Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
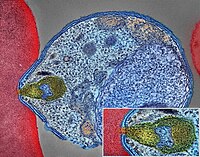
Malaria
Mayo Clinic
To reduce malaria infections, world health programs distribute preventive drugs and insecticide-treated bed nets to protect people from mosquito bites. The World Health Organization has recommended a malaria vaccine for use in children who live in countries with high numbers of malaria cases. Protective clothing, bed nets and insecticides can protect you while traveling. ... The World Health Organization estimates that about 94% of all malaria deaths occur in Africa — most commonly in children under the age of 5. Malaria deaths are usually related to one or more serious complications, including: Cerebral malaria. ... Do not use products with oil of lemon eucalyptus (OLE) or p-Menthane-3,8-diol (PMD) on children under age 3. Apply repellent to clothing. Sprays containing permethrin are safe to apply to clothing. Sleep under a net. Bed nets, particularly those treated with insecticides, such as permethrin, help prevent mosquito bites while you are sleeping.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
-
Epileptic Encephalopathy, Early Infantile, 45
OMIM
The patient had onset of seizures at age 12 months and showed developmental regression at age 35 months. ... Lien et al. (2016) reported a 32-month-old boy with severe developmental delay and hypotonia who developed refractory epilepsy at age 3 months. Brain imaging was normal. ... In vitro functional studies in HEK293 cells showed that the mutation altered the kinetic properties of the channel, resulting in the net loss of GABAergic inhibition. In a boy with EIEE45, Lien et al. (2016) identified a de novo heterozygous missense mutation in the GABRB1 gene (T287I; 137190.0002).
-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Gerontophobia
Wikipedia
Part of a series on Discrimination General forms Age Class ( Caste ) Physical Disability Education Economic Employment Genetics Hair texture Height Housing Language Looks Race / Ethnicity / Nationality Rank Religion Sanity Sex Sexual orientation Size Skin color Specific forms Social Acephobia Adultism Amatonormativity Anti-albinism Anti-autism Anti-homelessness Anti-intellectualism Anti-intersex Anti-left handedness Anti-Masonry Antisemitism (Judeophobia) Aporophobia Audism Biphobia Clannism Cronyism Drug use Elitism Ephebiphobia Fatism Gerontophobia Heteronormativity Heterosexism HIV/AIDS stigma Homophobia Leprosy stigma Lesbophobia Misandry Misogyny Nepotism Pedophobia Perpetual foreigner Pregnancy Reverse Sectarianism Supremacism Black White Transphobia Non-binary Transmisogyny Vegaphobia Xenophobia Religious Ahmadiyya Atheism Baháʼí Faith Buddhism Catholicism Christianity post–Cold War era Druze Falun Gong Hinduism Persecution Islam Persecution Jehovah's Witnesses Judaism Persecution LDS or Mormon Neopaganism Eastern Orthodox Oriental Orthodox Copts Protestantism Rastafarianism Shi'ism Sufism Sunnism Zoroastrianism Ethnic/national African Albanian American Arab Armenian Australian Austrian Azerbaijani British Canadian Catalan Chechen Chilean Chinese Croat Dutch English Estonian European Filipino Finnish French Georgian German Greek Haitian Hazara Hispanic Hungarian Igbo Indian Indonesian Iranian Irish Israeli Italian Japanese Jewish Khmer Korean Kurdish Malay Manchu Mexican Middle Eastern Mongolian Montenegrin Pakistani Pashtun Polish Portuguese Quebec Romani Romanian Russian Scottish Serb Slavic Somali Soviet Tatar Thai Tibetan Turkish Ukrainian Venezuelan Vietnamese Western Manifestations Blood libel Bullying Compulsory sterilization Counter-jihad Cultural genocide Defamation Democide Disability hate crime Dog-whistle politics Eliminationism Ethnic cleansing Ethnic conflict Ethnic hatred Ethnic joke Ethnocide Forced conversion Freak show Gay bashing Gendercide Genital modification and mutilation Genocide examples