Joseph Goldberger Pellagra was studied mostly in Europe until the late 19th century when it became an epidemic especially in the southern United States. [29] [30] In the early 1900s, pellagra reached epidemic proportions in the American South. [30] Between 1906 and 1940 more than 3 million Americans were affected by pellagra with more than 100,000 deaths, yet the epidemic resolved itself right after dietary niacin fortification. [31] Pellagra deaths in South Carolina numbered 1,306 during the first ten months of 1915; 100,000 Southerners were affected in 1916. At this time, the scientific community held that pellagra was probably caused by a germ or some unknown toxin in corn. [31] The Spartanburg Pellagra Hospital in Spartanburg, South Carolina , was the nation's first facility dedicated to discovering the cause of pellagra.
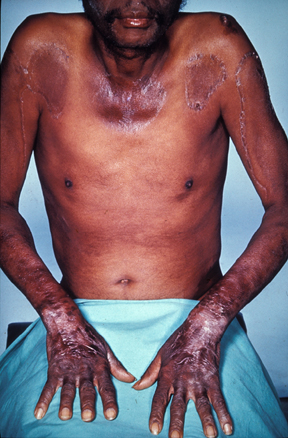
Pellagra is a nutritional disorder caused by a deficiency in niacin (vitamin B3) or its precursor (tryptophan) that is mainly observed in Asia and Africa where it is generally due to poor nutrition. It is characterized by dermatitis (symmetrical photodistributed erythema that may be accompanied by vesicles and bullae, and that develops into hyperkeratotic and hyperpigmented skin), gastrointestinal symptoms (diarrhea), and neuropsychiatric disorders (dementia). It can be life-threatening without a correct management.
Fast recognition and treatment of MH utilizes skills and procedures that are utilized with a low-frequency and high-risk. [35] Conducting MH crisis training for perioperative teams can identify system failures as well as improve response to these events. [36] Simulation techniques to include the use of cognitive aids have also been shown to improve communication in clinical treatment of MH. [37] [38] Prognosis [ edit ] Prognosis is poor if this condition is not aggressively treated. [5] In the 1970s, mortality was greater than 80%; with the current management, however, mortality is now less than 5%. [5] Epidemiology [ edit ] It occurs in between 1:5,000 and 1:100,000 in procedures involving general anaesthesia. [5] This disorder occurs worldwide and affects all racial groups.
Summary Clinical characteristics. Malignant hyperthermia susceptibility (MHS) is a pharmacogenetic disorder of skeletal muscle calcium regulation associated with uncontrolled skeletal muscle hypermetabolism. Manifestations of malignant hyperthermia (MH) are precipitated by certain volatile anesthetics (i.e., halothane, isoflurane, sevoflurane, desflurane, enflurane), either alone or in conjunction with a depolarizing muscle relaxant (specifically, succinylcholine). The triggering substances cause uncontrolled release of calcium from the sarcoplasmic reticulum and may promote entry of extracellular calcium into the myoplasm, causing contracture of skeletal muscles, glycogenolysis, and increased cellular metabolism, resulting in production of heat and excess lactate. Affected individuals experience acidosis, hypercapnia, tachycardia, hyperthermia, muscle rigidity, compartment syndrome, rhabdomyolysis with subsequent increase in serum creatine kinase (CK) concentration, hyperkalemia with a risk for cardiac arrhythmia or even cardiac arrest, and myoglobinuria with a risk for renal failure. In nearly all cases, the first manifestations of MH (tachycardia and tachypnea) occur in the operating room; however, MH may also occur in the early postoperative period.
For a phenotypic description and a discussion of genetic heterogeneity of malignant hyperthermia, see MHS1 (145600). Mapping In a single German pedigree with classic malignant hyperthermia, Sudbrak et al. (1995) found a maximum multipoint lod score of 3.22 for linkage to markers defining a 1-cM interval on 3q13.1. The malignant hyperthermia phenotype was determined by the in vitro contracture test (IVCT) performed on a sample of freshly obtained muscle. Muscle - Masseter or generalized muscle contracture - Rhabdomyolysis Misc - Triggered by certain anesthetics, such as halothane or succinylcholine - Rapid body temperature rise Inheritance - Autosomal dominant (3q13.1) Metabolic - Malignant hyperthermia - Acidosis - Hypoxia ▲ Close
Malignant hyperthermia (MH) is a severe reaction to certain gases used during anesthesia and/or a muscle relaxant used to temporarily paralyze a person during surgery. Signs and symptoms of MH include marked hyperthermia, a rapid heart rate, rapid breathing, acidosis, muscle rigidity, and breakdown of muscle tissue (rhabdomyolysis). Without prompt treatment, MH can be life-threatening. People who are at increased risk for this reaction are said to have MH susceptibility. Susceptibility to MH may be caused by mutations in any of several genes and is inherited in an autosomal dominant manner. People with certain inherited muscle diseases (e.g., central core disease and multiminicore disease ) also have MH susceptibility.
For a phenotypic description and a discussion of genetic heterogeneity of malignant hyperthermia, see MHS1 (145600). By linkage studies in 3 families, Sudbrak et al. (1993) excluded linkage either to chromosome 19 or 17q, thus suggesting the existence of a third locus for malignant hyperthermia susceptibility. In MHS families linked to neither chromosome 17 nor chromosome 19, Iles et al. (1994) found linkage with no recombination to markers flanking the CACNA2D1 gene (114204) on chromosome 7. Since this gene encodes a subunit of the L-type voltage-dependent calcium channel that is intimately associated at the skeletal muscle triadic junctions with the ryanodine receptor (RYR1; 180901), it is possible that the mutation is located in this gene. In affected members of a family linked to the MHS3 locus by Iles et al. (1994), Schleithoff et al. (1999) did not identify any pathogenic mutations in the coding region of the CACNA2D1 gene.
Overview Malignant hyperthermia is a severe reaction to certain drugs used for anesthesia. This severe reaction typically includes a dangerously high body temperature, rigid muscles or spasms, a rapid heart rate, and other symptoms. Without prompt treatment, the complications caused by malignant hyperthermia can be fatal. In most cases, the gene that puts you at risk of malignant hyperthermia is inherited, though sometimes it's the result of a random genetic change. Genetic testing can reveal whether you have an affected gene. This genetic disorder is called malignant hyperthermia susceptibility (MHS).
For a phenotypic description and a discussion of genetic heterogeneity of malignant hyperthermia, see MHS1 (145600). Mapping In a Belgian kindred with malignant hyperthermia, Robinson et al. (1997) performed a genomewide search for the locus responsible for this disorder. Disease status was classified according to the European IVCT (in vitro contracture tests) protocol. Multipoint linkage analysis showed linkage to the region on 5p between D5S419 and D5S398. They also found linkage of MHS to chromosome 1q (601887). Robinson et al. (1997) stated that studies in a third kindred suggested the existence of at least 1 more MHS locus.
Malignant hyperthermia (MH) is a pharmacogenetic disorder of skeletal muscle that presents as a hypermetabolic response to potent volatile anesthetic gases such as halothane, sevoflurane, desflurane and the depolarizing muscle relaxant succinylcholine, and rarely, to stresses such as vigorous exercise and heat. Epidemiology The incidence of MH reactions ranges from 1/5000 to 1/50,000-100,000 anesthesias. However, the genetic prevalence of the genetic abnormalities may be as great as 1/400 individuals. A significant male preponderance has been reported. Clinical description Clinical symptoms of MH are highly variable, ranging from self limiting courses with mild or moderate symptoms to fulminant MH crises. The classic signs of MH include marked hyperthermia, tachycardia, supraventricular and ventricular arrhythmia, tachypnea, increased carbon dioxide production, increased oxygen consumption, acidosis, isolated masseter spasm or generalized muscle rigidity and rhabdomyolysis, all of which are related to a hypermetabolic response.
For a phenotypic description and a discussion of genetic heterogeneity of malignant hyperthermia, see MHS1 (145600). Mapping In 3 unrelated families, Levitt et al. (1991) excluded linkage of the MHS phenotype to loci on 19q13.1, thus indicating genetic heterogeneity. Levitt et al. (1992) extended these studies to 16 MHS families. Four were found to be linked to chromosome 19; 5 were found to be closely linked to the anonymous marker NM23 (156490) on 17q11.2-q24 (maximum lod = 3.26 at theta = 0.0); and 2 families were clearly unlinked to either of these regions. In 5 additional families, there were insufficient data to determine their linkage status. Olckers et al. (1992) provided evidence for linkage of MHS to the SCN4A gene (603967), which encodes the adult sodium channel alpha subunit, in 3 informative families (cumulative lod score of 2.1 at theta = 0.0).
A number sign (#) is used with this entry because susceptibility to malignant hyperthermia-5 (MHS5) is caused by heterozygous mutation in the CACNA1S gene (114208) on chromosome 1q32. For a phenotypic description and a discussion of genetic heterogeneity of malignant hyperthermia, see MHS1 (145600). Mapping In a collaborative study in 3 pedigrees in Europe, in which disease status was classified according to the European in vitro contracture test (IVCT), Robinson et al. (1997) performed a genomewide screen and found that at least 2 further loci exist for MH susceptibility. One of these was located on 5p (601888). The other was located on 1q, between markers D1S422 and D1S1660. Between these 2 markers had already been localized a candidate gene, CACNL1A3 (CACNA1S; 114208), assigned to 1q32.
