Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Cerebral Amyloid Angiopathy, App-Related
OMIM
The proband's mother had died at age 83 with profound dementia; one sister, who was diagnosed with dementia with occipital calcifications and leukoencephalopathy at age 67, died 2 years later from intracranial hemorrhage; a brother had an occipital hemorrhage at age 58, at which time occipital calcifications and leukoencephalopathy were discovered; and another brother died after a minor stroke at age 70 with dementia, occipital calcifications, and external carotid artery dysplasia. ... The number of microbleeds correlated with increasing age, possibly reflecting disease progression. ... Revesz et al. (2003) reviewed the pathology and genetics of APP-related CAA and discussed the different neuropathologic consequences of different APP mutations. ... Human APP mRNA was detected in neurons and neuronal processes, but not in vessel walls. ... Herzig et al. (2006) extended their earlier studies by developing several murine models of APP-related CAA and APP-related parenchymal amyloid deposition.
-
Alzheimer Disease
OMIM
Bateman et al. (2012) used the participant's age at baseline assessment and the parent's age at the onset of symptoms of AD to calculate the estimated years from expected symptom onset (age of the participant minus parent's age at symptom onset). ... The mean age of onset of dementia was 43 years. ... In the 5 index cases with the combination of early-onset Alzheimer disease and CAA, they found evidence for a duplication of the APP locus (104760.0020). In the corresponding families, the APP locus duplication was present in affected subjects but not in healthy subjects over the age of 60 years. ... Revesz et al. (2003) reviewed the pathology and genetics of APP-related CAA and discussed the different neuropathologic consequences of different APP mutations. ... Di Fede et al. (2009) identified a homozygous mutation in the APP gene (A673V; 104760.0022) in a patient with early-onset progressive AD beginning at age 36 years.TOMM40, TREM2, ABCA7, APP, APOE, PSEN2, PSEN1, MAPT, SORL1, PRNP, CASP3, BACE1, GSK3B, NCSTN, IDE, IL1B, HFE, A2M, ACE, DHCR24, BIN1, ESR1, ADAM10, ADAMTS1, PGRMC1, VEGFA, ARC, CYP46A1, SLC30A4, VSNL1, PICALM, HMOX1, HLA-DRB5, IGF1R, IGF1, INPP5D, IGF2, MPO, NPY, NOS3, PLAU, PLCG2, PPARG, RELN, MTHFR, PYY, NECTIN2, SLC2A4, IGF2R, SOD2, MAOB, TF, LEP, TFAM, INSR, INS, TNF, TPI1, EPHA1, F2, ENO1, CR1, CASS4, ATP5F1A, CLU, CHRNB2, CHRNA7, MIR766, CD33, IQCK, EIF2S1, MIR505, APOC1, CALM1, MIR100, MIR146A, BDNF, BCL2, MIR375, MIR296, BCHE, MIR708, TPP1, SLC30A6, SNAR-I, DPYSL2, ACHE, CD2AP, GAPDHS, PCDH11X, CYP2D6, MIR4467, CRH, MIR3622B, BAX, AMFR, ABI3, CST3, MS4A4A, WWOX, BRCA2, FANCD2, TFF1, TAS2R64P, CTNNB1, SUCLA2, SNCA, CTSD, RNR2, NEFL, TAS2R62P, SOD1, ITPR3, ITPR2, ITPR1, FLAD1, PSENEN, TP53, CDK5R1, EIF2AK3, UBQLN1, ALG3, PIK3CG, PIK3CA, PIK3CD, SERPINA3, PIK3CB, DOCK3, APLP1, OGDH, CREB1, NOTCH1, CASP6, NGF, CCND1, FOS, DLX4, DLG4, DDIT3, RABGEF1, PEBP1, PYCARD, DAPK2, KCNIP3, CTSB, CSF2, CRMP1, CTSG, EHMT2, ENO2, ERBB4, TMED10, TERF2IP, PTK2B, FCN2, PTGES3, FGF2, ACKR1, DNM1L, SDC3, G6PD, GCHFR, ITM2B, CREBBP, MAP3K8, TRPM7, ADI1, MTCO2P12, UPK3B, ACTB, AKT1, AKT2, ANXA1, APBB1, DNLZ, STS, MIR34A, BRCA1, MIR137, C5AR1, DDR1, CAMK4, TMED10P1, MPEG1, C9orf72, ESCO1, CDCA5, PRRT2, MAP1LC3B, CAT, EHMT1, CNR2, SPPL2B, RAB9A, NRXN3, GFAP, SYNJ1, SERPINB5, CD99, MME, MNAT1, CCL2, RRAS, RPS27, RPS21, RAP1A, PYCR1, COX2, PTS, PTGS2, MTHFD1, MMUT, NCAM1, NFIA, NFIB, MAPK8, MAPK3, PRKCB, PRKCA, PPBP, MED1, NFIC, PPARA, NFIX, PKD1, NOTCH3, NRGN, MEOX2, MEF2A, SPRR2A, TTC3, GRIN2A, DENR, GRIN2B, RAB7A, LRP8, HPRT1, HSP90AA1, VIM, IDUA, UTRN, SUMO1, UBE2I, TTK, TPT1, SULT1E1, IL1A, IL6, IL12A, TSPAN6, TIE1, TGFB1, TG, KNG1, LAMC2, LGALS3, TERT, TERC, STIM1, H3P17
-
Early-Onset Alzheimer's Disease
Wikipedia
Alzheimer's disease developing before the age of 65 Early-onset Alzheimer's disease Specialty Neurology Early-onset Alzheimer's disease , also called early-onset Alzheimer's , younger-onset Alzheimer's [1] or early-onset AD , is Alzheimer's disease diagnosed before the age of 65. ... This loss of brain volume affects ones ability to live and function properly, ultimately being fatal. [5] Beta-amyloid is a small piece of a larger protein called the amyloid precursor protein (APP). Once APP is activated, it is cut into smaller sections of other proteins. ... Alpha-secretase cleavage of APP, which precludes the production of Aβ, is the most common processing event for APP. 21 allelic mutations have been discovered in the APP gene. ... "A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid". ... (PDF) . The International Journal of Ageing and Later Life . 10 (2): 9–29. doi : 10.3384/ijal.1652-8670.16302 .