Glass ceiling Hate crime Hate group Hate speech online Homeless dumping Indian rolling Lavender scare LGBT hate crimes Lynching Mortgage Murder music Occupational segregation Persecution Pogrom Purge Red Scare Religious persecution Religious terrorism Religious violence Religious war Scapegoating Segregation academy Sex-selective abortion Slavery Slut-shaming Trans bashing Victimisation Violence against women White flight White power music Wife selling Witch-hunt Policies Age of candidacy Blood purity Blood quantum Crime of apartheid Disabilities Catholic Jewish Ethnocracy Ethnopluralism Gender pay gap Gender roles Gerontocracy Gerrymandering Ghetto benches Internment Jewish quota Jim Crow laws Law for Protection of the Nation McCarthyism MSM blood donation restrictions Nonpersons Numerus clausus (as religious or racial quota) Nuremberg Laws One-drop rule Racial quota Racial steering Redlining Same-sex marriage (laws and issues prohibiting) Segregation age racial religious sexual Sodomy law State atheism State religion Ugly law Voter suppression Countermeasures Affirmative action Anti-discrimination law Cultural assimilation Cultural pluralism Diversity training Empowerment Feminism Fighting Discrimination Hate speech laws by country Human rights Intersex rights LGBT rights Masculism Multiculturalism Nonviolence Racial integration Reappropriation Self-determination Social integration Toleration Related topics Allophilia Anti-cultural, anti-national, and anti-ethnic terms Bias Christian privilege Civil liberties Cultural assimilation Dehumanization Diversity Ethnic penalty Eugenics Internalized oppression Intersectionality Male privilege Masculism Medical model of disability autism Multiculturalism Net bias Neurodiversity Oikophobia Oppression Police brutality Political correctness Polyculturalism Power distance Prejudice Prisoner abuse Racial bias in criminal news Racism by country Religious intolerance Second-generation gender bias Snobbery Social exclusion Social model of disability Social stigma Stereotype threat The talk White privilege v t e Gerontophobia is the fear of age-related self-degeneration (similar to Gerascophobia ), or a hatred or fear of the elderly due to memento mori . The term comes from the Greek γέρων – gerōn , "old man" [1] and φόβος – phobos , "fear". [2] Contents 1 Ageism 2 See also 3 References 4 External links Ageism [ edit ] Discriminatory aspects of ageism have been strongly linked to gerontophobia . [3] This irrational fear or hatred of the elderly is associated with the fact that someday all young people including oneself will be old inevitably and suffer from the irreversible health decline that comes with old age , which is associated with disability , disease and death . The sight of aged people is a reminder of death ( memento mori ) and inevitable biological vulnerability. ... External links [ edit ] AGEISM AND AGING UP: A Q and A with Mariah MedFriendly Age Wave v t e Discrimination General forms Age Caste Class Disability Education Economic Employment Genetic Hair texture Height Housing Language Looks Race / Ethnicity / Nationality Rank Sanity Sex Sexual orientation Size Skin color Social Acephobia Adultism Amatonormativity Anti-albinism Anti-autism Anti-homelessness Anti-intellectualism Anti-intersex Anti-left handedness Anti-Masonry Antisemitism (Judeophobia) Aporophobia Audism Biphobia Clannism Cronyism Drug use Elitism Ephebiphobia Fatism Gerontophobia Heteronormativity Heterosexism HIV/AIDS stigma Homophobia Leprosy stigma Lesbophobia Misandry Misogyny Nepotism Pedophobia Perpetual foreigner Pregnancy Reverse Sectarianism Supremacism Black White Transphobia Non-binary Transmisogyny Vegaphobia Xenophobia Religious Ahmadiyya Atheism Baháʼí Faith Buddhism Catholicism Christianity post–Cold War era Falun Gong Hinduism Persecution Islam Persecution Jehovah's Witnesses Judaism Persecution LDS or Mormon