Malignant hyperthermia is a severe reaction to particular anesthetic drugs that are often used during surgery and other invasive procedures. Specifically, this reaction occurs in response to some anesthetic gases, which are used to block the sensation of pain, either given alone or in combination with a muscle relaxant that is used to temporarily paralyze a person during a surgical procedure. If given these drugs, people at risk of malignant hyperthermia may experience a rapid increase in heart rate and body temperature (hyperthermia), abnormally fast breathing, muscle rigidity, breakdown of muscle fibers (rhabdomyolysis), and increased acid levels in the blood and other tissues (acidosis). Without prompt treatment and cessation of the drugs, the body's reaction can cause multiple organs to be unable to function, including the heart (cardiac arrest) and kidneys (renal failure), and it can cause a blood clotting abnormality called disseminated intravascular coagulation. These complications may be life-threatening. (In medicine, the term malignant refers to conditions that are dangerous to one's health.)
Spanish Minister of Health María Luisa Carcedo declared that the outbreak had expanded out of the Andalusian Autonomous Community , [45] with one confirmed case in Extremadura , [46] and five suspected cases in Extremadura and Madrid . [47] The Minister specified that, although new cases could appear as a consequence of the long incubation period , the contaminated product had been withdrawn totally. [48] As of 21 August 2019, 132 people were diagnosed and 23 pregnant woman in hospital. [49] On 22 August an international alert was declared. [50] See also [ edit ] List of United States foodborne illness outbreaks 2008 Canadian listeriosis outbreak 2014 Macedonia listeriosis outbreak Listeriosis in animals References [ edit ] ^ a b Ryan KJ, Ray CG, eds. (2003).
A listeria infection or listeriosis is an infection caused by a bacteria known as Listeria monocytogenes . It mainly affects newborn infants, elderly patients, pregnant women and patients who have low immunity. Listeria can be spread by several methods. A common cause is ingestion (food-borne transmission) of unpasteurized milk or contaminated vegetables. It can also be transmitted from mother to fetus during pregnancy or directly to the newborn at the time of delivery. Listeriosis can cause a variety of symptoms, depending on the person and the part of the body affected.
A rare bacterial infectious disease caused by the foodborne pathogen Listeria monocytogenes , characterized by a febrile gastroenteritis, which is usually mild and self-limiting in otherwise healthy persons, but can progress to severe illness in at-risk groups like pregnant women, elderly people, immunocompromised people, and neonates. Complications include sepsis, meningitis, and encephalitis. Listeriosis during pregnancy usually occurs during the third trimester and may lead to preterm labor, miscarriage, stillbirth, or intrauterine infection of the unborn child.
Overview Listeria infection is a foodborne bacterial illness that can be very serious for pregnant women, people older than 65 and people with weakened immune systems. It's most commonly caused by eating improperly processed deli meats and unpasteurized milk products. Healthy people rarely become ill from listeria infection, but the disease can be fatal to unborn babies, newborns and people with weakened immune systems. Prompt antibiotic treatment can help curb the effects of listeria infection. Listeria bacteria can survive refrigeration and even freezing. So people who are at higher risk of serious infections should avoid eating the types of food most likely to contain listeria bacteria.
The first two had acquired the Streptococcus pyogenes bacteria during surgery; the remaining four were community-acquired. [20] The cases generated much newspaper coverage, with lurid headlines such as "Flesh Eating Bug Ate My Face". [21] 1997 Ken Kendrick , former agent and partial owner of the San Diego Padres and Arizona Diamondbacks , contracted the disease.
Necrotizing fasciitis is a serious infection of the skin, the tissue just beneath the skin (subcutaneous tissue), and the tissue that covers internal organs (fascia). Necrotizing fasciitis can be caused by several different types of bacteria, and the infection can arise suddenly and spread quickly. Early signs include flu-like symptoms and redness and pain around the infection site. A prompt diagnosis and treatment are essential. If the infection is not treated promptly, it can lead to multiple organ failure and death. Treatment typically includes intravenous (IV) antibiotics and surgery to remove infected and dead tissue.
Pathogenic variant c.700A>G; p.Thr234Ala has been identified in approximately ten families with paraganglioma/pheochromocytoma with and without renal cell carcinoma [Author, personal communication]. Penetrance Penetrance is currently unknown, as most studies have focused on families with clinical manifestations. ... Human clinical trials using combination therapy for these tumors are currently in development [Author, personal communication]. Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a condition that causes benign tumors of smooth muscle tissue in the skin (cutaneous leiomyomas) and in the uterus in females (uterine leiomyomas, or fibroids). The condition also increases the risk of kidney cancer. Signs and symptoms usually begin in adulthood as skin growths appear on the torso, arms, legs, and occasionally on the face. They tend to increase in size and number over time. About 10% to 16% of people with HLRCC develop a type of kidney cancer called renal cell cancer; symptoms of this cancer may include lower back pain, blood in the urine, and/or a mass in the kidney that can be felt by a physician. Some people have no symptoms until the cancer is advanced. HLRCC is caused by mutations in the FH gene and is inherited in an autosomal dominant manner.
Reed’s syndrome Other names Familial leiomyomatosis cutis et uteri Micrograph showing the characteristic hyalinized papillary cores found in some hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cell carcinomas. H&E stain . Reed’s syndrome is a rare inherited condition characterised by multiple cutaneous leiomyomas and, in women, uterine leiomyomas . It predisposes for renal cell cancer , an association denominated hereditary leiomyomatosis and renal cell cancer , [1] [2] and it is also associated with increased risk of uterine leiomyosarcoma . [3] The syndrome is caused by a mutation in the fumarate hydratase gene, which leads to an accumulation of fumarate . The inheritance pattern is autosomal dominant . Contents 1 Signs and symptoms 1.1 Associated conditions 2 Cause 3 Pathogenesis 4 Diagnosis 4.1 Histology 4.2 Differential diagnosis 5 Treatment 6 Prognosis 7 History 8 Notes 9 References 10 External links Signs and symptoms [ edit ] Almost all women present with uterine fibroids, approximately 76% with dermal manifestations and 10-16% with renal tumors. [3] The uterine fibroids tend to occur at younger age and larger and more numerous than in general population. They may be distinguishable from sporadic fibroids by special histological features such as prominent nucleoli with perinucleolar halos. [4] The skin presentation is of asymmetrical, reddish-brown nodules or papules with a firm consistency, predominantly located on the limbs ( multiple cutaneous leiomyoma ), although they may occur anywhere, including the face.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a disorder in which affected individuals tend to develop benign tumors containing smooth muscle tissue (leiomyomas) in the skin and, in females, the uterus . This condition also increases the risk of kidney cancer. In this disorder , growths on the skin (cutaneous leiomyomas) typically develop in the third decade of life. Most of these growths arise from the tiny muscles around the hair follicles that cause "goosebumps". They appear as bumps or nodules on the trunk, arms, legs, and occasionally on the face. Cutaneous leiomyomas may be the same color as the surrounding skin, or they may be darker.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a hereditary cancer syndrome characterized by a predisposition to cutaneous and uterine leiomyomas and, in some families, to renal cell cancer. Epidemiology The prevalence is unknown. Over 200 families with HLRCC have been reported. Clinical description Disease onset can occur at any age, but is more common in young adults and elderly patients. Multiple or single benign cutaneous leiomyomas are common and usually present at around the age of 25 (range from 10-47 years) as firm papules or nodules that are skin colored to light brown. They are usually localized to the trunk and extremities but sometimes on the face.
A number sign (#) is used with this entry because multiple cutaneous and uterine leiomyomatosis with or without renal cell carcinoma, also referred to as hereditary leiomyatosis and renal cell cancer (HLRCC), is caused by heterozygous mutation in the gene encoding fumarate hydratase (FH; 136850) on chromosome 1q43. Homozygous mutation in the FH gene causes fumarase deficiency (FMRD; 606812). Description Hereditary leiomyomatosis and renal cell cancer is an autosomal dominant tumor predisposition syndrome characterized by the variable development of 3 tumors: cutaneous piloleiomyomata that develop in essentially all patients by age 40 years; leiomyomata (fibroids) of the uterus, and rarely leiomyosarcomas, at a mean age of 30 years (range, 18 to 52 years); and type 2 papillary renal cell carcinoma at a mean age of 46 years (range, 17 to 75 years), which occurs in about 20% of patients. Type 2 papillary renal cell carcinoma is a pathologic subtype characterized by large tumor cells with eosinophilic cytoplasm and pseudostratified nuclei; it shows an aggressive clinical course. Some patients with FH mutations may develop collecting duct renal cell carcinoma.
Amish individuals heterozygous for APOB p.Arg3500Gln (the most common pathogenic variant in the Amish community) have average LDL-C levels below the suggested minimum for a diagnosis of FH.