-
Alzheimer's Disease
Wikipedia
The hypothesis holds that an amyloid-related mechanism that prunes neuronal connections in the brain in the fast-growth phase of early life may be triggered by ageing-related processes in later life to cause the neuronal withering of Alzheimer's disease. [64] N-APP, a fragment of APP from the peptide's N-terminus , is adjacent to beta-amyloid and is cleaved from APP by one of the same enzymes. N-APP triggers the self-destruct pathway by binding to a neuronal receptor called death receptor 6 (DR6, also known as TNFRSF21 ). [64] DR6 is highly expressed in the human brain regions most affected by Alzheimer's, so it is possible that the N-APP/DR6 pathway might be hijacked in the ageing brain to cause damage. ... A β is a fragment from the larger amyloid precursor protein (APP). APP is a transmembrane protein that penetrates through the neuron's membrane. ... Since the incidence of AD increases with age, it is particularly important to include the mean age of the population of interest. ... These costs will probably increase with the ageing of society, becoming an important social problem .APP, ACE, TREM2, ADAM10, APOE, PSEN1, GSK3B, HFE, MAPT, PLAU, NPY, BCL2, CASP3, BDNF, IDE, INSR, IL1B, LEP, BACE1, IGF2, IGF1R, ATP5F1A, INS, BAX, CR1, A2M, ABCA7, TOMM40, CD2AP, BIN1, EPHA1, CLU, PICALM, NOS3, PSEN2, APOC1, MPO, SORL1, VSNL1, INPP5D, NECTIN2, MS4A4A, PCDH11X, CASS4, BCHE, MIR146A, CYP46A1, DHCR24, CHRNA7, NCSTN, VEGFA, DPYSL2, PRNP, ESR1, PPARG, RELN, HMOX1, ACHE, CST3, MAOB, TNF, MTHFR, IGF1, CD33, TFAM, IL6, CYP2D6, CRH, SOD2, UNC5C, PLCG2, TF, ABI3, WWOX, SLC30A6, CHRNB2, ARC, PGRMC1, F2, CALM1, EIF2S1, HLA-DRB5, ENO1, TPI1, IGF2R, SLC30A4, MIR296, SLC2A4, MIR100, IQCK, MIR375, AMFR, SNAR-I, ADAMTS1, MAPK14, PIN1, PYY, PTGS2, S100B, PPARGC1A, NOS2, NGFR, NGF, NFE2L2, SOD1, SYP, CDK5, NGB, MIR505, GAPDHS, MME, MAP2, CTNNB1, TPP1, LRP1, IRS1, CHAT, GAPDH, MIR4467, MIR3622B, AGER, MIR766, MIR708, CAV1, NTRK2, PTGS1, APLP2, ADAM17, MFN2, DNM1, HSF1, GSR, IL33, CCR5, HSPD1, HSPB1, CIB1, CASP8, IKBKB, SERPINF1, ATP7A, MT2A, ADAM9, INS-IGF2, BCL2L2, CASP9, GAB2, PTK2B, PLCB1, ABCA1, GRN, CASP12, SQSTM1, FERMT2, HLA-DRB1, NFIC, CSF1R, APOB, MARK4, HSPG2, MS4A6A, CELF1, VCP, SYNJ1, ZCWPW1, MS4A4E, APH1B, APOC2, F13A1, EXOC3L2, PLXNA4, ADAMTS4, AKAP9, MADD, DST, PILRA, FRMD4A, LAMP1, SLC24A4, GLIS3, SPON1, CADPS2, IL34, COL18A1, TRIP4, SPI1, TGFB2, BCL3, MTHFD1L, AICDA, IL6R, DCHS2, MEGF10, SLC16A7, EPHX2, NDUFAF6, DSG2, OSBPL6, CELF2, UBE2L3, SPPL2A, MAPK7, CDH13, LAMA1, SGK1, SUCLG2, LUZP2, PTPRG, ST6GAL1, AP2A2, RBFOX1, SORCS3, TSPOAP1-AS1, TLN2, ZAP70, ALDH1A2, TCF7L2, FMN2, OTOF, EXOC4, HSD17B10, DNM1L, ALOX5, GULOP, HTR2A, AHCYL1, SDR42E2, HTR6, GTF2H1, GRM5, IAPP, AHSA1, STAG3, ALB, FARP1, TSHZ1, HRES1, AGFG2, DCAF7, SIGMAR1, BCKDK, RTN3, TPPP, HSPA4, HCLS1, HSP90AA1, G3BP1, PGAM5P1, BACE1-AS, KHDRBS1, ALDH2, CFH, HCRT, GPC6, ABCA8, GLP1R, NR3C1, TNRC6A, IL19, PARVB, DDX25, BZW2, FCN2, FGF10, GOLIM4, LINC00476, HPGDS, FANCD2, PDE7B, SIGLEC7, TSPAN16, TRPC4AP, POLDIP2, CNTNAP2, RNF19A, BACE2, UBQLN1, ITSN2, GRIN2B, ZGLP1, BCAS3, EPDR1, TMED9, ENO2, CYCS, ANXA1, EPHB2, EPO, RAPGEF6, APH1A, LARS1, EYA4, SNX9, ESR2, WAC, SLC8A1-AS1, DCTN4, RMDN1, QPCT, MTOR, SGK3, FBXL7, SZT2, GIP, ACSL6, WWC1, CLEC16A, KAZN, LINC01672, PRRC2C, COLGALT2, PLD3, HECW1, ZNF292, MYO16, DKK1, SIRT2, ACOT7, PSIP1, CLASRP, LINC00271, SIRT3, SIRT1, GFAP, NUP62, FYN, CBLC, INHCAP, TRIM51CP, GABPA, GABRA2, GABRG3, DAPK2, SMUG1, GAP43, GATA1, GCG, GCHFR, TARDBP, NCS1, GDNF, HDAC6, RAB3D, MVP, OGG1, LINC02268, LINC02325, SOAT1, ACTG2, ACTG1, SNCG, SNCA, SNCB, SNAP25, NOS1, ACTB, MEF2C-AS1, SLC6A4, NPC1, SLC6A3, GEMIN7-AS1, SLC1A2, LINC01508, LINC01725, NEFL, COX2, TNFRSF1B, STAG3L5P, TLR4, TLR2, MSH2, THY1, MT3, TH, SST, STAG3L5P-PVRIG2P-PILRB, TGFB1, TFF1, RNR2, LINC02653, LINC01712, TCF3, NRGN, CX3CL1, ELMO1, CCL2, PIK3CG, ABCA2, PLA2G1B, EIF2AK2, SERPINA3, PLG, MAPK8, MAPK1, PRKCB, PRKCA, PRKAB1, PRKAA2, PRKAA1, PMS2P1, POLD1, PTPA, PON1, PIK3CD, PIK3CB, PIK3CA, MOK, UPK3B, SORT1, ROS1, SERPINE1, REST, REN, RELB, RAC1, LINC01965, PVR, PVALB, PTPRA, LINC00972, ABCB1, LINC02008, MTCO2P12, TP53, OVCH1-AS1, MOBP, MNAT1, SLC4A8, AGT, INSIG1, AZIN1-AS1, EIF3E, FHL5, IRF2, GSTO1, ITM2B, GRAP2, LIPG, ADIPOQ, KL, MSC, LRAT, AP4M1, CCRL2, IL18, IL17A, IL12A, ST18, IFNG, ARL17B, AKT1, AIF1, KRBOX1, IGFALS, HDAC9, PHF14, IL10, IL1A, ALOX12-AS1, MICAL2, IL2RB, IL4, CLOCK, CXCL8, KCNN2, MPZL1, HERC2, VDR, TFEB, MAOA, COX10-AS1, ZNF232, YWHAZ, VLDLR, PARP1, UTRN, BCAM, UCHL1, UBB, TYROBP, AFF1, TTR, TRPM1, MMP9, AIMP2, LTBP2, KNG1, CRADD, CACNA1G, LAMC2, RPSA, CDK5R1, SUCLA2, LCN2, LDLR, BECN1, LPL, PDE5A, ABCB11, APOC4-APOC2, KHSRP, DENR, AGPS, LPA, CDKAL1, PPARA, MEIKIN, COL4A4, CRK, CEACAM22P, SCIMP, CREB1, CR1L, ZNF862, SH2D4B, CP, PNPLA7, SIMC1, GGACT, COMT, COL12A1, FAM181A, BRCA2, PPP1R37, GPR141, TENM3-AS1, CNR2, L3MBTL4, UBXN11, ACKR2, TMEM132C, CASTOR3, CLPTM1, STRADA, FNIP1, CDCA5, NLRP3, APOA1, CRMP1, KAT8, CSMD1, CLMN, PINK1, CYP8B1, AHNAK, MIR132, MIR107, CUX1, POTEM, PPP1R3B, FAS, SAP30L, ANKRD55, CTSD, CTSB, GEMIN7, EHMT1, LINC01184, CTNNA2, LINC01185, TMC5, THSD4, CCDC134, SP6, LINC01567, PDCD1LG2, SETD7, APOD, BHMG1, CSF2, HYI, BLOC1S3, TSPO, CHRNA4, CHRNA2, TAS2R62P, C3orf67, C9orf72, CCDC83, CCDC89, KDM1B, CD14, TGM6, ATXN7L1, RSPO4, ADGRF2, STH, TAS2R64P, CALHM1, RUNX1T1, PPP1R42, ALPK2, PCSK9, CAT, ANKRD31, CASP6, NKAIN3, TRIQK, CALB1, STEAP1B, CASP1, EPHA1-AS1, CAPN1, APOC4, FAM181A-AS1, NKPD1, SPRED2, CD36, SCARB1, PLPP4, MED12L, ARAP2, CHN2, CHI3L1, ACTBL2, C10orf71, MCIDAS, LRRK2, AKR1C4, ANO4, AGBL1, CEACAM20, ZNF813, RMDN3, CETP, CDR1, LINC00343, TCAM1P, APOC1P1, IGSF23, RMDN2, CDK1, SLC25A48, NKAIN2, FSIP1, CD68, BMPER, C3, CD40, CYP19A1, CRP, NIT2, ANO3, DLG4, ARHGAP20, RCAN1, WDR41, NDUFA12, STK32B, EDEM2, DSCAML1, RNF165, SH3RF1, DYRK1A, MIR29A, SYBU, AQP4, APBB1, DLX5, DBN1, PALM2AKAP2, CEACAM19, DPP4, IL6-AS1, ARVCF, CDC42SE2, DMXL1, TULP4, DAPK1, PMS2CL, POTEKP, MIR34A, VAT1L, OLR1, HDAC2, LRP8, GSN, CCL11, S100A9, COL25A1, POTEF, KLK6, BLMH, HSD17B7, P2RX7, COX8A, ABCB6, PRRT2, IL2, SORCS1, NR1I2, MAPK3, ITGAM, CASR, ATP7B, VDAC1, EGR1, PDE4A, RAB5A, SUMO1, NRG1, OXER1, NTRK1, TFCP2, ANK1, CSNK1D, DLST, APLP1, BLVRA, NFIB, IL1RN, HTT, ACAT1, PLA2G4A, NFIX, NLRP1, GPRC6A, HMGCR, PPID, LPAR3, FZD4, REG1A, MRGPRX1, LRP2, DBH, PSENEN, VPS35, ESCO1, HSD11B1, VN1R17P, SOX2, AGTR1, XBP1, MIR155, MRGPRX4, MRGPRX3, GAL, GPR151, IL13, PAEP, OGDH, GPR166P, STAT3, SET, NFIA, PLB1, AR, LGR6, DHRS11, ABCG2, C4A, KCNIP3, HSD17B13, ABCG1, TTBK1, NOTCH1, EIF2AK3, SLCO6A1, CHMP2B, RBM45, CD44, RIPK1, APBA1, GSTK1, ADNP, ICAM1, BRCA1, APCS, TNFRSF1A, NFKB1, CNTF, MMP3, KLC1, LBP, CTNNA3, SGSM3, FGF2, C4B, HIF1A, CREBBP, SERPINA1, TMEM106B, GRIA1, GRIA2, ECE1, C4B_2, GSAP, OGA, TFRC, PLA2G6, ST3GAL4, PAWR, MFAP1, KAT5, GSTM1, APRT, COX1, HP, NTF3, MIR206, FPR2, CDC42, FUS, MARK1, FGF1, PREP, C5AR1, PON2, MIR29C, CALB2, PDIK1L, SYK, S100A1, CH25H, SREBF2, COX5A, GRIN2A, VCAM1, TMED10, GSTP1, KLK8, PHF1, CXCL10, MEFV, SP1, GJA1, IGFBP3, SLC17A7, CYP3A4, FOXO3, HMGA1, SLC11A2, XPR1, MARK2, PPIF, CRHR1, SHANK3, MYC, CD40LG, CPOX, FKBP5, ANPEP, CAST, C1D, FKBP4, HSPA1A, FLT1, MIF, PLA2G2A, CX3CR1, CSF3, IFNB1, KALRN, PLTP, STXBP3, DDR1, PWAR1, PRDX2, TP63, VIM, IL23A, F2RL3, MMP14, MEF2C, TREM1, TMED10P1, NAT2, MIR342, SAMD9, RAB7A, PGR-AS1, TRPM2, EGFR, ADRB2, CLDN5, ETS2, SYT1, TIMP1, NME8, ELANE, F2R, CD59, EPHA4, CBS, MSMB, APLN, MMP2, MYCL, CALML5, SYN1, XRCC1, TGM2, EEF2, PLA2G7, ELAVL2, EDN1, TMEM97, HMGB1, MIR455, HTRA1, BPIFA2, SLC52A2, NQO1, TUBA1B, FOS, CRYAB, SLC2A1, GPR3, LGMN, SLC2A3, RIDA, FN1, ABCA4, HSPA1B, PECAM1, PTEN, HSPA8, HLA-A, PTPN1, HAMP, TXNIP, GRM2, P4HB, LIN28A, PSPH, GSTT1, CCN2, DECR1, CPLX1, BCYRN1, NES, POU5F1P4, MIR21, GRK5, POU5F1, NTSR1, MIR212, PRKN, LINC02210-CRHR1, HSPA5, DLD, DAB1, HTRA2, POU5F1P3, MIR137, DNAH8, MAPK10, GH1, SERPING1, ADAMTS2, EEF2K, GSTO2, ROCK2, NEDD9, SPTBN1, NTN1, CEBPD, GDF2, CEBPB, PWAR4, SYNM, IGFBP2, GLUL, ABCC9, ATM, PSPN, PTPRC, MIR29B1, RANBP9, NDRG2, CNR1, RTN4R, PTBP1, AQP1, PDK1, MIR106B, PDE4D, ARNTL, PRDX1, ADM, RENBP, POMC, PTPN4, MS, PDGFRB, P2RY2, MIR142, PDE9A, SSTR4, KLK3, BCL2A1, C2, SRPK2, NFATC2, ADCYAP1, LRRTM3, PLD1, NUBP1, MIR424, ATF4, BSG, MIR29B2, PPY, BMP4, TBP, SLC18A3, POLB, NOTCH3, SLC18A2, NPTX2, MTR, SI, MIR222, SH3GL2, ND2, NR4A2, SELENOP, CXCL12, CCL5, ATXN1, CALM3, ITGAX, IFNA13, DISC1, OPTN, HTR1F, HTR4, WNT3A, COL11A2, C20orf181, IFNA1, KEAP1, HDAC4, KLK4, SEMA6A, LRRC4, CRTC1, IL9, DAO, ALOX15, AGTR2, IDO1, SLC25A27, ABCG4, CD55, APOA4, MCOLN1, REM1, ATCAY, EBPL, HSPB2, HSPA9, PLK2, GAD1, NANOG, DCX, COASY, UBE2K, DDIT3, TREML2, APBB2, MAP1LC3B, SRRM2, GZMB, FXN, HNRNPA1, HPSE, RAB10, CIP2A, FOLH1, STIM2, DIO2, MMP24, CEBPZ, GBA, CDR2, ITPR3, CDKN2A, MELTF, SLC30A3, ADRA2B, FTO, FNDC5, GGA3, XPNPEP1, VGF, NR1H2, UGCG, MFGE8, MGAT3, CXCR4, TLR9, APOC3, GPT, ELK3, NEAT1, ADORA2A, MMEL1, TRPC6, EIF4E, CAMK2A, MS4A6E, SRR, HSPA14, IRS2, MGAM, C1orf52, HDAC3, PABPC4, ACKR3, LGALS3, FAM20C, WNK1, DRD4, CYP2C9, MBTPS1, DRD1, LRP6, GRK2, CYP2B6, OGT, LIPA, AD11, GORASP1, PTGDS, SPEN, MIR200B, NPTXR, DNMBP, MIR200A, NCOA6, MIR181C, RBP4, RELA, OPN1LW, EFHD2, MIR188, TPH1, HNRNPA1P10, CTNNBL1, SLC40A1, PNO1, CHCHD2, SDF4, RETN, GOLM1, PPP3R1, PYCARD, PAG1, CCR2, DDIT4, RCBTB1, SBNO1, PPARD, CD274, PCBP4, ACE2, PROS1, PRND, PPP1R15A, CIZ1, MIR26B, TPSG1, GGA1, CFAP97, MAP2K1, AATF, SHANK2, PRL, RBMS3, LOC107987479, MAP2K2, PAXIP1, CHCHD10, SBNO2, PTGES, SPHK1, LPAR2, PPIG, NRXN3, MED23, SPP1, MAPK8IP1, BAG3, APBA3, TAP2, PRDX6, CARTPT, SNAP91, SV2A, MALAT1, MAK16, SYVN1, GDF11, TAC1, TAT, SLC6A2, TRPV1, CISD3, TPT1, TSC2, TM7SF2, TXN, UBE2I, TLE1, TIMP2, XK, CNTN2, TGFBR2, YY1, GOLGA6A, ANP32A, TAM, TERT, DCP1B, TNFSF10, PPP1R1B, GPHN, RGS2, ADAM30, SAA1, METAP2, IMMT, SDS, ADAP1, RYR3, ECHDC3, RYR2, RXRA, SCD, RPS6KB1, CIT, RPS6, CTXN3, LMTK2, NLGN1, ROCK1, RGS4, ATXN2, SRL, STUB1, SHBG, LILRB2, AKR1A1, OLFM1, SKIL, SLC9A6, CREB3, PITRM1, SCGN, CXCR6, OCM, RIN3, SGCA, SFPQ, NCKAP1, CPLX2, TP73, EDAR, CCL3, NPS, BEST1, HPS1, CXCL1, GLO1, LIF, CDH1, LHCGR, CDK4, PCNA, PCK1, L1CAM, GPR42, GPX1, GRB2, ANGPT1, ANGPT2, CETN1, MYD88, GLB1, AKT2, LMNA, DNMT3B, TSC22D3, CD38, PER1, CD69, DRD3, LOX, CD74, LMNB1, DNM2, GAS6, AVP, GC, DMRT1, SARDH, GRIN1, ANXA5, BACH1, P2RY1, INPPL1, CSF1, CS, NEFM, NTS, APEX1, STS, HTC2, NEUROD1, NPTX1, NPPA, ATF2, HTR2C, ARRB2, IGFBP7, HMGCS2, CLK1, CKB, APBA2, CYP17A1, GSTM3, CHGA, CYBB, JUN, CTSS, ORM1, ITPR1, CHRM1, CHRM2, OPRK1, OPRD1, CTRL, HK1, ADRB1, LAMP2, CASP2, EDNRA, PLD2, CAPN2, F2RL1, ACO1, ERBB4, FAT1, FGFR3, CALM2, BRS3, CALCA, CAD, EGF, CASP4, FCGR3B, FDPS, LYZ, FCGR3A, FLNA, MECP2, FABP3, ADRA1A, PLXNA2, MBP, FAAH, ENPEP, F12, BAG1, MEOX2, HOMER1, ITGB2, DBA2, ITGAL, ITGB1, AIM2, AZIN2, CD80, ITIH4, CD46, VIP, CHRNA3, ATG5, TREML1, MCL1, GPRASP2, VWF, APOA5, TMEM119, KLF4, SOCS6, WNT1, XBP1P1, FOXQ1, C3AR1, OPN4, USF1, C1QA, CXCR2, VPS26A, MOGAT3, CCR3, IL6ST, IL5, IL1RAP, LMF2, IL1R1, CREB3L1, UNG, MCU, CLSTN3, SNPH, IL9R, NPEPPS, NAPSA, USF2, MEF2A, ING1, CGB8, FTMT, CHM, VAV1, IMPA1, C1R, ILK, ADAMTS3, IL16, MAP3K5, IL15, GDF15, CGB5, CA2, KCNB1, TSPOAP1, SMAD2, DPPA2, SGO1, LIG3, IFNL3, TNK1, CP20, APCDD1, HSD17B6, CDKN1A, TTBK2, CDKN1B, SLC2A14, CFLAR, STMN1, LIPC, TAB3, CHIT1, BRAP, SPARCL1, MLKL, PTCRA, CD47, LGR5, CD8A, CCT, NR4A3, USP9X, MSRB3, CDR3, CCK, LNPEP, CASP7, CHRFAM7A, CAMP, PER3, YES1, CGA, MARK3, CGB3, SYNGR1, CALCR, IL1RL1, ARHGEF2, PER2, SLC33A1, TPH2, CHEK1, RAB7B, NOG, MBL2, CFL2, HSPB6, AHSA2P, SLC30A1, CD200R1, SOCS3, KDR, KIF5A, HAP1, CALR, CES1, TRPA1, HSPB3, WASF1, SLC32A1, ARHGEF7, CAMK4, COL3A1, H3P40, SCRN1, PTCD1, SPHK2, ATN1, PPIL2, POU2F1, FOSB, GCA, FLT4, SH2B1, APPL1, DNMT1, FLG, FOXO1, DUSP1, DUSP6, HHAT, HSPB8, E2F1, PLXNA3, DOCK2, PNPLA2, ADI1, GMFB, GPI, GPC1, KIF21B, NMNAT2, TRIB3, ALS2, DLG2, DLG3, GLS, PDSS2, ASTN2, MCF2L, GLI2, KIDINS220, CBLIF, SYNE1, DMD, GGT1, FKBP1A, SIT1, SV2C, GDE1, FOXP3, ASCC1, TMED7, FIS1, PRLH, CRYL1, ADIPOR1, LSR, F11, MBL3P, SIRT6, TRMO, NRN1, LCMT1, PRRX2, ERN1, BIN2, UBR5, HEBP1, GEMIN4, PDCD4, TBK1, SLC25A38, FGF14, PCSK1N, TRPM7, DLL1, FLVCR1, AHI1, SETD2, ELK1, IL22, NCAPH2, ELN, PADI1, BPTF, NRBF2, FABP5, EP300, PLA2G3, GRHL3, CXCR3, NAV3, SIRPB1, FLOT1, MET, KCNMB2, CRISPLD2, ARHGAP24, HNMT, SNX27, NPL, BHLHB9, TRIM13, HMOX2, KLF2, CSNK1E, LPAL2, CPQ, PPP1R2C, RAPGEF3, TET1, CRYZ, SORBS3, CTBP1, BCL2L11, COL17A1, NCAPD2, COX10, UBASH3B, NR1I3, ACOT8, PTPN5, PPP1R9B, TOM1, MINDY4, CPE, HTR7, NR1H3, HTR1B, HTR1A, LRPPRC, PDIA6, RHBDD1, NAA25, HLA-C, TPX2, BCAN, ADAMTS13, MOAP1, TNMD, GRIA3, NEUROD6, CHEK2, PADI2, HRH3, PHB2, SIL1, MGLL, FFAR1, GADD45A, MMRN1, DEFA1, IL21, NLRC4, GPR17, AZI2, HHIP, CTF1, HCRTR2, HLA-B, CTNND2, HHEX, HGF, CTSG, HDAC1, CTCF, DHX40, PTGES3, STIP1, CTSZ, CXADR, CARD14, CYP1A2, PDE10A, LILRB1, EHMT2, PDIA2, UMOD, ANGPT4, MIR339, SYN2, MSD, ACP3, APEH, ST8SIA1, AZU1, PI4KA, NCAM1, MIR144, SMIM10L2B, MSI1, NAP1L1, PRSS3, PEBP1, MASP1, SIM2, TIA1, MIR15B, ATD, RPL29, ABCC1, NCAM2, MIR125A, TLR5, SERPINF2, TNFAIP1, CXADRP1, MAPK9, ZFHX3, GGTLC4P, AD10, SGCG, MIR451A, CDR1-AS, TPTEP2-CSNK1E, MIR384, ITSN1, CBSL, MPZ, NFATC4, PKM, NCL, NTF4, LOC643387, SLC1A3, APOA2, THAS, PSMB6, SERPINB6, PSMB9, RHOA, ARMCX5-GPRASP2, SMPD1, REG3A, ATP4A, MIR193B, RFC1, NOTCH4, SLPI, PGF, AEBP1, MIR214, MIR219A1, SLC19A1, MIR22, PCSK1, ALPP, AMD1, MTNR1A, TGFBR1, COX3, ATP12A, NM, ADD3, PSD, TGM1, ARR3, NPM1, PAK1, TMED7-TICAM2, PNP, MIR195, MTHFD1, ADH1B, AMPH, ND4, AMD1P2, ARG1, SULT2A1, RRAS, PDE7A, TTPA, TYK2, TXNRD1, PPP1R1A, PPP1R10, SPG7, SPAST, RAB4A, PPP2CA, OTC, PPP2R2B, MMP1, ARMS2, RAB3A, GGTLC3, RTL1, P2RX4, ST13, SPARC, ALAS1, PPP3CA, NEFH, SEL1L, TYR, RAB6A, MICB, PPIA, CCL4, BST1, TICAM2, BNIP3, MIR326, OPRL1, PON3, BMP6, OPRM1, AHSG, H3P17, SDC2, AGRN, TYRP1, PPP1CA, BMI1, TYRO3, PNMT, DEFA1B, GGT2, ORI6, SMIM10L2A, CISD2, ARSA, EIF2AK4, PRKAR1A, LRP1-AS, SRSF2, MIR98, MIRLET7B, CD200, PDCD1, RAP1A, GGTLC5P, CCND1, ANXA6, FXYD1, S100A6, NFATC3, PLK1, ABO, PTGER3, APC, S100A12, ASL, HSP90B2P, SETMAR, PRRX1, PZP, STAT1, ODC1, CFB, CDNF, ZBTB4, PARK16, SUGP1, DIO1, SORCS2, MAGEE1, ALDH1A1, MIR1306, DES, DIAPH1, LSM2, MIR1229, XPO5, HCN3, CFD, MIR664A, KIF17, WDR48, MTRNR2L12, PRX, DHFR, EPG5, SEPTIN1, FAS-AS1, RNF213, MIR320E, MIR1908, HECW2, LINC00672, NUFIP2, ABCD1, DLG1, QRFP, ZNF410, AOC2, NBEAL1, CYP11A1, OPN1MW2, AD6, P2RY12, NMNAT1, DEPTOR, TNS3, CYP2D7, FAM72A, NUCKS1, CLEC7A, CYP26A1, ARAP3, GREM2, CDKN2B-AS1, CYP2J2, UBE2Z, MIR1246, TSPY3, MIR632, CTSK, GTDC1, CTSL, MIR650, MIR660, SNORD118, TNFAIP8L2, LYNX1, MUL1, PAGR1, CYLD, MAPKAP1, APOF, PDCL3, CYP2C19, NOC3L, CYP27A1, DPEP2, MFT2, PROK2, HPSE2, AD14, AKR1C2, ALPI, TRPV4, NTN4, PRM3, PDF, JAM2, ALOX5AP, TSPY10, DEFB4A, DEFB4B, ALOX12, PTBP2, DCN, NECAB3, FKBPL, NEUROG2, DGKQ, SLC25A4, MIR873, MIR301B, CENPK, DAXX, GFRA4, GOLPH3, ERVK-6, MTUS1, DBI, MIR937, ANG, ACE3P, SOD2-OT1, ANK3, DRD2, ZNF608, NAT10, DYM, LOC102724334, TRIT1, EIF4G2, TET2, EIF5, SERPINB1, ELAVL4, PDP1, ACO2, THRA1/BTR, UGT1A1, CCHCR1, CPVL, SMOX, TOLLIP, TERF2IP, SNTG1, EMP1, LOC102723407, EIF4EBP1, MIR6845, EIF2S3, ADCY2, NUDT11, EGR2, MSTO1, ADARB1, SLC6A15, ADA, TAPBPL, TESC, MIR6840, ACVRL1, FOCAD, EIF4A1, CASZ1, QRICH1, PGPEP1, EIF4A2, NDE1, ASIC2, CTTN, ACADVL, GSKIP, LNCRNA-ATB, ATP6V1H, H3P7, TDP2, ERBB2, CINP, ZCCHC17, H3P13, DTL, GPRC5B, ERCC1, DCDC2, NAT8B, GULP1, H3P23, ERG, H3P28, H3P11, PPIL1, STIN2-VNTR, NANS, EPHA8, H2BS1, POLE3, ACACA, SLC29A1, FXYD6, LRP1B, CST12P, SIRT1-AS, INPP5K, MSRB1, ARID4B, EPOR, ABL1, AAVS1, NR2F6, ERVK-32, LOC110366354, MNS16A, EFNA5, SLC47A1, ALAD, EEF1A1, SNHG19, MICA, DNTT, SOX21-AS1, DOCK3, DPYSL3, MIR626, XAB2, MFF, DUSP22, ARNTL2, SPPL2B, MCCC1, TMX2-CTNND1, ANKS1B, DPYSL5, FXYD6-FXYD2, BARHL1, DSC1, TWSG1, TLE5, DNASE1, DNA2, OCLN, NLN, AMIGO1, AHR, PLEKHG5, SLC24A3, SPC25, TTC7A, PELI1, JAG1, TMEM159, RTN4, APMAP, CD177, CAMK1D, PLAAT1, NR0B1, TIGAR, P2RX5-TAX1BP3, PARD3, GKN1, ADH6