Neopaganism Eastern Orthodox Oriental Orthodox Protestantism Rastafarianism Shi'ism Sufism Zoroastrianism Ethnic/National African Albanian American Arab Armenian Australian Austrian British Canadian Catalan Chilean Chinese Croat Dutch English Estonian European Filipino Finnish French Georgian German Greek Haitian Hazara Hindu Hispanic Hungarian Igbo Indian Indonesian Iranian Irish Israeli Italian Japanese Jewish Khmer Korean Kurdish Malay Manchu Mexican Middle Eastern Mongolian Pakistani Pashtun Polish Portuguese Quebec Romani Romanian Russian Scottish Serb Slavic Somali Soviet Tatar Thai Turkish Ukrainian Venezuelan Vietnamese Western Manifestations Blood libel Bullying Compulsory sterilization Counter-jihad Cultural genocide Defamation Democide Disability hate crime Dog-whistle politics Eliminationism Enemy of the people Ethnic cleansing Ethnic conflict Ethnic hatred Ethnic joke Ethnocide Forced conversion Freak show Gay bashing Gendercide Genital modification and mutilation Genocide examples Glass ceiling Hate crime Hate group Hate speech Homeless dumping Indian rolling Lavender scare LGBT hate crimes Lynching Mortgage Murder music Native American sports mascots Occupational segregation Persecution Pogrom Purge Red Scare Religious persecution Religious terrorism Religious violence Religious war Scapegoating Segregation academy Sex-selective abortion Slavery Slut-shaming Trans bashing Victimisation Violence against women White flight White power music Wife selling Witch-hunt Discriminatory policies Age of candidacy Blood purity Blood quantum Crime of apartheid Disabilities Catholic Jewish Ethnocracy Ethnopluralism Gender pay gap Gender roles Gerontocracy Gerrymandering Ghetto benches Internment Jewish quota Jim Crow laws Law for Protection of the Nation McCarthyism MSM blood donation restrictions Nonpersons Numerus clausus (as religious or racial quota) Nuremberg Laws One-drop rule Racial quota Racial steering Redlining Same-sex marriage (laws and issues prohibiting) Segregation age racial religious sexual Sodomy law State atheism State religion Ugly law Voter suppression Countermeasures Affirmative action Anti-discrimination law Cultural assimilation Cultural pluralism Diversity training Empowerment Feminism Fighting Discrimination Hate speech laws by country Human rights Intersex rights LGBT rights Masculism Multiculturalism Nonviolence Racial integration Reappropriation Self-determination Social integration Toleration Related topics Allophilia Anti-cultural, anti-national, and anti-ethnic terms Bias Christian privilege Civil liberties Cultural assimilation Dehumanization Diversity Ethnic penalty Eugenics Internalized oppression Intersectionality Male privilege Masculism Medical model of disability autism Multiculturalism Net bias Neurodiversity Oikophobia Oppression Police brutality Political correctness Polyculturalism Power distance Prejudice Prisoner abuse Racial bias in criminal news Racism by country Religious intolerance Second-generation gender bias Snobbery Social exclusion Social model of disability Social stigma Stereotype threat The talk White privilege Category
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Maple Syrup Urine Disease
GeneReviews
Acute metabolic decompensation is corrected by treating the precipitating stress while delivering sufficient calories, insulin, free amino acids, isoleucine, and valine to achieve sustained net protein synthesis in tissues. Some centers use hemodialysis/hemofiltration to remove BCAAs from the extracellular compartment, but this intervention does not alone establish net protein accretion. ... Signs of deepening encephalopathy including lethargy, intermittent apnea, opisthotonus, and stereotyped movements such as "fencing" and "bicycling" are evident by age four to five days. Coma and central respiratory failure may occur by age seven to ten days, sometimes before newborn screening