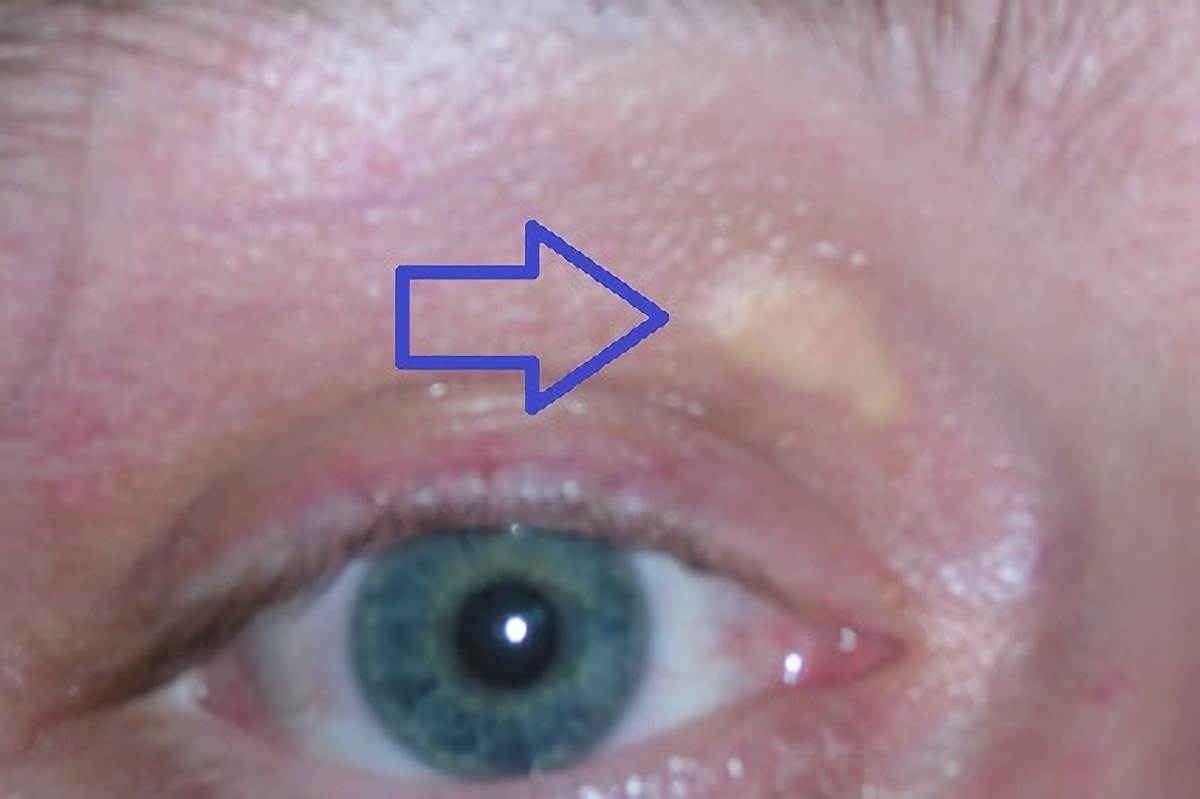
Genetic disorder characterized by high cholesterol levels Familial hypercholesterolemia Other names Familial hypercholesterolaemia Xanthelasma palpebrarum , yellowish patches consisting of cholesterol deposits above the eyelids. These are more common in people with FH. Specialty Endocrinology Familial hypercholesterolemia ( FH ) is a genetic disorder characterized by high cholesterol levels , specifically very high levels of low-density lipoprotein (LDL, "bad cholesterol"), in the blood and early cardiovascular disease .The most common mutations diminish the number of functional LDL receptors in the liver. [ citation needed ] Since the underlying body biochemistry is slightly different in individuals with FH, their high cholesterol levels are less responsive to the kinds of cholesterol control methods which are usually more effective in people without FH (such as dietary modification and statin tablets). Nevertheless, treatment (including higher statin doses) is usually effective. FH is classified as a type 2 familial dyslipidemia. [1] There are five types of familial dyslipidemia (not including subtypes), and each are classified from both the altered lipid profile and by the genetic abnormality. For example, high LDL (often due to LDL receptor defect) is type 2. Others include defects in chylomicron metabolism, triglyceride metabolism, and metabolism of other cholesterol-containing particles, such as VLDL and IDL.
Overview Familial hypercholesterolemia affects the way the body processes cholesterol. As a result, people with familial hypercholesterolemia have a higher risk of heart disease and a greater risk of early heart attack. The genetic changes that cause familial hypercholesterolemia are inherited. The condition is present from birth, but symptoms may not appear until adulthood. People who inherit the condition from both parents usually develop symptoms in childhood.
External links [ edit ] Arterial Gas Embolism Classification D ICD - 10 : O88.0 , T79.0 ICD - 9-CM : 673.0 , 999.1 MeSH : D004618 DiseasesDB : 313 SNOMED CT : 271376002 External resources eMedicine : emerg/787 v t e Pathology of pregnancy , childbirth and the puerperium Pregnancy Pregnancy with abortive outcome Abortion Ectopic pregnancy Abdominal Cervical Interstitial Ovarian Heterotopic Embryo loss Fetal resorption Molar pregnancy Miscarriage Stillbirth Oedema , proteinuria and hypertensive disorders Gestational hypertension Pre-eclampsia HELLP syndrome Eclampsia Other, predominantly related to pregnancy Digestive system Acute fatty liver of pregnancy Gestational diabetes Hepatitis E Hyperemesis gravidarum Intrahepatic cholestasis of pregnancy Integumentary system / dermatoses of pregnancy Gestational pemphigoid Impetigo herpetiformis Intrahepatic cholestasis of pregnancy Linea nigra Prurigo gestationis Pruritic folliculitis of pregnancy Pruritic urticarial papules and plaques of pregnancy (PUPPP) Striae gravidarum Nervous system Chorea gravidarum Blood Gestational thrombocytopenia Pregnancy-induced hypercoagulability Maternal care related to the fetus and amniotic cavity amniotic fluid Oligohydramnios Polyhydramnios Braxton Hicks contractions chorion / amnion Amniotic band syndrome Chorioamnionitis Chorionic hematoma Monoamniotic twins Premature rupture of membranes Obstetrical bleeding Antepartum placenta Circumvallate placenta Monochorionic twins Placenta accreta Placenta praevia Placental abruption Twin-to-twin transfusion syndrome Labor Amniotic fluid embolism Cephalopelvic disproportion Dystocia Shoulder dystocia Fetal distress Locked twins Nuchal cord Obstetrical bleeding Postpartum Pain management during childbirth placenta Placenta accreta Preterm birth Postmature birth Umbilical cord prolapse Uterine inversion Uterine rupture Vasa praevia Puerperal Breastfeeding difficulties Low milk supply Cracked nipples Breast engorgement Childbirth-related posttraumatic stress disorder Diastasis symphysis pubis Postpartum bleeding Peripartum cardiomyopathy Postpartum depression Postpartum psychosis Postpartum thyroiditis Puerperal fever Puerperal mastitis Other Concomitant conditions Diabetes mellitus Systemic lupus erythematosus Thyroid disorders Maternal death Sexual activity during pregnancy Category v t e Trauma Principles Polytrauma Major trauma Traumatology Triage Resuscitation Trauma triad of death Assessment Clinical prediction rules Revised Trauma Score Injury Severity Score Abbreviated Injury Scale NACA score Investigations Diagnostic peritoneal lavage Focused assessment with sonography for trauma Management Principles Advanced trauma life support Trauma surgery Trauma center Trauma team Damage control surgery Early appropriate care Procedures Resuscitative thoracotomy Pathophysiology Injury MSK Bone fracture Joint dislocation Degloving Soft tissue injury Resp Flail chest Pneumothorax Hemothorax Diaphragmatic rupture Pulmonary contusion Cardio Internal bleeding Thoracic aorta injury Cardiac tamponade GI Blunt kidney trauma Ruptured spleen Neuro Penetrating head injury Traumatic brain injury Intracranial hemorrhage Mechanism Blast injury Blunt trauma Burn Penetrating trauma Crush injury Stab wound Ballistic trauma Electrocution Region Abdominal trauma Chest trauma Facial trauma Head injury Spinal cord injury Demographic Geriatric trauma Pediatric trauma Complications Posttraumatic stress disorder Wound healing Acute lung injury Crush syndrome Rhabdomyolysis Compartment syndrome Contracture Volkmann's contracture Embolism air fat Chronic traumatic encephalopathy Subcutaneous emphysema v t e Underwater diving Diving modes Atmospheric pressure diving Freediving Saturation diving Scuba diving Snorkeling Surface oriented diving Surface-supplied diving Unmanned diving Diving equipment Cleaning and disinfection of personal diving equipment Human factors in diving equipment design Basic equipment Diving mask Snorkel Swimfin Breathing gas Bailout gas Bottom gas Breathing air Decompression gas Emergency gas supply Heliox Nitrox Oxygen Travel gas Trimix Buoyancy and trim equipment Buoyancy compensator Power inflator Dump valve Diving weighting system Ankle weights Integrated weights Trim weights Weight belt Decompression equipment Decompression buoy Decompression cylinder Decompression trapeze Dive computer Diving shot Jersey upline Jonline Diving suit Atmospheric diving suit Dry suit Sladen suit Standard diving suit Rash vest Wetsuit Dive skins Hot-water suit Helmets and masks Anti-fog Diving helmet Free-flow helmet Lightweight demand helmet Orinasal mask Reclaim helmet Shallow water helmet Standard diving helmet Diving mask Band mask Full-face mask Half mask Instrumentation Bottom timer Depth gauge Dive computer Dive timer Diving watch Helium release valve Pneumofathometer Submersible pressure gauge Mobility equipment Diving bell Closed bell Wet bell Diving stage Swimfin Monofin PowerSwim Towboard Diver propulsion vehicle Advanced SEAL Delivery System Cosmos CE2F series Dry Combat Submersible Human torpedo Motorised Submersible Canoe Necker Nymph R-2 Mala-class swimmer delivery vehicle SEAL Delivery Vehicle Shallow Water Combat Submersible Siluro San Bartolomeo Wet Nellie Wet sub Safety equipment Alternative air source Octopus regulator Pony bottle Bolt snap Buddy line Dive light Diver's cutting tool Diver's knife Diver's telephone Through-water communications Diving bell Diving safety harness Emergency gas supply Bailout block Bailout bottle Lifeline Screw gate carabiner Emergency locator beacon Rescue tether Safety helmet Shark-proof cage Snoopy loop Navigation equipment Distance line Diving compass Dive reel Line marker Surface marker buoy Silt screw Underwater breathing apparatus Atmospheric diving suit Diving cylinder Burst disc Diving cylinder valve Diving helmet Reclaim helmet Diving regulator Mechanism of diving regulators Regulator malfunction Regulator freeze Single-hose regulator Twin-hose regulator Full face diving mask Open-circuit scuba Scuba set Bailout bottle Decompression cylinder Independent doubles Manifolded twin set Scuba manifold Pony bottle Scuba configuration Sidemount Sling cylinder Diving rebreathers