refer to 3:1 Mobitz block or 4:3 Mobitz block, for example, without creating ambiguity, except in the case of 2:1 block. 2:1 AV block [ edit ] In the case of 2:1 block (2 P waves for every QRS complex) it is impossible to differentiate type I from type II Mobitz block based solely on the P:QRS ratio or on a pattern of lengthening PR intervals. [12] :182 In this case, a lengthened PR interval with a normal QRS width is most likely indicative of a type I-like pathology, and a normal PR interval with a widened QRS is most likely indicative of a type II-like pathology. [12] :182 See also [ edit ] Electrocardiogram (ECG or EKG) SA node AV node Atrioventricular block First-degree AV block Third-degree AV block References [ edit ] ^ "Lesson VI - ECG Conduction Abnormalities" .

-
Ventricular Escape Beat
Wikipedia
Contents 1 Causes 2 Diagnosis 3 Management 3.1 Cilostazol 3.2 Ouabain 4 References Causes [ edit ] Ventricular escape beats occur when the rate of electrical discharge reaching the ventricles (normally initiated by the heart's sinoatrial node (SA node), transmitted to the atrioventricular node (AV node), and then further transmitted to the ventricles) falls below the base rate determined by the rate of Phase 4 spontaneous depolarisation of ventricular pacemaker cells. [1] An escape beat usually occurs 2–3 seconds after an electrical impulse has failed to reach the ventricles. [2] This phenomenon can be caused by the sinoatrial node (SA node) failing to initiate a beat, by a failure of the conductivity from the SA node to the atrioventricular node (AV node), or by atrioventricular block (especially third degree AV block ). Normally, the pacemaker cells of the sinoatrial node discharge at the highest frequency and are thus dominant over other cells with pacemaker activity. The AV node normally has the second fastest discharge rate. When the sinus rate falls below the discharge rate of the AV node, this becomes the dominant pacemaker, and the result is called a junctional escape beat . If the rate from both the SA and AV node fall below the discharge rate of ventricular pacemaker cells, a ventricular escape beat ensues. ... Ventricular pacemaker cells discharge at a slower rate than the SA or AV node. While the SA node typically initiates a rate of 70 beats per minute (BPM), the atrioventricular node (AV node) is usually only capable of generating a rhythm at 40-60 BPM or less.
-
Atrial Septal Defect 7 With Or Without Atrioventricular Conduction Defects
OMIM
His mother had the same mutation and ASD with atrial fibrillation; her elder sister had ASD secundum with AV block and sick sinus syndrome requiring permanent pacemaker implantation, and a nephew had ASD with AV block. ... In 3 members of the other family (see 600584.0010), surgical closure of a secundum ASD had been performed; all 3 had ECG evidence of first- or second-degree AV block. In 1 member of the family, first-degree AV block was the only manifestation of heart disease. Hirayama-Yamada et al. (2005) reported that 4 sibs from a family with ASD and AV conduction defects had the same deletion mutation (600584.0012) in the NKX2E gene. The proband was diagnosed with ASD secundum and first-degree AV block that progressed to second-degree block; in 2 of her sibs, AV block also progressed to second degree, although 1 of those sibs had no cardiac malformation. ... One apparent asymptomatic carrier was found to have first-degree AV block on Holter monitoring. The conduction defect in this family was at the AV node, manifesting as first-degree block and evolving toward second-degree block; 3 patients also had atrial fibrillation, and 1 had unexplained ventricular tachycardia seen on Holter monitoring.
-
Junctional Ectopic Tachycardia
Wikipedia
(See electrical conduction system of the heart ). The AV node acts as a gatekeeper, limiting the electrical activity that reaches the ventricles of the heart. This function of the AV node is important, because if the signals generated in the atria of the heart were to increase in rate (as they do during atrial fibrillation or atrial flutter ), the AV node will limit the electrical activity that conducts to the ventricles. For instance, if the atria are electrically activated at 300 beats per minute, half those electrical impulses are blocked by the AV node, so that the ventricles are activated at 150 beats per minute (giving a pulse of 150 beats per minute). Another important property of the AV node is that it slows down individual electrical impulses. ... Tachycardia (from the Greek takhys , meaning "swift", and kardia , meaning heart) means a swift heart rate. [11] By this definition, junctional ectopic tachycardia is an abnormally swift heart rhythm due to cells firing within the heart near the AV node. References [ edit ] ^ Sarubbi B, Vergara P, D'Alto M, Calabro R (2003).