results are available. ... Following the neonatal period, acute metabolic intoxication (leucinosis) and neurologic deterioration can develop rapidly at any age as a result of net protein degradation precipitated by infection, surgery, injury, or psychological stress (see Figure 1). ... Plasma leucine levels rise predictably as a result of net protein catabolism provoked by a variety of physiologic stresses, including (more...) ... Coronal T 2 -weighted MRI from a Mennonite boy age five years during an acute metabolic crisis.DBT, BCKDHB, BCKDHA, BCAT2, PPM1K, DLD, ARID4B, BDNF, CTSD, SERPINE1, TNS3, CACNA2D2, MKRN3, UMOD, SPN, NME1, PAH, NBN, MEA1, IL1B, GPR4, GLI2, F2, MECP2

-
Rahman Syndrome
OMIM
Clinical Features Tatton-Brown et al. (2017) reported 5 unrelated patients, ranging in age from 1.9 to 16 years, with mild to severe intellectual disability associated with variable somatic overgrowth, including height, weight, and/or head circumference. ... Additional features, each found only in 1 or 2 patients, included kyphoscoliosis, camptodactyly, talipes equinovarus, advanced bone age, dental anomalies, skin nevi, strabismus, astigmatism, and amblyopia. ... The truncated proteins were predicted to have a reduced net charge compared to the wildtype protein, rendering them likely to be less effective in neutralizing negatively charged linker DNA. ... INHERITANCE - Autosomal dominant GROWTH Height - Increased birth length - Increased height Weight - Increased birth weight - Increased weight HEAD & NECK Head - Large head circumference Face - Full cheeks Eyes - Telecanthus - Strabismus - Amblyopia - Astigmatism ABDOMEN Gastrointestinal - Poor feeding in the neonatal period SKELETAL - Advanced bone age Spine - Kyphoscoliosis Hands - Camptodactyly Feet - Talipes equinovarus SKIN, NAILS, & HAIR Skin - Nevi MUSCLE, SOFT TISSUES - Hypotonia, neonatal - Hypertonia, neonatal NEUROLOGIC Central Nervous System - Delayed development - Intellectual disability, mild to severe MISCELLANEOUS - Highly variable features - De novo mutation MOLECULAR BASIS - Caused by mutation in the histone gene cluster 1, H1 histone family, member E gene (HIST1H1E, 142220.0001 ) ▲ Close
-
Neuroendocrine Tumor
Wikipedia
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Mutyh Polyposis
GeneReviews
Surveillance: Colonoscopy with polypectomy every one to two years beginning at age 25-30 years; upper endoscopy and side viewing duodenoscopy every three months to four years beginning at age 30-35 years with subsequent follow up based on initial findings. ... The risk was found to be higher in one study, with median age at diagnosis 53 years (range 45-76) [Vogt et al 2009]. ... Two of 15 probands with familial NET of the small intestine and four of 215 individuals with nonfamilial NET of the small intestine were heterozygous for MUTYH pathogenic variant p.Gly396Asp [Dumanski et al 2017]. It is unclear if a heterozygous MUTYH pathogenic variant is a risk factor for NET or ACC, as the risk of NET or ACC in individuals with biallelic MUTYH pathogenic variants appears to be quite low. ... NCCN [2019] guidelines propose that MUTYH heterozygotes with a first-degree relative with CRC (who does not have MAP) undergo colonoscopy every five years beginning at age 40 years or ten years prior to the age of the first-degree relative's age at CRC diagnosis.
-
Epilepsy, Progressive Myoclonic, 10
OMIM
The proband, who was the most severely affected, developed school difficulties, dysarthria, and myoclonus at age 5 years. She then developed seizures, ataxia, bladder incontinence; she became mute by age 12 and was wheelchair-bound by age 14. At age 34, she was bedridden and unresponsive, with spastic tetraplegia. ... Studies in patient tissues showed no net change in glycogen synthase (see, e.g., GYS1, 138570) activity, indicating that polyglucosan formation was not related to glycogen synthase.