, INAVA, CDK5RAP2, OGDHL, LINC01080, ATF7IP, IPO9, VAC14, DVL1, PPP4R3A, OPN1MW3, EBM, OTUB1, SOX6, SLC30A10, SMPD3, MEG3, PLIN2, FBXW7, TDP1, ADORA1, DSC3, ACSS2, BTNL2, KIAA1217, ZNF253, CFC1, MIR4668, DSG1, APOM, MYO5C, MIR4487, NOTCH2NLC, USE1, SELENOS, GDNF-AS1, DSPP, ADCY10, ADRA2A, ZNF415, LINC-ROR, NARS2, CSF2RB, MIR616, MIR20A, CDC25C, PROM2, ATP6V1E1, IL23R, GLIS1, PM20D1, PHF13, CDH2, ZNF569, MIR191, CDK9, MIR192, PRIMA1, CDKN2D, MIR196A1, OR2AG1, LAYN, PIWIL4, MIR19B1, GPBAR1, CDC25B, GDF7, ZDHHC15, MIR139, CD63, SGMS2, MIR140, TMPRSS6, RHBDL3, AVPR2, MIR15A, CBLL2, MIR186, PRUNE2, AMOTL1, CD81, MIR18A, SLC2A12, CDA, MIR181A2, ATP5PO, SESN3, ATP6V1B2, UBR1, ATP5PF, PPME1, MIR224, LYZL4, KCNH8, MTERF4, MIR23A, CPO, ACMSD, MIR23B, BHLHE23, MIR25, CFL1, OSCAR, SPNS2, SEZ6, SLC38A10, MSI2, CFTR, MIR27A, MIR223, ALDH7A1, MIR221, SELENOM, ATP5MC2, CACUL1, HECTD2, SREK1, CTCFL, CBLN4, CDSN, ATP5MC1, DEFB104A, OCIAD2, MAGEC3, MIR210, CEACAM5, CECR, PPARGC1B, ATP5F1B, IL31RA, GNPDA2, SCARB2, NSMCE1, SOCS4, UBE2L1, BNC1, CACNA1C, SLC25A20, SERPINA13P, BLM, SREK1IP1, MIF-AS1, C20orf203, SYPL2, ZNF763, CCL4L1, BID, BGN, ZSCAN1, ZADH2, SMIM20, MILR1, PGP, GOLGA6L2, TMEM189-UBE2V1, TMEM189, IL31, C4BPA, BTK, AMIGO2, HCN1, NHLRC2, ATP9B, SBSN, BMP1, OSTN, C5, CFAP410, BARHL2, NANOS3, C9, STING1, GADL1, ARMH1, VPS51, HCAR2, CAPG, LINC00639, TMEM201, LIN28B, CD5L, MIR127, CD19, KIF6, MS4A1, MS4A3, HYLS1, STOX1, FOLH1B, OR8J1, TRIML2, CD28, KHDRBS2, GAPT, CENPV, KLHDC8B, MIR134, CD86, PIKFYVE, SLC29A4, CCNC, KRIT1, PTF1A, DAOA-AS1, BDKRB2, HCA1, BRD3OS, ASPM, BCS1L, SGMS1, BCR, MIRLET7D, MIR122, RUNX1, ANKK1, BCL6, PHYHD1, BAK1, MIR10A, EBF3, CCKAR, MIR28, FRMD6, MIR613, ADAMTS10, TMEM175, XIAP, MAF1, BIRC3, SLA2, ANTXR1, ASCC2, CRHBP, EVA1A, QRFPR, MAGT1, NCALD, LOC646506, ROPN1L, L3MBTL2, GMNC, SCFV, CSE1L, RNF146, PHF6, HOOK3, BRSK1, MBOAT4, COX15, MIR497, MFSD2A, MIR501, ACCS, ARG2, FAM126A, CPB1, CPN1, ECSCR, MAP1LC3A, MIR484, AQP9, CPS1, SNORD35B, CPT1A, ABLIM2, FASLG, NETO1, DOCK8, RNFT2, C1QBP, TM2D3, ASRGL1, PTGES2, PANK2, MIR590, SCD5, CSNK2A1, VCAN, ZC3H14, CSPG4, CTBS, MIR592, MIR598, CAMKMT, CTNS, SLTM, PTCD2, CTNND1, MIR603, NUBPL, WDR26, SPHKAP, GSTT2B, TMEM163, NCF1, LBH, SPAG11A, SFTPA1, CSF3R, ZNF436, CSN2, NDFIP1, MIR545, SLC44A4, SLC19A3, FAM72B, AIRE, IQCJ, CSNK1G2, DNAJC5, CSNK1G3, LINGO1, ATG4C, ATP2B4, MIR346, SERPINC1, CISH, ASPA, CLC, UCN3, TMEM54, ASS1P1, CLCN3, NACC1, ASIP, STX1B, IFT43, MIR133B, MIR151A, CLK2, TP53INP1, MIR330, MIR335, MIR338, MIR93, SLC26A7, OMA1, CHGB, PLD4, TDRD9, CHD1, MIR299, ATIC, ATHS, H4-16, LRIG3, EXOSC6, CHRM3, MIR30B, MIR30E, AGAP2, MIR31, MIR34C, MIR9-1, GRIN3B, GRIN3A, ASAH1, MIR369, H2BC12, KPRP, LRSAM1, MIR429, H4C15, SHF, GADD45GIP1, COL11A1, ZNF628, MIR431, HNP1, NAV2, MIR409, SLC31A1, RPPH1, SNORD14E, SNORD14D, SNORD14C, SNORD14B, COX6B1, MIR485, CNTN1, DNM1P33, CNTFR, CHRDL1, CLN3, MIR361, MYOCD, PRDM6, DNER, SPECC1, MIR377, CLN5, EXOC3L4, CNP, MIR425, ARRB1, ZNF804A, BDNF-AS, NLRP12, CCR6, ABCC2, DEFB104B, LRRC15, POU3F4, HOOK1, TERC, NAT1, MCM2, EZR, MDH1, VEGFC, MDH2, MDM4, MEF2D, UVRAG, UROD, UQCRC1, UGT1A, SLC35A2, UCP2, UBTF, UBP1, MID1, UBE3A, UBE2V1, CXCL9, ATXN3, UBE2D2, UBE2A, UBC, MAP3K10, MC1R, WARS1, WAS, ZMYM2, MANF, SCG2, FZD5, SMAD7, MAG, MAP3K12, MAP1A, MAP1B, ZNF236, ZNF224, ZNF217, RNF112, WEE1, MZF1, ZIC1, MARS1, MAS1, MAT1A, MAT2A, MAZ, XIST, WT1, WNT2B, WNT5A, UBA52, KMT2A, MLLT3, THBS1, TLR3, TLE3, MSH3, TKT, TIMP4, TIMP3, MSR1, MSRA, THRA, THOP1, THBS4, CYTB, MRE11, NUDT1, TGFBI, TGFB3, ND1, TFF3, TFDP1, MTNR1B, MTRR, TRNL1, MUC1, TERF2, TSPAN7, MRC1, TWIST1, TRAF2, TUBA4A, NR3C2, TTN, TSPY1, TSHR, TSG101, TSC1, MMP7, MMP8, TRPC1, TRH, NR2C2, TNFAIP6, MMP13, TPM1, MOG, MOV10, TP53BP2, TP53BP1, MPG, TNR, TNNI3, MPI, MPST, SLBP, REEP5, DEK, SNX3, TNFRSF6B, RAB11A, LDHA, LEPR, LGALS4, LGALS9, GPAA1, RNMT, GBF1, ADAM19, URI1, TRADD, LCK, B3GALT4, LIFR, LIG1, SOCS1, NUMB, LIMS1, AOC3, PDE8B, USO1, TNFSF11, STK16, LCT, CES2, KMO, KLRC1, BRSK2, KCNQ1, KIR2DL2, NOL3, ATP6V0E1, SELENBP1, USP13, KLKB1, CDK5R2, RAB29, MBD2, KIF11, TMEM11, ENDOU, EIF2S2, TAX1BP1, NAE1, KRT14, KRT18, LAMC1, LBR, PROM1, LCAT, SOCS2, PRKRA, DEGS1, DDX39B, PABPN1, EOMES, BAP1, LTF, H4C9, COLQ, DYSF, CHAF1B, LYN, NRIP1, COIL, SLC7A5, AD5, H4C1, FGF23, ADAM12, BRD3, PSCA, ARHGEF5, TFPI2, FZD3, GHS, M6PR, MARCKS, SMAD1, LTC4S, H4C4, BHLHE40, IRS4, MAPKAPK5, LMO4, LOXL1, CST7, DDO, DGKZ, GAS7, PIK3R3, PKP4, PPFIA1, LRPAP1, SORBS2, H4C6, CUL4A, GNPAT, LTB, H4C14, H4C13, H4C5, H4C2, H4C8, H4C3, H4C11, H4C12, TERF1, MUC4, KCNMA1, TDO2, PCP4, CDK18, RBM3, RBL2, RBBP6, RB1, RASGRF1, RASA1, RARRES2, RAN, RAF1, RAD52, PCYT1A, RAD23B, RAC2, PDB1, RAB27B, RAB27A, PDC, PDE2A, PURA, PDGFB, ENPP2, PDYN, PTPN13, PC, PAX6, PARN, RPL15, P2RX1, S100A8, P2RX3, P2RX5, P2RY4, RREB1, RPS23, RPS21, RPS6KB2, P2RY6, RPS3A, RPL13, RET, RPA1, PAFAH1B2, RORA, ROM1, SNORD15A, BRD2, RNASE1, RHO, RHD, PAK3, TRIM27, PTPN11, PENK, PTN, PMM2, PLAUR, PLCL1, PLEK, PRKCE, PRKCD, PLP1, PRKAR1B, PRKACB, PRKACA, PLXNB1, PML, PRG2, PLAT, PMP22, PRB1, PPT1, PPP2R5E, POLG, PPP2R1A, PPP1CB, PPL, PPIC, PPIB, POR, MAP2K3, PLAG1, PFDN5, PSMD2, PFKFB3, PTGER2, PTGER1, PGD, PTGDR, PTCH1, PGR, PHB, PSMD9, PSMD7, PSMD3, PSMB2, PITX2, SERPINE2, SERPINI1, KLK10, PIK3C3, PIK3R1, KLK7, PIK3R2, PRS, PROS2P, PIN4, PROC, S100A10, OXT, OXA1L, SQLE, STAR, ST14, ST2, NDUFA6, SSTR3, SSTR2, NDUFA9, NDUFB8, SRM, SRF, NEDD4, SEPTIN2, STC1, SP4, NEU1, SOX5, SOX3, SOS2, SOS1, SOD3, NFE2L1, NFKB2, SNRPG, SNRNP70, NDUFA5, STIM1, NME1, MYH9, TRBV20OR9-2, TCP1, TCN2, MMUT, MUTYH, TCF4, ELOC, TBX2, MX1, MYH6, TARBP2, MAP3K7, STK11, MYO6, TACR2, NACA, VAMP2, VAMP1, SURF1, ABCC8, SUOX, NDP, STXBP1, STX1A, NINJ2, NME2, SAA2, OMP, SFTPC, TRA2B, SRSF6, SRSF5, SRSF3, SRSF1, SFRP1, OCA2, MAP2K4, ODF1, SELE, OPA1, OAS3, CXCL11, CCL21, CCL20, CCL19, CCL8, CCL1, SCP2, SCN1A, ORM2, OSM, TSPAN31, OAT, SGSH, SUMO2, SLC8A3, SMPD2, SLN, SLIT3, SLC22A5, SLC22A2, NQO2, SLC18A1, SLC16A1, SLC12A3, SLC11A1, SLC10A2, SLC8A1, NUP98, SLC6A12, NPHP1, SLC6A1, SLC5A2, NRCAM, NRDC, SLC1A1, NRF1, PMEL, YBX1, NT5E, NAT8, SEMA5A, ESRRA, RAB31, MACF1, HEY2, BRD4, TRAM1, CBX5, ANGPTL2, OPN1MW, GRIP1, KCNH4, MSTN, GFER, GFRA1, SIRT5, COTL1, GFRA3, GHR, ZNF629, UBR4, GHSR, WASHC4, GLI1, NUP160, CLUH, GLI3, SCFD1, KCTD2, CLCF1, SEC14L2, NR5A1, SLC24A2, PRPF6, TFIP11, MAFF, EID1, RAB38, FSHR, TMEFF2, PLA2G15, SLC7A11, FTH1, SNHG1, TSPAN15, GAST, ACKR1, G6PD, GAB1, NTSR2, GABBR1, GAD2, GALNS, DDAH1, PADI4, GART, NBEAL2, GMPR, PLEKHM2, WDHD1, GPR39, CARD8, ARHGEF15, AAK1, SYNPO, GPX4, ECD, PARK7, KLF8, TREX1, WIF1, WDR45, ATF6, CORO1A, TBC1D8, SLC7A9, GRIA4, FAF1, GRIK4, RER1, GRM1, STMN2, RAPGEF4, ADRM1, MSRB2, RAB3GAP1, GNA12, ZNF423, ATG4B, UBXN4, RCOR1, SEPTIN8, STAB1, GNAI1, MAPK8IP3, GRAMD4, GNB3, NFASC, KIF1B, GOLGA2, GPER1, SETX, GOLGA4, PDZD2, SAMD4A, KDM1A, GPM6A, RAB21, GPR6, P2RX2, GPR20, SNW1, FRK, FBXO7, BRI3, SNX12, SOCS7, CD209, FABP6, TBX21, NOP53, FABP7, SLC2A8, UBQLN2, A1CF, PSAT1, FANCG, HOOK2, DELEC1, FASN, SNX8, NPC1L1, BLNK, MS4A2, FCGR1A, FCGR2A, MYLIP, SCG3, DROSHA, TMEM230, NOX4, PCA3, SPCS1, VRK3, ETFA, SLC25A37, TLR8, SLC22A17, ECSIT, CD320, EZH2, DNAJC27, CLEC1B, NT5C3A, MZB1, F2RL2, HP1BP3, F3, F9, IRAK4, SAR1B, UTP11, F13B, SH3GLB1, SIDT2, SHANK1, TRAT1, F11R, RGCC, TMEM176B, NOCT, FBXO2, NPTN, TPK1, VCX, GREM1, FKBP1AP2, FKBP1AP3, AGO1, SEZ6L2, FKBP1AP4, FGF21, CLDN17, NOC2L, SND1, FOXM1, EPC2, ATRNL1, LRP10, FLNB, FMR1, POU2F3, TXN2, FBXL2, FOLR1, FOLR2, FKBP1AP1, B3GAT1, IGHV1-68, TNFRSF21, FEB1, FES, FGF9, RABGEF1, PRPF19, FGF13, FGFR1, FGFR4, PDLIM3, RND1, KLHL20, COQ2, CACYBP, HCAR1, FHL2, VPS4A, IL37, GLS2, NAAA, CYTH4, DKK2, DKK3, BBC3, SDCBP2, GRM3, IL24, SPAG9, CXCL2, PCLAF, IGFBP1, ACAP1, IGFBP5, SART3, KDM4A, IGHG3, SDC3, SH3PXD2A, RGS6, SNCAIP, TCL1B, IL4R, IL7, BCAR1, CCL4L2, CXCR1, BAG2, IL12B, BAG5, TMEM59, TBPL1, GAL3ST1, AKAP5, STX8, PIEZO1, BMS1, PTDSS1, IDH1, SH2D3C, GNE, SH2B3, IRF8, SRA1, TANK, KCNE3, HNRNPDL, CCS, NUP153, MED12, HS3ST1, TOMM20, RBM8A, IDH