-
Aortic Valve Stenosis
GARD
Aortic valve stenosis (AVS) is a condition characterized by narrowing of the heart's aortic valve opening. This narrowing prevents the valve from opening fully, which obstructs blood flow from the heart into the aorta, and onward to the rest of the body. AVS can range from mild to severe. Signs and symptoms typically develop when the narrowing of the opening is severe and may include chest pain (angina) or tightness; shortness of breath or fatigue (especially during exertion); feeling faint or fainting; heart palpitations; and heart murmur. Individuals with less severe congenital AVS (present at birth) may not develop symptoms until adulthood. ... The condition can eventually lead to heart failure. AVS can have several causes including abnormal development before birth (such as having 1 or 2 valve leaflets instead of 3); calcium build-up on the valve in adulthood; and rheumatic fever .TNF, NPPC, VWF, ENO3, NOTCH1, GATA4, EBP, GATA5, FLI1, CCDC22, ROBO4, GLB1, BCOR, GATA6, FOXF1, NPHP3, FGFR1, FBN1, ARHGAP31, IFIH1, CRELD1, IDUA, TAB2, HAAO, WASHC5, NR2F2, WT1, CTSA, CHST3, ADAMTSL2, NOTCH2, SMAD6, CYP24A1, SMAD4, LTBP2, LPA, LMNA, B4GALT7, KRAS, SNIP1, ACTB, CBL, SLC2A10, ADAMTS10, NEK8, CLIC2, GNPTG, NKX2-5, EHMT1, ESCO2, AMER1, REN, NLRP6, CRP, LGALS3, ACE, APOE, AGT, NPPB, TGFB1, TNFRSF11B, APOB, PALMD, SPP1, MMP2, MAS1, FN1, OGN, NR1I2, IL10, CAD, APOA1, MMRN1, PON1, MLC1, SIRT1, MME, CD86, STS, IGF1, ACE2, RETN, MUC16, FCGR3A, FCGR3B, AHSG, SDC4, MIR424, TNNI3, TNNT2, TTN, AKAP12, GDF15, CILP, OGT, TTR, APLN, VDR, PDA1, PDE5A, CNBP, PTDSS1, TNFSF11, SCG2, LINC00273, ADIPOQ, MIR10B, MIR423, CRTAC1, SYPL2, KLF15, TLR2, KLF3, PCSK9, CRYGEP, RNLS, IL17RA, CHDH, MFN1, METTL3, MYL7, TRPV4, FTO, C1QTNF9, CRTC1, HDAC6, MIR19B1, ATP6AP2, PDPN, MIR34A, OGA, PPARGC1A, MIR210, MIR146A, STING1, MIR126, UTS2, ADAMTS13, WIF1, TEX41, DKK1, TBX22, PIK3CB, THOP1, CRYGC, CTH, CTSS, CYP11B2, DMD, ECE1, LPAR1, EDN1, EDNRA, ELN, ENG, ERBB4, ESR1, FH, FLNC, FOS, FOSB, GAPDH, MSTN, GH1, CCN2, CPOX, GUCY2D, CPB2, AGTR2, AKT1, ALOX5, ARSD, BCL2, CFB, BGN, BMP7, BSG, VPS51, DDR1, RUNX2, CBS, CD14, ENTPD1, CHGB, CLTA, CMA1, CNP, GLS, HSPA4, THBS2, NINJ1, SERPINE1, PIK3CA, JAG1, PIK3CD, PIK3CG, PITX1, PLTP, PPARG, PPP3CB, PSAP, PTPN1, PTPRC, S100A9, S100B, CCL2, SDC1, SLC2A1, SLC22A4, SOD3, NT5E, MYH7, HSPD1, MPO, TNC, IL6, IL6R, ITGAM, ITGB3, JARID2, JUN, JUNB, JUND, KDR, LDLR, LEP, LGALS4, LOX, MMP3, MMP9, MMP13, MMP14, MPG, CBSL
-
Bradycardia
Wikipedia