-
Yellow Fever
Orphanet
Management and treatment As there is presently no antiviral drug available for YF, treatment is supportive, following the guidelines for treatment of severe septicemia. Insecticide-treated bed nets and/or room screens should be used in open-air settings to prevent further transmission. ... YF-17D is indicated for persons over 9 months of age who are traveling to or living in YF endemic areas. Vaccination is contraindicated in children <4 months old and pregnant women and caution is advised in persons with egg allergy, the immunocompromised, children 4-9 months of age and elderly persons, especially if the risk is minimal, such as trips restricted to attending conferences in modern urban hotels with no rural exposure.ERVK-32, ERVK-6, DDOST, PARTICL, TRIM56, CYP20A1, EMC3, TRAV12-2, BACE1, AMACR, TRPA1, TFPI, RPL19, RAF1, CRP, MRC1, IL10, IL6, IL1RN, IFNG, IFNAR1, GYPC, GPT, GLS, FCGR3B, FCGR3A, F10, MAP2K1

-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Sneddon Syndrome
Orphanet
The disease predominantly affects women in young adulthood. Clinical description The mean age of onset of neurological symptoms is 39 years, though the livedo is generally observed up to 10 years earlier and sometimes since childhood. Livedo racemosa is a persistent net-like violaceous-cyanotic, mottled discoloration of the skin affecting primarily the legs and arms, but also involving the buttocks and the trunk, and that is exacerbated by cold or pregnancy. ... It has to be considered in cases of unexplained stroke at a young age, cognitive decline without stroke, and assumed autoimmune-related vasculitis in which immunosuppressive therapy has proven ineffective.

-
Retinal Dystrophy, Reticular Pigmentary, Of Posterior Pole
OMIM
Description Reticular pigmentary retinal dystrophy is a form of patterned dystrophy (see MDPT1, 169150) characterized by a reticular pattern of pigmentation that likely appears in infancy and may be fully developed at age 15 years. Indirect funduscopy has shown that the condition is bilateral and symmetric and that the pigmentary deposits are localized below the neuroepithelium, very likely in the pigment epithelium. ... Schauwvlieghe et al. (2013) described 3 children with reticular dystrophy, 2 North American sisters, aged 10 years and 14 years, and an unrelated 12-year-old Belgian boy. ... Variation in the presence of autofluorescent chromophores was observed, with the older sister and the unrelated boy exhibiting a milder and more punctiform hyperautofluorescence of the net, whereas the younger sister showed a more intense hyperautofluorescent pattern.
-
Epithelial Recurrent Erosion Dystrophy
OMIM
The disorder became manifest between 4 and 6 years of age. Recurring ulcerations are also seen in macular and lattice types of classic dystrophy. ... Affected individuals presented within the first decade of life with corneal erosions that occurred approximately every 2 to 3 months. With increasing age, the erosions became much less frequent, and the episodes appeared to cease in the third decade of life. ... Most patients had onset of symptoms between 6 and 7 years of age, although 3 patients reported late onset and 2 family members were asymptomatic despite characteristic corneal changes. ... Examination revealed characteristic diffuse subepithelial opacities in the paracentral cornea, sometimes showing a net-like pattern with raised areas of Salzmann degeneration, and the opacities tended to increase with age. ... Patients presented between 5 and 7 years of age, and episodes of erosions decreased in frequency over time, subsiding in the third to fourth decade.