Carbon dioxide scrubber Carleton CDBA CDLSE Cryogenic rebreather CUMA DSEA Dolphin Electro-galvanic oxygen sensor FROGS Halcyon PVR-BASC Halcyon RB80 IDA71 Interspiro DCSC KISS LAR-5 LAR-6 LAR-V LARU Porpoise Ray Siebe Gorman CDBA Siva Viper Surface-supplied diving equipment Air line Diver's umbilical Diving air compressor Gas panel Hookah Scuba replacement Sea Trek Snuba Standard diving dress Escape set Davis Submerged Escape Apparatus Momsen lung Steinke hood Submarine Escape Immersion Equipment Diving equipment manufacturers AP Diving Apeks Aqua Lung America Aqua Lung/La Spirotechnique Beuchat René Cavalero Cis-Lunar Cressi-Sub Dacor DESCO Dive Xtras Divex Diving Unlimited International Drägerwerk Fenzy Maurice Fernez Technisub Oscar Gugen Heinke HeinrichsWeikamp Johnson Outdoors Mares Morse Diving Nemrod Oceanic Worldwide Porpoise Sub Sea Systems Shearwater Research Siebe Gorman Submarine Products Suunto Diving support equipment Access equipment Boarding stirrup Diver lift Diving bell Diving ladder Diving platform (scuba) Diving stage Downline Jackstay Launch and recovery system Messenger line Moon pool Breathing gas handling Air filtration Activated carbon Hopcalite Molecular sieve Silica gel Booster pump Carbon dioxide scrubber Cascade filling system Diver's pump Diving air compressor Diving air filter Water separator High pressure breathing air compressor Low pressure breathing air compressor Gas blending Gas blending for scuba diving Gas panel Gas reclaim system Gas storage bank Gas storage quad Gas storage tube Helium analyzer Nitrox production Membrane gas separation Pressure swing adsorption Oxygen analyser Oxygen compatibility Decompression equipment Built-in breathing system Decompression tables Diving bell Bell cursor Closed bell Clump weight Launch and recovery system Wet bell Diving chamber Diving stage Recreational Dive Planner Saturation system Platforms Dive boat Canoe and kayak diving Combat Rubber Raiding Craft Liveaboard Subskimmer Diving support vessel HMS Challenger (K07) Underwater habitat Aquarius Reef Base Continental Shelf Station Two Helgoland Habitat Jules' Undersea Lodge Scott Carpenter Space Analog Station SEALAB Tektite habitat Remotely operated underwater vehicles 8A4-class ROUV ABISMO Atlantis ROV Team CURV Deep Drone Épaulard Global Explorer ROV Goldfish-class ROUV Kaikō ROV Kaşif ROUV Long-Term Mine Reconnaissance System Mini Rover ROV OpenROV ROV KIEL 6000 ROV PHOCA Scorpio ROV Sea Dragon-class ROV Seabed tractor Seafox drone Seahorse ROUV SeaPerch SJT-class ROUV T1200 Trenching Unit VideoRay UROVs Safety equipment Diver down flag Diving shot Hyperbaric lifeboat Hyperbaric stretcher Jackstay Jonline Reserve gas supply General Diving spread Air spread Saturation spread Hot water system Sonar Underwater acoustic positioning system Underwater acoustic communication Freediving Activities Aquathlon Apnoea finswimming Freediving Haenyeo Pearl hunting Ama Snorkeling Spearfishing Underwater football Underwater hockey Underwater ice hockey Underwater rugby Underwater target shooting Competitions Nordic Deep Vertical Blue Disciplines Constant weight (CWT) Constant weight without fins (CNF) Dynamic apnea (DYN) Dynamic apnea without fins (DNF) Free immersion (FIM) No-limits apnea (NLT) Static apnea (STA) Skandalopetra diving Variable weight apnea (VWT) Variable weight apnea without fins Equipment Diving mask Diving suit Hawaiian sling Polespear Snorkel (swimming) Speargun Swimfins Monofin Water polo cap Freedivers Deborah Andollo Peppo Biscarini Sara Campbell Derya Can Göçen Goran Čolak Carlos Coste Robert Croft Mandy-Rae Cruickshank Yasemin Dalkılıç Leonardo D'Imporzano Flavia Eberhard Şahika Ercümen Emma Farrell Francisco Ferreras Pierre Frolla Flavia Eberhard Mehgan Heaney-Grier Elisabeth Kristoffersen Loïc Leferme Enzo Maiorca Jacques Mayol Audrey Mestre Karol Meyer Stéphane Mifsud Alexey Molchanov Natalia Molchanova Dave Mullins Patrick Musimu Guillaume Néry Herbert Nitsch Umberto Pelizzari Annelie Pompe Michal Risian Stig Severinsen Tom Sietas Aharon Solomons Martin Štěpánek Walter Steyn Tanya Streeter William Trubridge Devrim Cenk Ulusoy Danai Varveri Alessia Zecchini Nataliia Zharkova Hazards Barotrauma Drowning Freediving blackout Deep-water blackout Shallow-water blackout Hypercapnia Hypothermia Historical Ama Octopus wrestling Swimming at the 1900 Summer Olympics – Men's underwater swimming Organisations AIDA International Scuba Schools International Australian Underwater Federation British Freediving Association Confédération Mondiale des Activités Subaquatiques Fédération Française d'Études et de Sports Sous-Marins Performance Freediving International Professional diving Occupations Ama Commercial diver Commercial offshore diver Hazmat diver Divemaster Diving instructor Diving safety officer Diving superintendent Diving supervisor Haenyeo Media diver Police diver Public safety diver Scientific diver Underwater archaeologist Military diving Army engineer diver Clearance diver Frogman List of military diving units Royal Navy ships diver Special Boat Service United States military divers U.S. ... Vandenberg HMS Ghurka Glen Strathallan SAS Good Hope Gothenburg Herzogin Cecilie Hilma Hooker Hispania HMS Hood HMAS Hobart Igara James Eagan Layne Captain Keith Tibbetts King Cruiser SMS Kronprinz Kyarra HMS Laforey USAT Liberty Louis Sheid USS LST-507 SMS Markgraf Mikhail Lermontov HMS M2 Maine Maloja HMS Maori Marguerite SS Mauna Loa USAT Meigs Mendi USCGC Mohawk Mohegan RMS Moldavia HMS Montagu MV RMS Mulheim Nagato Oceana USS Oriskany Oslofjord P29 P31 Pedernales Persier HMAS Perth SAS Pietermaritzburg Piłsudski Pool Fisher HMS Port Napier Preußen President Coolidge PS Queen Victoria Radaas Rainbow Warrior RMS Rhone Rondo Rosehill Rotorua Royal Adelaide Royal Charter Rozi HMS Safari Salem Express USS Saratoga USS Scuffle HMS Scylla HMS Sidon USS Spiegel Grove Stanegarth Stanwood Stella HMAS Swan USS Tarpon Thesis Thistlegorm Toa Maru Torrey Canyon SAS Transvaal U-40 U-352 U-1195 Um El Faroud Varvassi Walter L M Russ Washingtonian (1913) HMNZS Wellington USS Yancey Yongala Zenobia Zealandia Zingara Cave diving sites Blauhöhle Chinhoyi Caves Devil's Throat at Punta Sur Engelbrecht Cave Fossil Cave Jordbrugrotta Piccaninnie Ponds Pluragrotta Pollatoomary Sistema Ox Bel Ha Sistema Sac Actun Sistema Dos Ojos Sistema Nohoch Nah Chich Freshwater dives Dutch Springs Ewens Ponds Little Blue Lake Training sites Capernwray Dive Centre Deepspot National Diving and Activity Centre Stoney Cove Open ocean diving Blue-water diving Black-water diving Diving safety Human factors in diving equipment design Human factors in diving safety Life-support system Safety-critical system Scuba diving fatalities Diving hazards List of diving hazards and precautions Environmental Current Delta-P Entanglement hazard Overhead Silt out Wave action Equipment Freeflow Use of breathing equipment in an underwater environment Failure of diving equipment other than breathing apparatus Single point of failure Physiological Cold shock response Decompression Nitrogen narcosis Oxygen toxicity Seasickness Uncontrolled decompression Diver behaviour and competence Lack of competence Overconfidence effect Panic Task loading Trait anxiety Willful violation Consequences Barotrauma Decompression sickness Drowning Hypothermia Hypoxia Hypercapnia Hyperthermia Diving procedures Ascending and descending Emergency ascent Boat diving Canoe and kayak diving Buddy diving buddy check Decompression Decompression practice Pyle stop Ratio decompression Dive briefing Dive log Dive planning Scuba gas planning Diver communications Diving hand signals Diving line signals Diver voice communications Diver rescue Diver training Doing It Right Drift diving Gas blending for scuba diving Night diving Solo diving Water safety Risk management Checklist Hazard identification and risk assessment Hazard analysis Job safety analysis Risk assessment Risk control Hierarchy of hazard controls Incident pit Lockout–tagout Permit To Work Redundancy Safety data sheet Situation awareness Diving team Bellman Chamber operator Diver medical technician Diver's attendant Diving supervisor Diving