This usually appears on an electrocardiogram (EKG) with a normal QRS complex accompanied with an inverted P wave either before, during, or after the QRS complex. [5] An AV-junctional escape beat is a delayed heartbeat originating from an ectopic focus somewhere in the AV junction . It occurs when the rate of depolarization of the SA node falls below the rate of the AV node . [5] This dysrhythmia also may occur when the electrical impulses from the SA node fail to reach the AV node because of SA or AV block . [7] This is a protective mechanism for the heart, to compensate for an SA node that is no longer handling the pacemaking activity, and is one of a series of backup sites that can take over pacemaker function when the SA node fails to do so. ... Those above the bundle of His, also known as junctional, will typically range between 40 and 60 BPM with a narrow QRS complex. [8] [9] In a third-degree heart block , about 61% take place at the bundle branch-Purkinje system, 21% at the AV node, and 15% at the bundle of His. [9] AV block may be ruled out with an EKG indicating "a 1:1 relationship between P waves and QRS complexes." [8] Ventricular bradycardias occurs with sinus bradycardia, sinus arrest, and AV block. ... Second-degree sinoatrial blocks can be detected only by use of a 12-lead EKG. [11] It is difficult and sometimes impossible to assign a mechanism to any particular bradycardia, but the underlying mechanism is not clinically relevant to treatment, which is the same in both cases of sick sinus syndrome: a permanent pacemaker . AV conduction disturbances (AV block; primary AV block , secondary type I AV block , secondary type II AV block , tertiary AV block ) may result from impaired conduction in the AV node, or anywhere below it, such as in the bundle of His. ... Clinical Sports Medicine. p. 259. ^ "AV Junctional Rhythm Disturbances (for Professionals)" .TAC1, CACNA1D, ADORA1, PDYN, HTR1A, KNG1, LHB, AGT, NTS, NISCH, POMC, GCG, PRL, UTS2, TACR1, TRH, GNRH1, GNAI2, EDN2, EDN1, DRD2, CYP2D6, EDN3, CYP1A1, CRH, CHRM2, MC4R, TRPM4, BRAT1, SGO1, TMEM43, GNB5, COQ9, QRICH1, LIPT1, MAGEL2, USP18, BSCL2, PPA2, HTRA2, PAX8, PRKAG2, AGPAT2, NPPA, GLUL, TSPYL1, DST, CACNA1C, CACNA2D1, SLC25A20, CASQ2, CAV1, FOS, CAVIN1, KCNH2, KCNJ2, KCNQ1, LMNA, PPARG, SCN9A, HCN4, SCN5A, ANK2
-
Partial Atrioventricular Septal Defect
Orphanet
Shunting is restricted to the atrial level because of fusion of the leaflets of the common AV valve with the crest of the ventricular septum. ... An elevated pulmonary arterial pressure and moderate to severe left atrioventricular valve regurgitation may be observed. First-degree AV block, right bundle branch block, and a superior QRS axis may be noted on electrocardiogram. ... A pacemaker insertion may be required for complete AV block, which may spontaneously develop after repair. ... High morbidity and need for reoperation, often related to residual problems of the systemic AV valve or LAVV regurgitation, may be observed. ... Norwood-type intervention in infancy can be required, as well as prosthetic replacement of the left AV valve.