-
Mosquito-Borne Disease
Wikipedia
Depending on the mosquito vector, and the affected community, a variety of prevention methods may be deployed at one time. Insecticidal nets and indoor residual spraying [ edit ] The use of insecticide treated mosquito nets (ITNs) are at the forefront of preventing mosquito bites that cause malaria. ... Because the Anopheles gambiae feeds indoors (endophagic) and rests indoors after feeding (endophilic), insecticide treated nets (ITNs) interrupt the mosquito's feeding pattern. The ITNs continue to offer protection, even after there are holes in the nets, because of their excito-repellency properties which reduce the number of mosquitoes that enter the home. ... The risk of complications from the vaccine are greater for individuals over 60 years of age. In addition, the vaccine is not usually administered to babies under nine months of age, pregnant women, people with allergies to egg protein, and individuals living with AIDS/HIV . ... "The Impact of Pyrethroid Resistance on the Efficacy of Insecticide-Treated Bed Nets against African Anopheline Mosquitoes: Systematic Review and Meta-Analysis" .

-
West Nile Fever
Wikipedia
Prevention [ edit ] Low-cost, ceiling hung mosquito netting for a bed Many of the guidelines for preventing occupational West Nile virus exposure are common to all mosquito-borne diseases . [57] Public health measures include taking steps to reduce mosquito populations. ... DEET formulations as high as 30% are recommended for children over two months of age. [58] The CDC also recommends the use of: IR3535, oil of lemon eucalyptus, para-menthane-diol, or 2-undecanone. [59] Protect infants less than two months of age by using a carrier draped with mosquito netting with an elastic edge for a tight fit. ... Repellents containing permethrin ( e.g. , Permanone) or other insect repellents may be applied to clothing, shoes, tents, mosquito nets, and other gear. (Permethrin is not suitable for use directly on skin.) ... Retrieved 28 October 2017 . ^ Gompf, Sandra. "West Nile Virus" . Medicine Net . MedicineNet Inc . Retrieved 15 January 2019 . ^ "Symptoms, Diagnosis, & Treatment" . ... S2CID 31695008 . ^ Glass, WG; Lim JK; Cholera R; Pletnev AG; Gao JL; Murphy PM (October 17, 2005).CCR5, ERVK-32, ROBO3, MAVS, DDX58, PLAAT4, IFIT2, ERVK-6, STAT1, SPP1, OAS1, IL1B, IFNB1, RNASEL, CASP8, HLA-DRB1, PELI1, SELENBP1, ARHGEF2, LRRFIP1, NAMPT, TRAIP, RIPK3, SEC14L2, CSF1R, LAMP3, ERVW-1, FOXP3, ZMYND10, DDX56, CCR7, VCP, CDKN2A, IFIH1, DHX58, ZBP1, HAVCR2, PIK3IP1, NLRP3, TNFRSF13C, TRIM6, RBM45, CCR2, ERVK-20, ERVK-18, VAMP8, TNFRSF1A, IFNA1, TNF, IFNA13, HLA-DQA1, IL1A, HLA-C, IL10, IL17A, IL18, IRF3, IRF5, KIR2DL2, KIR3DL1, KIR3DS1, LSAMP, CD180, SMAD4, MMP9, HLA-A, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PZP, GLS, CASP1, SNCA, GEM, DDX3X, TAP1, TLR3, ATF4

-
Cryohydrocytosis
OMIM