systems technician Gas man Life support technician Stand-by diver Equipment safety Breathing gas quality Testing and inspection of diving cylinders Hydrostatic test Sustained load cracking Diving regulator Breathing performance of regulators Occupational safety and health Approaches to safety Job safety analysis Risk assessment Toolbox talk Housekeeping Association of Diving Contractors International Code of practice Contingency plan Diving regulations Emergency procedure Emergency response plan Evacuation plan Hazardous Materials Identification System Hierarchy of hazard controls Administrative controls Engineering controls Hazard elimination Hazard substitution Personal protective equipment International Marine Contractors Association Occupational hazard Biological hazard Chemical hazard Physical hazard Psychosocial hazard Occupational hygiene Exposure assessment Occupational exposure limit Workplace health surveillance Safety culture Code of practice Diving safety officer Diving superintendent Health and safety representative Operations manual Safety meeting Standard operating procedure Diving medicine Diving disorders List of signs and symptoms of diving disorders Cramp Motion sickness Surfer's ear Pressure related Alternobaric vertigo Barostriction Barotrauma Air embolism Aerosinusitis Barodontalgia Dental barotrauma Pulmonary barotrauma Compression arthralgia Decompression illness Dysbarism Oxygen Freediving blackout Hyperoxia Hypoxia Oxygen toxicity Inert gases Avascular necrosis Decompression sickness Isobaric counterdiffusion Taravana Dysbaric osteonecrosis High-pressure nervous syndrome Hydrogen narcosis Nitrogen narcosis Carbon dioxide Hypercapnia Hypocapnia Breathing gas contaminants Carbon monoxide poisoning Immersion related Asphyxia Drowning Hypothermia Immersion diuresis Instinctive drowning response Laryngospasm Salt water aspiration syndrome Swimming-induced pulmonary edema Treatment Demand valve oxygen therapy First aid Hyperbaric medicine Hyperbaric treatment schedules In-water recompression Oxygen therapy Therapeutic recompression Personnel Diving Medical Examiner Diving Medical Practitioner Diving Medical Technician Hyperbaric nursing Screening Atrial septal defect Effects of drugs on fitness to dive Fitness to dive Psychological fitness to dive Research Researchers in diving physiology and medicine Arthur J.
Uropathogenic E. coli from the gut is the cause of 80–85% of community-acquired urinary tract infections, [23] with Staphylococcus saprophyticus being the cause in 5–10%. [4] Rarely they may be due to viral or fungal infections. [24] Healthcare-associated urinary tract infections (mostly related to urinary catheterization ) involve a much broader range of pathogens including: E. coli (27%), Klebsiella (11%), Pseudomonas (11%), the fungal pathogen Candida albicans (9%), and Enterococcus (7%) among others. [6] [25] [26] Urinary tract infections due to Staphylococcus aureus typically occur secondary to blood-borne infections. [9] Chlamydia trachomatis and Mycoplasma genitalium can infect the urethra but not the bladder. [27] These infections are usually classified as a urethritis rather than urinary tract infection. [28] Intercourse In young sexually active women, sexual activity is the cause of 75–90% of bladder infections, with the risk of infection related to the frequency of sex. [4] The term "honeymoon cystitis" has been applied to this phenomenon of frequent UTIs during early marriage. ... PMID 20927755 . ^ "FDA Drug Safety Communication: FDA updates warnings for oral and injectable fluoroquinolone antibiotics due to disabling side effects" .
It occurs almost exclusively in India and Indian communities living abroad. Pathophysiology [ edit ] Main article: Carcinogenesis Oral squamous cell carcinoma is the end product of an unregulated proliferation of mucous basal cells.
Description Diffuse panbronchiolitis (DPB) is a rare chronic inflammatory obstructive pulmonary disease primarily affecting the respiratory bronchioles. 'Diffuse' refers to the distribution of the lesions throughout both lungs, and 'pan-' refers to the involvement of inflammation in all layers of the respiratory bronchioles. Onset of the disorder occurs in the second to fifth decade of life, and is clinically manifest by chronic cough, exertional dyspnea, and sputum production. Most patients also have chronic paranasal sinusitis. If untreated, the disorder progresses to bronchiectasis, respiratory failure, and death (summary by Poletti et al., 2006). Clinical Features Yamanaka et al. (1969) first described a chronic airway disease in Japan.
Diffuse panbronchiolitis is a rare chronic inflammatory obstructive pulmonary disease primarily affecting the respiratory bronchioles throughout both lungs and inducing sinobronchial infection. Onset occurs in the second to fifth decade of life and manifests by chronic cough, exertional dyspnea, and sputum production. Most patients also have chronic paranasal sinusitis
Diffuse panbronchiolitis (DPB) is a rare condition characterized by inflammation of the small airways of the lungs ( bronchiolitis ) and chronic sinusitis . It mainly occurs among the Japanese but has been reported in other populations. Symptoms typically develop anywhere from the teenage years to the fifth decade of life and are slowly progressive over months to years. Common symptoms include chronic sinusitis, a productive cough (producing mucus), breathlessness with exertion, wheezing, and weight loss (especially as symptoms worsen). The exact cause of DPB is not known, but a variety of genetic, environmental, and systemic factors appear to contribute to the condition.
. ^ a b c d e f g h i j k l m n o p q r s t u v w x y Liaison, Ray Fleming, Office of Communications and Public (May 2016). "Questions and Answers About Alopecia Areata" .
Risk factors There are many risk factors for heart diseases: age, sex, tobacco use, physical inactivity, excessive alcohol consumption, unhealthy diet, obesity, genetic predisposition and family history of cardiovascular disease, raised blood pressure ( hypertension ), raised blood sugar ( diabetes mellitus ), raised blood cholesterol ( hyperlipidemia ), undiagnosed celiac disease , psychosocial factors, poverty and low educational status, and air pollution . [14] [15] [16] [17] [18] While the individual contribution of each risk factor varies between different communities or ethnic groups the overall contribution of these risk factors is very consistent. [19] Some of these risk factors, such as age, sex or family history/genetic predisposition, are immutable; however, many important cardiovascular risk factors are modifiable by lifestyle change, social change, drug treatment (for example prevention of hypertension, hyperlipidemia, and diabetes). [20] People with obesity are at increased risk of atherosclerosis of the coronary arteries . [21] Genetics Genetic factors influence the development of cardiovascular disease in men who are less than 55 years old and in women who are less than 65 years old. [20] Cardiovascular disease in a person's parents increases their risk by 3 fold. [22] Multiple single nucleotide polymorphisms (SNP) have been found to be associated with cardiovascular disease in genetic association studies, [23] [24] but usually, their individual influence is small, and genetic contributions to cardiovascular disease are poorly understood. [24] Age Calcified heart of an older woman with cardiomegaly Age is the most important risk factor in developing cardiovascular or heart diseases, with approximately a tripling of risk with each decade of life. [25] Coronary fatty streaks can begin to form in adolescence. [26] It is estimated that 82 percent of people who die of coronary heart disease are 65 and older. [27] Simultaneously, the risk of stroke doubles every decade after age 55. [28] Multiple explanations are proposed to explain why age increases the risk of cardiovascular/heart diseases. ... Sex difference in ischaemic heart disease mortality and risk factors in 46 communities: an ecologic analysis. Cardiovasc Risk Factors. 1999; 7:43–54. ^ Doll R, Peto R, Boreham J, Sutherland I (June 2004).