-
Cimino Fistula
Wikipedia
An AV fistula. A Cimino fistula , also Cimino-Brescia fistula , surgically created arteriovenous fistula and (less precisely) arteriovenous fistula (often abbreviated AV fistula or AVF ), is a type of vascular access for hemodialysis . It is typically a surgically created connection between an artery and a vein in the arm, although there have been acquired arteriovenous fistulas which do not in fact demonstrate connection to an artery. [1] Surgically created AV fistula Contents 1 Theoretical basis 2 History 3 Anatomy 4 See also 5 References 6 External links Theoretical basis [ edit ] Surgically created AV fistulas work effectively because they: Have high volume flow rates (as blood takes the path of least resistance ; it prefers the (low resistance) AV fistula over traversing (high resistance) capillary beds). ... Cimino, MD, and the Development of the AV Fistula

-
Atrioventricular Reentrant Tachycardia
Wikipedia
Not to be confused with AV nodal reentrant tachycardia . Atrioventricular reentrant tachycardia Conduction pathway in atrioventricular reentrant tachycardia, a form of supraventricular tachycardia Specialty Cardiology Atrioventricular reentrant tachycardia ( AVRT ), or atrioventricular reciprocating tachycardia , is a type of abnormal fast heart rhythm and is classified as a type of supraventricular tachycardia (SVT). ... During AVRT, the electrical signal passes in the normal manner from the AV node into the ventricles. Then, the electrical impulse pathologically passes back into the atria via the accessory pathway, causing atrial contraction, and returns to the AV node to complete the reentrant circuit (see figure). ... In the absence of shock, inhibition at the AV node is attempted. This is achieved first by a trial of specific physical maneuvers such as holding a breath in or bearing down. If these maneuvers fail, using intravenous adenosine [4] causes complete electrical blockade at the AV node and interrupts the reentrant electrical circuit. Long-term management includes beta blocker therapy and radiofrequency ablation of the accessory pathway. See also [ edit ] AV nodal reentrant tachycardia Electrical conduction system of the heart Wolff-Parkinson-White syndrome Permanent junctional reentrant tachycardia (PJRT) References [ edit ] ^ Josephson ME.

-
Third-Degree Atrioventricular Block
Wikipedia
Contents 1 Presentation 2 Cause 3 Treatment 4 Intervention 5 Prognosis 6 See also 7 References 8 External links Presentation [ edit ] People with third-degree AV block typically experience severe bradycardia (an abnormally low measured heart rate), hypotension , and at times, hemodynamic instability. [2] Cause [ edit ] Leads I and II demonstrating complete AV block. ... This may be preceded by first-degree AV block , second-degree AV block , bundle branch block , or bifascicular block . In addition, acute myocardial infarction may present with third-degree AV block. An inferior wall myocardial infarction may cause damage to the AV node, causing third-degree heart block. ... The risk factors for asystole include 1) previous asystole, 2) complete heart block with wide complexes, and 3) ventricular pause for > 3 seconds. Mobitz Type 2 AV block is another indication for pacing. ... Patients with 1st and 2nd degree heart block are usually asymptomatic. [15] See also [ edit ] Cardiac pacemaker Electrical conduction system of the heart Electrocardiogram (ECG) Atrioventricular block First-degree AV block Second-degree AV block References [ edit ] ^ "ECG Conduction Abnormalities" .IFNL3, TRIM21, IFNA1, PAEP, TRNL1, ATP8, RRM2B, TTN, FBL, TTN-AS1, PTPN11, GPT, IFNG, AFP, IFNA13, HLA-DPB1, TNF, MIR122, ISG20, IL17A, HLA-DRB1, IL2RA, TLR3, TLR2, CALR, IL10, TGFB1, FOXP3, HAVCR2, IL21, STAT4, SSB, LGALS3BP, IFNA2, TLR7, KLRC1, CXCL10, IL18, SLC10A1, HLA-A, HLA-C, IL6, PNPLA3, TLR4, NCAM1, GSTP1, CXCL8, TLR9, IL1B, ALB, KLRC4-KLRK1, IFNAR1, IL22, TBX18, ESR1, KLRK1, RO60, SLC17A5, HMGB1, CLTC, HLA-B, SPP1, SOCS1, SLC4A1, STAT3, RBM45, SLCO6A1, KIR2DL3, SRY, IL7R, PDCD1, TSC1, PLAAT4, TSC2, GSTK1, TNFAIP8L2, MIR146A, HCN4, TERT, IL4, TNFSF10, TRBV20OR9-2, TCF19, TAP1, ROBO3, IGFBP7, APOBEC3G, CD274, ADIPOQ, SIGLEC1, CXCL13, TLR5, TNFSF13B, EHMT2, VDR, NCR1, MEG3, KIR3DL1, USP18, CD244, DDX58, FGF21, AIM2, MMP9, TLR1, DLEU2, IFI16, HIF1A, GSTM3, CD14, CD28, GEM, GPBAR1, CD69, MIR224, CNR1, CNR2, MIR210, CP, ACE, MIR192, MIR155, CCN2, CXCR5, IL23R, OPN1SW, HLA-G, APOA1, IFNL4, MIR22, KLRG1, CCL26, BATF, CD163, IL32, RGN, ARFGEF1, MIR21, FGL2, GGTLC4P, DSTN, RIPK3, LOC102724197, ADAMTS13, TP53COR1, MIR182, ATF6, NOL3, KIF1B, MIR150, MBD2, TBC1D9, MIR195, MIR7855, MIR222, HAR1B, HDAC9, HDAC4, HAR1A, PPIP5K1, GGT2, GGTLC3, MIR92B, GGTLC5P, HLA-DPA2, POU5F1P4, MIR4804, MIR30C1, POU5F1P3, RAPGEF5, JMJD6, MIR99A, COX5A, MIR34A, NAMPT, MIR33A, EBI3, MIR30C2, ZCCHC14, TMEFF2, SIRT1, CHDH, UBE2Q1, PAG1, RETN, ARNTL2, TRIM69, CD248, VARS2, APOA5, CTHRC1, NDRG2, NLRP3, MAVS, IL17F, ATP5IF1, GGTLC1, MBD4, RPTOR, EP400, ADO, GPT2, FSD1L, GOLPH3, SMOC1, IFIH1, CD276, PPP1R2C, XPO4, FSD1, MBOAT7, TICAM1, VPS53, ANGPTL2, KCTD9, RASD2, MAPKAP1, GCA, SH2B1, MIR148A, MIR144, MIR130A, VCX, SIGLEC9, MIR106A, MIRLET7I, MIRLET7G, ST20, ICOS, TBX21, SFTA2, IL21R, ZACN, STING1, NEAT1, GOLM1, FOLH1B, TLR8, IL23A, APOBEC3A, TDP2, GHRL, ARID2, PTCRA, ACVR2B, SOX9, TMEM11, F11, GJA5, GGT1, FOLH1, FCN2, FCGR3B, FCGR3A, FABP4, F2, NR3C1, EXTL3, ERV3-1, EPO, EPHA1, EPAS1, ATN1, DNMT3A, CXCR3, PDIA3, IFNGR1, HNRNPH1, IFNAR2, HTR4, HSPA1L, HSF4, HP, HOXD13, HOXA10, HLA-J, GSN, HLA-E, HLA-DRB5, HLA-DQB1, HLA-DQA1, HLA-DPA1, HLA-DMA, GZMB, DNMT1, DES, DCK, HCN2, CAT, CASP1, CALCR, VPS51, C3, C2, BGN, BCL2, CYP27B1, STS, ARHGAP4, AQP4, FAS, APOC3, ANGPT2, ADCYAP1, CD86, CD34, CD36, CD40, CYBB, CUX1, CTNNB1, NKX2-5, CST3, CRP, CR1, CPOX, CPB2, COX8A, CCR8, CISH, CHI3L1, CDO1, CDKN2A, IFNB1, IFNGR2, IL18R1, S100A4, CXCL12, CXCL6, CCL24, CCL11, CCL5, S100A12, S100A9, RORA, SLC8A1, RAG1, PSMB9, MAP2K7, MAPK9, PPARG, POU5F1, PON1, SLC6A8, SOD1, IGF1R, UBE2L3, TNFRSF6B, CDR3, XRCC4, VWF, VIM, VHL, UQCRB, TXN, ADAR, TNFRSF1B, TNFRSF1A, TAPBP, TAP2, ADAM17, STAT1, SOX11, PMM2, PNOC, PLAGL1, ITGAE, TNPO1, KLRC2, KIR3DL2, KIR2DS1, KIR2DL1, KCNQ1, KCNH2, IRF7, PLA2G2A, IRF5, IL15, CXCR1, IL2, IL1RN, IL1A, IGFBP2, KRT5, KRT18, LCN2, LGALS3, PIN1, ABCB1, PCYT1A, OPRL1, NUCB1, MT1M, MT1G, MPI, MOV10, CXCL9, CIITA, MBL2, MBD1, MAL, LGALS9, CERNA3

-
Supraventricular Tachycardia
Wikipedia