Clinical Features Miller et al. (1965) described a 19-year-old man who presented with jaundice and splenomegaly at age 13 years and was found to have moderately severe hemolytic anemia. ... The proband, who had already undergone splenectomy, presented at age 40 years with shortness of breath and was found to have multiple pulmonary emboli. ... The original proband, who was the only member of the pedigree to be splenectomized, died at age 51 of pulmonary hypertension, which the authors stated was likely due to postsplenectomy thrombotic complications that are typical of the hereditary stomatocytoses. ... The Darlington proband, a farmer who had undergone splenectomy at age 22 years for a presumed diagnosis of 'atypical spherocytosis,' developed severe thrombosis of the superficial femoral vein at age 40. ... Unidirectional K(+) influx measurements showed that the patient's cells had abnormally high activities of the K(+)Na(+)/H(+) exchanger (KNHE) and the K(+),Cl(-) cotransporter (KCC), which could account for the observed net movements of cations. Neither chloride nor cation conductance in patient RBCs differed from that of healthy donors.SLC4A1, IFNA1, IFNL3, PAEP, IL10, IFNA13, IFNA2, GPT, PNPLA3, IFNL4, CXCL10, MIR122, ADIPOQ, FBL, TNF, IFNG, AFP, HFE, IL6, TLR3, LEP, STOM, LGALS3BP, TLR7, MTHFR, TM6SF2, RBP4, HLA-C, VDR, TP53, HAMP, IL21, ITPA, IGF1, CLTC, SCARB1, SOCS1, KIR3DL1, IL22, IL15, MIR146A, IFNB1, APOB, TLR4, HLA-DRB1, HLA-DQA1, MIR145, HLA-B, HLA-A, SOCS3, UGT1A1, SLC2A1, TLR2, GEM, TLL1, SEMA3C, RNF7, NR1D2, MIR21, MIR324, AKT3, MIR27A, WDHD1, EIF2AK3, POLG2, USP18, MIR19A, MIR199A2, KLF12, GDF15, TNFRSF1A, MIR221, DDX58, MIR224, IER3, NR0B2, SEMA5A, VEGFA, MIR296, PGR-AS1, ABCB11, VCAM1, SLC33A1, TNFSF4, TRAF6, MIR34A, HEIH, SPEN, IFNL2, GALNT8, SIKE1, MAVS, MIR106A, CISD3, IFIH1, MBOAT7, CYBRD1, SEMA6D, RNF34, SLC17A5, IFNL1, GGT2, GGTLC3, SLC4A11, CYP2R1, RBM45, A2ML1, AICDA, GGTLC4P, RALGAPB, MIR130A, TBK1, TBX21, MIR199A1, SLC40A1, IFNLR1, IL21R, GOLM1, MIR195, MIR1307, TLR8, MIR192, GHRL, RTEL1, GGTLC5P, CD24, MIR602, PLIN2, TGFB1, CHI3L1, CMKLR1, CNR1, CNR2, CPT1A, CRP, CSF2, CST3, CTLA4, CYP2E1, DBP, DDX5, DPP4, EPO, ETFA, ETV6, FBP1, FCAR, FOXM1, GGT1, CCR5, CD69, TGFA, CD38, AGTR1, ANGPT2, APOA1, APOE, APP, AQP4, ARG1, ATM, BCL2, BCL2A1, BCL6, BGN, VPS51, CAD, CASP1, CD247, CD14, CD27, CD34, GPC3, GOT1, GOT2, CXCR3, NUDT1, MTTP, MUC1, MX1, NCAM1, OGG1, PECAM1, PIK3R2, PPP2CA, PTPA, PLAAT4, RIT2, CCL4, CCL22, CXCL11, SLC6A4, SPP1, STAT1, TAPBP, MBL2, SMAD4, LPL, IL17A, HBB, HLA-DQA2, IGF1R, IL4, IL5, CXCR2, IL10RA, TNFRSF9, IMPDH2, LGALS9, ITGAE, JUN, KDR, L1CAM, LAG3, LALBA, LAMC2, LBP, RARRES2
-
Nephrolithiasis, Calcium Oxalate
OMIM
Patients With Confirmed SLC26A1 Variants Gee et al. (2016) reported a boy of Macedonian descent with onset of acute renal failure due to calcium oxalate nephrolithiasis at 5 years of age. He also had ureteropelvic junction obstruction necessitating surgery. ... The inverse association was found independently of age, gender, race/ethnicity, region, and antibiotic use. ... In vitro flux studies indicated that mice lacking Slc26a6 have a defect in intestinal oxalate secretion resulting in enhanced net absorption of oxalate. Jiang et al. (2006) concluded that the anion exchanger, SLC26A6, has a major constitutive role in limiting net intestinal absorption of oxalate, thereby preventing hyperoxaluria and calcium oxalate urolithiasis.