In males, there can be deformities in the seminiferous tubule as in Klinefelter syndrome (most common cause in males), [37] defects in the production of testicular steroids, receptor mutations preventing testicular hormones from working, chromosomal abnormalities such as Noonan syndrome , or problems with the cells making up the testes. [30] Females can also have chromosomal abnormalities such as Turner syndrome (most common cause in girls), [37] XX gonadal dysgenesis , and XY gonadal dysgenesis , problems in the ovarian hormone synthesis pathway such as aromatase deficiency [30] or congenital anatomical deformities such as Müllerian agenesis . [36] Acquired disorders [ edit ] Acquired diseases include mumps orchitis , Coxsackievirus B infection, irradiation, chemotherapy , or trauma; all problems causing the gonads to fail. [2] [36] Genetic or acquired defect of the hormonal pathway of puberty (hypogonadotropic hypogonadism) [ edit ] The hypothalamic–pituitary–gonadal axis can also be affected at the level of the brain. [36] The brain does not send its hormonal signals to the gonads (low gonadotropins ) causing the gonads to never be activated in the first-place resulting in hypogonadotropic hypogonadism . [38] The HPG axis can be altered in two places, at the hypothalamic or at the pituitary level. [38] CNS disorders such as childhood brain tumors ( e.g. craniopharyngioma , prolactinoma , germinoma , glioma ) can disrupt the communication between the hypothalamus and the pituitary. [30] Pituitary tumors, especially prolactinomas , can increase the level of dopamine causing an inhibiting effect to the HPG axis. [1] Hypothalamic disorders include Prader-Willi syndrome and Kallmann syndrome , [2] but the most common cause of hypogonadotropic hypogonadism is a functional deficiency in the hormone regulator produced by the hypothalamus, the gonadotropin-releasing hormone or GnRH. [7] Diagnosis [ edit ] Pediatric endocrinologists are the physicians with the most training and experience evaluating delayed puberty.
Overview Tinnitus is when you experience ringing or other noises in one or both of your ears. The noise you hear when you have tinnitus isn't caused by an external sound, and other people usually can't hear it. Tinnitus is a common problem. It affects about 15% to 20% of people, and is especially common in older adults. Tinnitus is usually caused by an underlying condition, such as age-related hearing loss, an ear injury or a problem with the circulatory system. For many people, tinnitus improves with treatment of the underlying cause or with other treatments that reduce or mask the noise, making tinnitus less noticeable.
Rare pathogenic variants, such as c.297_300delAAGA, are also likely the consequence of gene conversion with SBDSP [Carvalho et al 2014; J Rommens, personal communication]. 10. Rare whole-exon deletions [Costa et al 2007], extended conversions of exon 2 and flanking introns, or gene rearrangements involving exon 2 have been observed. 11.
A number sign (#) is used with this entry because Shwachman-Diamond syndrome-1 (SDS1), also known as the Shwachman-Bodian-Diamond syndrome, is caused by compound heterozygous or homozygous mutations in the SBDS gene (607444) on chromosome 7q11. Heterozygous mutations in the SBDS gene have been associated with predisposition to aplastic anemia (609135). Description Shwachman-Diamond syndrome is a multisystem autosomal recessive disorder characterized by exocrine pancreatic dysfunction, bony metaphyseal dysostosis, and varying degrees of marrow dysfunction with cytopenias. Myelodysplastic syndrome and acute myeloid leukemia occur in up to one third of patients (summary by Dror and Freedman, 1999). For a review of Shwachman-Diamond syndrome, see Dror and Freedman (2002).
A number sign (#) is used with this entry because of evidence that Shwachman-Diamond syndrome-2 (SDS2) is caused by homozygous mutation in the EFL1 gene (617538) on chromosome 15q25. Description Shwachman-Diamond syndrome-2 (SDS2) is characterized by exocrine pancreatic dysfunction, hematopoietic abnormalities, short stature, and metaphyseal dysplasia (Stepensky et al., 2017). For a discussion of genetic heterogeneity of Shwachman-Diamond syndrome, see SDS1 (260400). Clinical Features Stepensky et al. (2017) reported a 6-year-old Mexican boy and his 4-year-old sister (family A) who had short stature, mild global developmental delay, severe myopia, exocrine pancreatic insufficiency, and bilateral genu varum. Skeletal survey showed metaphyseal widening and irregularity, especially in the ribs and femurs.
Shwachman–Diamond syndrome Other names Shwachman–Bodian–Diamond syndrome Specialty Medical genetics Shwachman–Diamond syndrome ( SDS ), or Shwachman–Bodian–Diamond syndrome , is a rare congenital disorder characterized by exocrine pancreatic insufficiency , bone marrow dysfunction, skeletal abnormalities and short stature . After cystic fibrosis (CF), it is the second most common cause of exocrine pancreatic insufficiency in children. Contents 1 Signs and symptoms 2 Genetics 3 Mechanisms 4 Diagnosis 5 Management 6 Epidemiology 7 History 7.1 Eponym 8 References 9 Further reading 10 External links Signs and symptoms [ edit ] The syndrome shows a wide range of abnormalities and symptoms. The main characteristics of the syndrome are exocrine pancreatic dysfunction, hematologic abnormalities and growth retardation. Only the first two of these are included in the clinical diagnostic criteria. [1] Hematologic abnormalities: neutropenia may be intermittent or persistent and is the most common hematological finding.
Shwachman-Diamond syndrome is an inherited condition that affects many parts of the body, particularly the bone marrow, pancreas, and bones. The major function of bone marrow is to produce new blood cells. These include red blood cells, which carry oxygen to the body's tissues; white blood cells, which fight infection; and platelets, which are blood cells that are necessary for normal blood clotting. In Shwachman-Diamond syndrome, the bone marrow malfunctions and does not make some or all types of white blood cells . A shortage of neutrophils, the most common type of white blood cell, causes a condition called neutropenia. Most people with Shwachman-Diamond syndrome have at least occasional episodes of neutropenia, which makes them more vulnerable to infections, often involving the lungs (pneumonia), ears (otitis media), or skin.
Shwachman-Diamond syndrome (SDS) affects many parts of the body, particularly the bone marrow, pancreas, and skeletal system. Symptoms include the inability to digest food due to missing digestive enzymes, low muscle tone, and anemia. Other symptoms include skeletal findings and intellectual disability. Children with SDS may have feeding difficulties, slow growth, and frequent infections. People with SDS are at increased risk for blood cancers. Shwachman-Diamond syndrome can be caused by the SBDS , DNAJC21 , EFL1 , or SRP54 gene not working correctly.
Shwachman-Diamond syndrome (SDS) is a rare multisystemic syndrome characterized by chronic and usually mild neutropenia, pancreatic exocrine insufficiency associated with steatorrhea and growth failure, skeletal dysplasia with short stature, and an increased risk of bone marrow aplasia or leukemic transformation. Epidemiology Worldwide prevalence is estimated at about 1/350,000 and birth-prevalence at around 1/200,000 live births. Clinical description SDS shows a variable clinical picture, even within families. It generally manifests during infancy or early childhood. The most common anomaly is usually intermittent and moderate neutropenia that is associated with recurrent infections. Mild anemia and thrombocytopenia may also occur. Exocrine pancreatic insufficiency results in failure to thrive, growth retardation, and chronic steatorrhea.
In this case a surgical procedure can be used to affect nerves supplying the heart that communicate using catecholamines. [2] A collection of nerves known as the sympathetic nervous system supply the heart as well as other organs.
Summary Clinical characteristics. Catecholaminergic polymorphic ventricular tachycardia (CPVT) is characterized by episodic syncope occurring during exercise or acute emotion in individuals without structural cardiac abnormalities. The underlying cause of these episodes is the onset of fast ventricular tachycardia (bidirectional or polymorphic). Spontaneous recovery may occur when these arrhythmias self-terminate. In other instances, ventricular tachycardia may degenerate into ventricular fibrillation and cause sudden death if cardiopulmonary resuscitation is not readily available. The mean age of onset of symptoms (usually a syncopal episode) is between age seven and twelve years; onset as late as the fourth decade of life has been reported.
A number sign (#) is used with this entry because catecholaminergic polymorphic ventricular tachycardia-1 (CPVT1) is caused by heterozygous mutation in the cardiac ryanodine receptor gene (RYR2; 180902) on chromosome 1q43. Description Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an arrhythmogenic disorder of the heart characterized by a reproducible form of polymorphic ventricular tachycardia induced by physical activity, stress, or catecholamine infusion, which can deteriorate into ventricular fibrillation. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. Typically, clinical cardiologic examinations, such as baseline ECG and echocardiogram, reveal mostly normal findings, and postmortem examinations, when carried out, have not disclosed any significant morphologic alterations in the fine structure of the heart, with the exception of mild fatty myocardial infiltration in a few patients. The hallmark of CPVT comprises ventricular arrhythmias of varying morphology not present under resting conditions but appearing only with physical exercise, excitement, or catecholamine administration.