AV nodal reentrant tachycardia (AVNRT) involves a reentry circuit forming next to, or within, the AV node. ... Classification [ edit ] Impulse arising in SA node , traversing atria to AV node , then entering ventricle. Rhythm originating at or above AV node constitutes SVT. ... Cryoablation is a newer treatment for SVT involving the AV node directly. SVT involving the AV node is often a contraindication for using radiofrequency ablation due to the small (1%) incidence of injuring the AV node, requiring a permanent pacemaker. ... SVTs can be classified by whether the AV node is involved in maintaining the rhythm. If so, slowing conduction through the AV node will terminate it. If not, AV nodal blocking maneuvers will not work, although transient AV block is still useful as it may unmask an underlying abnormal rhythm.CACNA1D, KCNJ2, HCN4, RANGRF, KCNE5, GPD1L, PKP2, AKAP9, ABCC9, RYR1, KCNJ8, SCN1B, SCN2B, SCN5A, SCN10A, SLMAP, LMNA, NAA10, KCNE3, CACNA2D1, JAK2, TRPM4, CACNA1C, SCN3B, CACNA1S, KCND3, CALM2, CACNB2, CALR, PRSS27, FSD1L, FSD1, CAP2, TNNI3K, ACTB, PROC, PTPN11, PROS1, SERPINC1, NPPC, MTHFR, MPL, MPI, IL6, IFNB1, GJA1, CPB2, CHI3L1, BCS1L, ZNF469

-
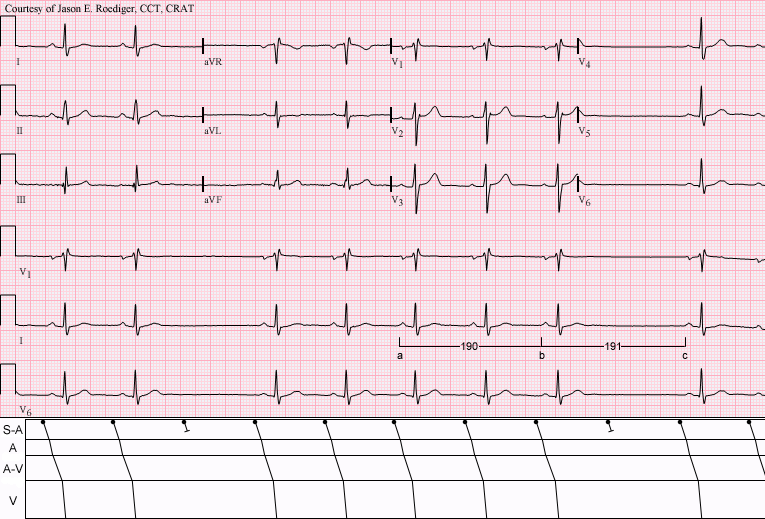
Sinoatrial Block
Wikipedia
The initial action impulse in a heart is usually formed in the sinoatrial node (SA node) and carried through the atria, down the internodal atrial pathways to the atrioventricular node (AV) node. [1] In normal conduction, the impulse would travel across the bundle of His (AV bundle), down the bundle branches , and into the Purkinje fibers . ... In an SA block, the electrical impulse is delayed or blocked on the way to the atria, thus delaying the atrial beat. [2] (An AV block , occurs in the AV node and delays ventricular depolarisation). ... Contents 1 Symptoms 2 Types 3 Treatment 4 References 5 External links Symptoms [ edit ] Sinoatrial blocks are typically well tolerated. They are not as serious as an AV block and most often do not require treatment. ... Second degree SA blocks are broken down into two subcategories just like AV blocks are: The first is a second degree type I, or Wenckebach block. [ citation needed ] This rhythm is irregular, and the R-R interval gets progressively smaller, while the P-R interval remains constant, until a QRS segment is dropped. Note that this is quite different from the Wenckebach AV block, in which the PR interval gets progressively longer, before the dropped QRS segment.
-
Re-Entry Ventricular Arrhythmia
Wikipedia
Conditions necessary for re-entry include a combination of unidirectional block and slowed conduction. [2] Circus movement may also occur on a smaller scale within the AV node (dual AV nodal physiology), a large bypass tract is not necessary. [3] Re-entry is divided into two major types: [Anatomically Defined] re-entry and [Functionally Defined] re-entry. ... WPW syndrome is an atrioventricular re-entrant tachycardia (AVRT), secondary to an accessory pathway that connects the epicardial surfaces of the atrium and ventricle along the AV groove. [4] The majority of time symptomatic WPW fits the definition of AVRT ( Supraventricular tachycardia ) however AVNRT (dual AV nodal physiology) exist in ~10% of patients with WPW syndrome creating the possibility of spontaneous atrial fibrillation degenerating into ventricular fibrillation (VF). ... For reentry to occur, the path length of circuit should be greater than the wave length (ERP × conduction velocity) of impulse. See also [ edit ] AV reentrant tachycardia AV nodal reentrant tachycardia References [ edit ] ^ "Cardiac Arrhythmias" .