A rare, severe genetic arrhythmogenic disorder of the structurally normal heart characterized by catecholamine-induced ventricular tachycardia (VT) manifesting as syncope and sudden death in young individuals. Epidemiology The prevalence of catecholaminergic polymorphic ventricular tachycardia (CPVT) is estimated to be 1/10,000. Both sexes are equally affected. Clinical description Typical age of onset of CPVT is between 7 and 15 years of age. Exercise- or emotion-induced syncopal spells are frequently the first symptom. In a subset of patients (10-20%), the disease is clinically silent, presenting only in the event of sudden death.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a genetic disorder that causes an abnormally fast and irregular heart rhythm in response to physical activity or emotional stress. Signs and symptoms include light-headedness, dizziness, and fainting. Symptoms most often develop between 7 to 9 years of age. If untreated CPVT can cause a heart attack and death. CPVT is caused by mutations in the RYR2 or CASQ2 genes. When a RYR2 gene mutation is involved, the condition is passed through families in an autosomal dominant fashion. When CASQ2 gene mutations are involved, the condition is inherited in an autosomal recessive fashion.
A number sign (#) is used with this entry because catecholaminergic polymorphic ventricular tachycardia-2 (CPVT2) is caused by homozygous or compound heterozygous mutation in the gene encoding calsequestrin-2 (CASQ2; 114251) on chromosome 1p13. For a general phenotypic description and a discussion of genetic heterogeneity of CPVT, see 604772. Clinical Features Lahat et al. (2001) studied 41 members of 7 families from a highly inbred Bedouin tribe in northern Israel in which 9 children had unexplained sudden death, 7 during vigorous exercise and 2 during excitement. In addition, 12 other children had onset of recurrent syncope and seizures at around 6 years of age, with 70% of the episodes occurring during vigorous physical activity and 30% following sudden excitement. The parents of affected children were all related and were all asymptomatic.
A number sign (#) is used with this entry because of evidence that catecholaminergic polymorphic ventricular tachycardia-4 (CPVT4) is caused by heterozygous mutation in the calmodulin gene (CALM1; 114180) on chromosome 14q32. For a general phenotypic description and a discussion of genetic heterogeneity of CPVT, see 604772. Clinical Features Nyegaard et al. (2012) studied a large 4-generation Swedish family with a history of ventricular arrhythmias, syncope, and sudden death, predominantly in association with physical exercise or stress. The proband was a 42-year-old man who first developed syncope at 12 years of age while playing football; electrocardiogram (ECG) at that time showed bradycardia with a prominent U-wave in leads V2 and V3, without evidence of QT prolongation. He had a history of loss of consciousness on at least 4 occasions during physical activity and once in connection with a fire alarm.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a condition characterized by an abnormal heart rhythm (arrhythmia). As the heart rate increases in response to physical activity or emotional stress, it can trigger an abnormally fast heartbeat called ventricular tachycardia. Episodes of ventricular tachycardia can cause light-headedness, dizziness, and fainting (syncope). In people with CPVT, these episodes typically begin in childhood. If CPVT is not recognized and treated, an episode of ventricular tachycardia may cause the heart to stop beating (cardiac arrest), leading to sudden death. Researchers suspect that CPVT may be a significant cause of sudden death in children and young adults without recognized heart abnormalities.
A number sign (#) is used with this entry because of evidence that catecholaminergic polymorphic ventricular tachycardia-3 (CPVT3) is caused by homozygous mutation in the TECRL gene (617242) on chromosome 4q13. Description Catecholaminergic polymorphic ventricular tachycardia-3 (CPVT3) is characterized by overlapping features of long QT syndrome (see 192500) and CPVT. Affected individuals exhibit adrenergic ventricular tachycardia associated with a high prevalence of cardiac arrest and sudden cardiac death, with recurrent atrial tachycardia sometimes triggering the ventricular arrhythmias. In addition, affected individuals have a normal or mildly prolonged QTc on baseline electrocardiography, with a paradoxical QT increase during adrenergic simulation (summary by Devalla et al., 2016). For a general phenotypic description and a discussion of genetic heterogeneity of CPVT, see 604772.
A number sign (#) is used with this entry because of evidence that catecholaminergic polymorphic ventricular tachycardia-5 with or without muscle weakness (CPVT5) is caused by homozygous or compound heterozygous mutation in the triadin gene (TRDN; 603283) on chromosome 6q22. For a general phenotypic description and a discussion of genetic heterogeneity of CPVT, see 604772. Clinical Features Roux-Buisson et al. (2012) studied 2 families with cardiac arrhythmias. In the first family, which originated from the French West Indies, the 2-year-old proband experienced syncope followed by cardiac arrest after a shock while playing with his 7-year-old brother. Resting electrocardiogram (ECG) after resuscitation showed numerous polymorphic or bidirectional ventricular extra beats and runs of polymorphic ventricular tachycardia.
Arbour et al. (2008) identified a missense mutation (607542.0040) in the KCNQ1 gene causing long QT syndrome-1 among a First Nations community of northern British Columbia.
A number sign (#) is used with this entry because of evidence that long QT syndrome-13 (LQT13) is caused by heterozygous mutation in the KCNJ5 gene (600734) on chromosome 11q24. Description Congenital long QT syndrome is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Clinical Features Yang et al. (2010) studied a large 4-generation Chinese family segregating autosomal dominant long QT syndrome (LQTS).
A number sign (#) is used with this entry because long QT syndrome-2 (LQT2) is caused by heterozygous mutation in the HERG gene (KCNH2; 152427) on chromosome 7q36. Digenic inheritance has also been reported; see MOLECULAR GENETICS. There is evidence that mutation in the KCR1 gene (ALG10B; 603313) on chromosome 12q12 confers reduced susceptibility to acquired long QT syndrome-2. Description Congenital long QT syndrome is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500).
A number sign (#) is used with this entry because of evidence that long QT syndrome-10 (LQT10) and familial atrial fibrillation-17 (ATFB17) are caused by heterozygous mutation in the SCN4B gene (608256) on chromosome 11q23. For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). For a discussion of genetic heterogeneity of familial atrial fibrillation, see ATFB1 (608583). Clinical Features Long QT Syndrome 10 Medeiros-Domingo et al. (2007) reported a 5-year-old Mexican mestizo girl who at 21 months of age was found to have asymptomatic bradycardia with rates less than 60 bpm. An ECG revealed profound QT prolongation with a QTc of 712 ms and intermittent 2:1 AV block; during 1:1 conduction, macroscopic T-wave alternans was observed.
A number sign (#) is used with this entry because long QT syndrome-3 (LQT3) is caused by heterozygous mutation in the gene encoding the alpha polypeptide of voltage-gated sodium channel type V (SCN5A; 600163) on chromosome 3p22. Digenic inheritance has also been reported; see MOLECULAR GENETICS. Description Congenital long QT syndrome is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Clinical Features Wang et al. (1995) cited preliminary data suggesting that in chromosome 3-linked LQT the onset of the T wave is later and duration of the QT interval longer than in other forms.
A number sign (#) is used with this entry because a cardiac arrhythmia syndrome with variable manifestations can be caused by heterozygous mutation in the ankyrin-B gene (ANK2; 106410) on chromosome 4q25-q26. Long QT syndrome-4 can also be caused by mutation in the ANK2 gene. For a general phenotypic description and a discussion of genetic heterogeneity of long QT syndrome, see 192500. Description Loss-of-function mutations in ANK2 can result in a broad spectrum of clinical cardiac phenotypes. Carriers of some mutations (e.g., E1425G, 106410.0001) display QT interval prolongation, stress- and/or exercise-induced polymorphic ventricular arrhythmia, syncope, and sudden cardiac death. Patients with other variants show clinical phenotypes, sometimes mild, extending beyond LQTS, leading to the label 'ankyrin-B syndrome.'
A number sign (#) is used with this entry because of evidence that long QT syndrome-14 (LQT14) is caused by heterozygous mutation in the CALM1 gene (114180) on chromosome 14q32. For a general phenotypic description and discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Clinical Features Crotti et al. (2013) reported an Italian girl who underwent cardiac arrest due to ventricular fibrillation (VF) at age 6 months. Electrocardiogram (ECG) after defibrillation showed a markedly prolonged QTc interval (630 ms), frequent episodes of T-wave alternans, and intermittent 2:1 atrioventricular block. Echocardiogram showed normal cardiac anatomy and contractile function.
A number sign (#) is used with this entry because of evidence that long QT syndrome-12 (LQT12) is caused by heterozygous mutation in the alpha-1 syntrophin gene (SNTA1; 601017) on chromosome 20q11. Description Congenital long QT syndrome (LQTS) is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Molecular Genetics Ueda et al. (2008) analyzed the SNTA1 gene in 50 unrelated patients with long QT syndrome who were negative for mutations in the 11 known LQTS genes and identified a heterozygous missense mutation (A390V; 601017.0001) in 1 patient.
A number sign (#) is used with this entry because long QT syndrome-9 (LQT9) is caused by heterozygous mutation in the CAV3 gene (601253), which encodes caveolin-3, on chromosome 3p25. Digenic inheritance has also been reported; see MOLECULAR GENETICS. Description Congenital long QT syndrome is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Molecular Genetics Vatta et al. (2006) analyzed the CAV3 gene (601253) in 905 unrelated patients with long QT syndrome who had previously been tested for mutations in known LQT genes; in 6 patients, they identified 4 heterozygous missense mutations (601253.0016-601253.0019, respectively) that were not found in more than 1,000 control alleles.
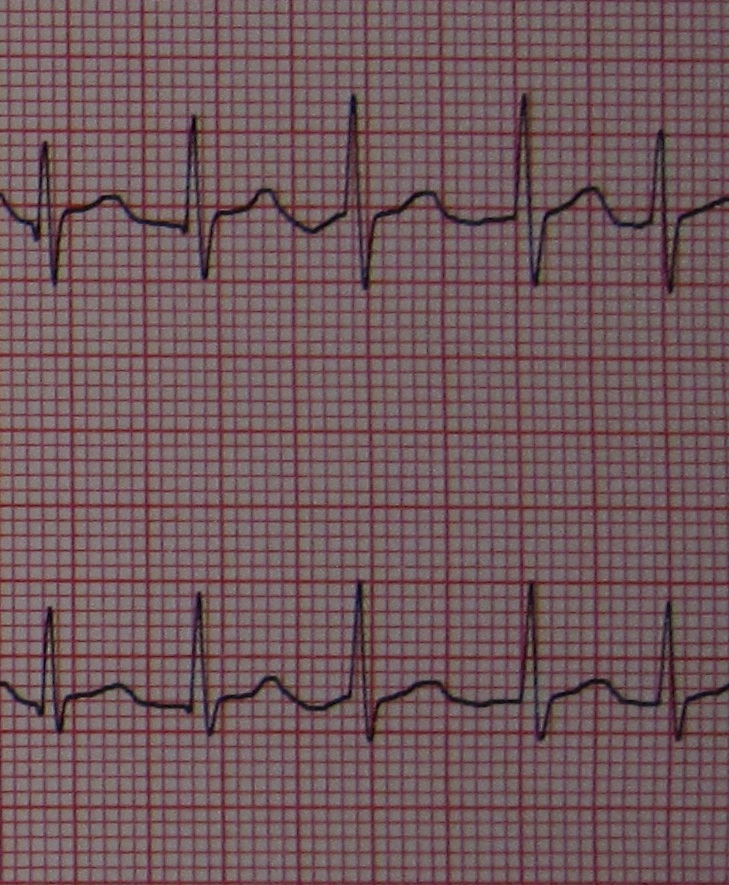
Congenital long QT syndrome (LQTS) is a hereditary cardiac disease characterized by a prolongation of the QT interval at basal ECG and by a high risk of life-threatening arrhythmias. Epidemiology Disease prevalence is estimated at close to 1 in 2,500 live births. Clinical description The two cardinal manifestations of LQTS are syncopal episodes, which may lead to cardiac arrest and sudden cardiac death, and electrocardiographic abnormalities, including prolongation of the QT interval and T wave abnormalities. Etiology The genetic basis of the disease was identified in the mid-nineties and all the LQTS genes identified so far encode cardiac ion channel subunits or proteins involved in modulating ionic currents. Mutations in these genes ( KCNQ1 , KCNH2 , KCNE1 , KCNE2 , CACNA1c , CAV3 , SCN5A , SCN4B ) cause the disease by prolonging the duration of the action potential.
A number sign (#) is used with this entry because of evidence that long QT syndrome-15 (LQT15) is caused by heterozygous mutation in the CALM2 gene (114182) on chromosome 2p21. For a general phenotypic description and discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Clinical Features Crotti et al. (2013) reported a Hispanic girl in whom fetal bradycardia was first noted at 21 weeks' gestation; fetal echocardiogram showed normal cardiac anatomy and function except for bradycardia. Two hours after birth she exhibited sinus bradycardia, T-wave alternans, markedly prolonged QTc (690 ms), and 2:1 AV block. At 3 weeks of age, she underwent cardiac arrest with multiple episodes of ventricular fibrillation (VF), during which time she also suffered a cerebral infarction of the right parietal lobe.
A number sign (#) is used with this entry because of evidence that long QT syndrome-5 (LQT5) is caused by heterozygous mutation in the KCNE1 gene (176261) on chromosome 21q22. Digenic inheritance has also been reported; see MOLECULAR GENETICS. Description Congenital long QT syndrome is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Molecular Genetics In affected members of 2 families with long QT syndrome, Splawski et al. (1997) identified heterozygosity for different missense mutations in the KCNE1 gene (176261.0003-176261.0004).
A number sign (#) is used with this entry because of evidence that long QT syndrome-6 (LQT6) is caused by heterozygous mutation in the KCNE2 gene (603796) on chromosome 21q22. Digenic inheritance has also been reported; see MOLECULAR GENETICS. Description Congenital long QT syndrome is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Clinical Features Abbott et al. (1999) reported a healthy 38-year-old Caucasian female who had had ventricular fibrillation while jogging.
A number sign (#) is used with this entry because of evidence that long QT syndrome-11 (LQT11) is caused by heterozygous mutation in the gene encoding the A-kinase anchor protein-9 (AKAP9; 604001) on chromosome 7q21. One such family has been reported. For a discussion of genetic heterogeneity of long QT syndrome, see LQT1 (192500). Description Congenital long QT syndrome is electrocardiographically characterized by a prolonged QT interval and polymorphic ventricular arrhythmias (torsade de pointes). These cardiac arrhythmias may result in recurrent syncope, seizure, or sudden death (Jongbloed et al., 1999). Molecular Genetics In a 13-year-old Caucasian girl with long QT syndrome, who was negative for mutation in the known LQT genes, Chen et al. (2007) identified heterozygosity for a ser1570-to-leu (S1570L; 604001.0001) substitution in the AKAP9 gene.
"The face of postural tachycardia syndrome - insights from a large cross-sectional online community-based survey" . Journal of Internal Medicine . 286 (4): 438–448. doi : 10.1111/joim.12895 . ... Cleveland Clinic . ^ "Most common conditions reported with POTS based on the experiences of 1,227 diagnosed members of the POTS research community" . Stuff That Works . ^ Fedorowski A, Li H, Yu X, Koelsch KA, Harris VM, Liles C, et al.
Human disease Orthostatic intolerance ( OI ) is the development of symptoms when standing upright which are relieved when reclining . [1] There are many types of orthostatic intolerance. OI can be a subcategory of dysautonomia , a disorder of the autonomic nervous system [2] occurring when an individual stands up. [3] There is a substantial overlap between syndromes of orthostatic intolerance on the one hand, and either chronic fatigue syndrome (CFS) or fibromyalgia (FM) on the other. [4] It affects more women than men (female-to-male ratio is at least 4:1), usually under the age of 35. [5] Orthostatic intolerance occurs in humans because standing upright is a fundamental stressor and requires rapid and effective circulatory and neurologic compensations to maintain blood pressure , cerebral blood flow , and consciousness . When a human stands, approximately 750 mL of thoracic blood is abruptly translocated downward. People who suffer from OI lack the basic mechanisms to compensate for this deficit. [1] Changes in heart rate , blood pressure, and cerebral blood flow that produce OI may be caused by abnormalities in the interactions between blood volume control, the cardiovascular system , the nervous system and circulation control system . [6] Contents 1 Signs and symptoms 1.1 Acute OI 1.2 Chronic OI 2 Causes 3 Diagnosis 4 Management 5 Notable case 6 See also 7 References 8 External links Signs and symptoms [ edit ] Orthostatic intolerance is divided, roughly based on patient history, in two variants: acute and chronic . Acute OI [ edit ] Patients who suffer from acute OI usually manifest the disorder by a temporary loss of consciousness and posture , with rapid recovery (simple faints , or syncope ), as well as remaining conscious during their loss of posture.
A number sign (#) is used with this entry because of evidence that orthostatic intolerance is caused by heterozygous mutation in the gene encoding the norepinephrine transporter (SLC6A2; 163970) on chromosome 16q12. One such family has been reported. Clinical Features Orthostatic intolerance is a syndrome characterized by adrenergic symptoms that occur when an upright posture is assumed: the heart rate increases by at least 30 beats per minute, without orthostatic hypotension (Jacob et al., 1997). Most patients with orthostatic intolerance are women between the ages of 20 and 50 years (Low et al., 1995). This syndrome, first described by Da Costa (1871), has been called soldiers heart (Fraser and Wilson, 1918), neurocirculatory asthenia (Wooley, 1976), and mitral valve prolapse syndrome (Boudoulas et al., 1980). It is similar in many respects to chronic fatigue syndrome (Schondorf and Freeman, 1999).
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.
Have been also identified new genes involved in tachycardia ( CASQ2 ) or associated with an alteration in cardiomyocyte communication ( PKP2 ). [46] Rare mutations in the cardiomyopathy gene TTN may also increase the risk of AF, even in individuals without signs of heart failure. [47] [48] Small genetic deletions on the X chromosome around the STS ( steroid sulfatase ) gene are associated with increased rates of AF in males [49] Sedentary lifestyle [ edit ] A sedentary lifestyle increases the risk factors associated with AF, such as obesity , hypertension , or diabetes mellitus .
Overview Atrial fibrillation (AFib) is an irregular and often very rapid heart rhythm. An irregular heart rhythm is called an arrhythmia. AFib can lead to blood clots in the heart. The condition also increases the risk of stroke, heart failure and other heart-related complications. During atrial fibrillation, the heart's upper chambers — called the atria — beat chaotically and irregularly. They beat out of sync with the lower heart chambers, called the ventricles.