-
Aerobic Vaginitis
Wikipedia
Aerobic vaginitis Aerobic vaginitis (in a 14-week pregnant woman): parabasal cells, absent lactobacilli and overgrowth of other bacilli, inflammation Specialty Gynecology Aerobic vaginitis ( AV ) is a form of vaginitis first described by Donders et al. in 2002. [1] [2] It is characterized by a more or less severe disruption of the lactobacillary flora , along with inflammation , atrophy , and the presence of a predominantly aerobic microflora , composed of enteric commensals or pathogens . [3] It can be considered the aerobic counterpart of bacterial vaginosis . ... Ideally, phase-contrast microscopy is used with a magnification of 400x ( high-power field ). [9] For scoring purposes, along with relative number of leucocytes, percentage of toxic leucocytes, background flora and proportion of epitheliocytes, lactobacillary grade must be evaluated: grade I numerous pleiomorphic lactobacilli; no other bacteria grade IIa mixed flora, but predominantly lactobacilli grade IIb mixed flora, but proportion of lactobacilli severely decreased because of an increased number of other bacteria grade III lactobacilli severely depressed or absent because of overgrowth of other bacteria AV score Lactobacillary grades Number of leukocytes Proportion of toxic leucocytes Background flora Proportion of parabasal epitheliocytes 0 I and IIa <10/ hpf None or sporadic Unremarkable or cytolysis None or <1% 1 IIb >10/hpf and; <10/ epithelial cell <50% of leukocytes Small coliform bacilli ≤10% 2 III >10/epithelial cell >50% of leukocytes Cocci or chains >10% The "AV score" is calculated according to what is described in the table. AV score <3: no signs of AV AV score 3 or 4: light AV AV score 5 or 6: moderate AV AV score ≥7:severe AV. pH measurement alone is not enough for the diagnosis. ... In some cases, systemic antibiotics can be helpful, such as amoxyclav or moxifloxacin . [10] Vaginal rinsing with povidone iodine can provide rapid relief of symptoms but does not provide long-term reduction of bacterial loads. [11] Dequalinium chloride can also be an option for treatment. [12] Epidemiology [ edit ] About 5 to 10% of women are affected by aerobic vaginitis. [13] Reports in pregnant women point to a prevalence of 8.3–10.8%. [14] [15] When considering symptomatic women, the prevalence of AV can be as high as 23%. [16] [17] [18] References [ edit ] ^ Donders, Gilbert G.G.; Vereecken, Annie; Bosmans, Eugene; Dekeersmaecker, Alfons; Salembier, Geert; Spitz, Bernard (2002).

-
Familial Progressive Cardiac Conduction Defect
Orphanet
A genetic cardiac rhythm disease that may progress to complete atrioventricular (AV) block. The disease is either asymptomatic or manifests as dyspnea, dizziness, syncope, abdominal pain, heart failure or sudden death. ... Diagnostic methods Diagnosis of familial PCCD relies on family history of syncope, pacemaker implantation, and sudden death as well as on echocardiogram (ECG) findings showing a major conduction defect (complete right bundle branch block, complete left bundle branch block, left anterior fascicular block / hemiblock or left posterior hemiblock, prolonged PR interval or complete AV block with broad QRS complexes). In most cases, normal cardiac structure and contractile function are observed, but in some, complete AV block can lead to dilation of the left ventricle and heart failure. Ventricular tachycardia and torsade de pointes may be recorded during the recovery phase of an exercise stress test or during complete AV block. Potential congenital heart disease or cardiomyopathy is investigated by ECG or cardiac MRI. ... A high incidence of sudden death is observed in patients with either first-degree AV block in association with bifascicular block or in those with symptomatic advanced AV block.