2, CFI, SV2B, TECPR2, IFI27, TOMM70, KIAA0319, IFIT3, IFNAR1, IGBP1, INSRR, PCYT1B, IL27RA, CCNE2, NOLC1, TIAF1, JUND, ZMYM3, KCNC4, KCNJ13, HGS, SYNGR3, ATG12, CBFA2T2, RABEP1, P2RX6, RAB11B, SLC16A3, SLC16A4, ATP6V0D1, RGN, USP10, USP2, USP14, DNAJA3, CLDN1, CLDN8, ARTN, JUNB, GPR50, PICK1, ITPKB, IRAK1, HOMER2, IREB2, IRF3, IRF6, IRF7, ITGAV, CYP7B1, ITGB3, NRXN1, SLC22A8, ITPR2, AIMP1, JAG2, ITGBL1, LHX2, SLIT2, TAOK2, CD163, JAK2, GPR37L1, PIWIL1, MAPKAPK2, PDLIM7, IARS1, TNC, IL18BP, CCT2, HCK, NRG3, USP39, DCTN6, CD226, HDC, HDLBP, CAMKK2, HIP1, TXNRD2, NPC2, SLC35A1, HBG2, PRDX4, ZNRD2, HYOU1, HLA-DQA1, SEMA4D, HLA-DQB1, NXF1, HLA-DRA, ATP5PD, COG5, GPNMB, CHL1, HAS3, FAM3C, GYPA, GSK3A, COPS5, GSM1, GSTM2, EBNA1BP2, PRSS21, PRDX3, GSTZ1, GTS, GUSB, C1QL1, GYPB, HAS1, GYPC, CCL27, ALDH1L1, GYPE, HAGH, WASF3, HSPH1, GJB6, HARS1, ARPP19, DHS, HLA-DRB4, HLA-G, PQBP1, MPHOSPH6, STAM2, GLYAT, HOXA@, RABEPK, HPCA, CALCOCO2, HRC, OLIG2, DDX39A, TOPORS, EIF1, HSD17B1, RAMP2, WASF2, HSD17B4, HSPA2, HSP90AB1, DNAJB1, NAMPT, BCAP31, CTDSP2, TSPAN3, ACTR2, CERT1, ABCC4, HNRNPK, HMBS, NPM3, CFDP1, PRMT5, HMGB2, YAP1, IFITM3, SPAG11B, PEMT, RACK1, SYCP2, TUBB4B, SEMA3A, WARS2, HNRNPC, FOXA1, CCL26, TLR6, FOXA2, LAMC3, LANCL1, HNF4A, TCIRG1, APBB3, APC2, HNRNPA2B1, MTCH2

-
Inclusion Body Myositis
OMIM
Description Sporadic inclusion body myositis (IBM) is the most common age-related muscle disease in the elderly that results in severe disability. ... All 5 affected males had significant clinical findings with age of onset at 20 to 30 years. The only affected female was clinically asymptomatic but on muscle biopsy showed mild changes consistent with IBM. ... The authors discussed the abnormalities of APP processing, the role of abnormal intracellular protein folding, oxidative stress, and the potential role of cholesterol in the pathogenic cascade of IBM. ... To elucidate the possible role of beta-APP mismetabolism in the pathogenesis of IBM, Sugarman et al. (2002) selectively targeted beta-APP overexpression to skeletal muscle in transgenic mice, using the muscle creatine kinase promoter. They reported that older (more than 10 months) transgenic mice exhibited intracellular immunoreactivity to beta-APP and its proteolytic derivatives in skeletal muscle.GNE, NT5C1A, APP, TARDBP, HLA-DRB1, SQSTM1, APOE, KHDRBS1, NUP62, DCTN4, GTF2H1, SDC1, CDR3, GSN, MAPT, TRBV20OR9-2, HLA-C, PLAAT4, FYCO1, MSTN, TNFRSF12A, NFAT5, CCR2, UBB, MALAT1, VCP, RBM45, AOC3, DCD, UCN2, DNAJB6, OPTN, KLRG1, MAP1LC3A, LILRB1, KDELR1, ICOSLG, SYNM, ROBO3, DDX58, CHMP1B, PABPC1, TIMP1, RRM2B, TWNK, FOXP3, KRT20, TTR, ACTB, THBS1, CST3, HLA-DQA1, HK1, H1-0, NR3C1, EPHB2, EMD, DES, CD47, TGFB1, CD38, CD36, CD34, MS4A1, CAPN3, BCL2, AOC2, HLA-DRB3, HMGB1, IFN1@, IFNG, TRIM21, AGER, MOK, PTPRC, PSME1, PSMB10, MAPK1, POLG, PMP22, MMP9, MMP1, MLF1, LMNA, IL6, IL1B, LOC102723996
-
Problematic Smartphone Use
Wikipedia
The recommendations are: For children in age less than one year: 30 minute physical activity, 0 hours screen time and 14 – 17 hours of sleep time per day. For children in age 1 year: 180 minutes physical activity, 0 hours screen time, 11–14 hours of sleep time per day. For children in age 2 year: 180 minutes physical activity, 1 hour screen time, 11–14 hours of sleep time per day. ... In Android a similar feature called "digital wellbeing" has been implemented to keep track of cell phone usage. [85] These apps usually work by doing one of two things: increasing awareness by sending user usage summaries, or notifying the user when he/she has exceeded some user-defined time-limit for each app or app category. ... The researchers implement an Android app that combined these three intervention types and found that users reduced their time with the apps they feel are a poor use of time by 21% while their use of the apps they feel are a good use of time remained unchanged. [86] AppDetox allows users to define rules that limit their usage of specific apps. [87] PreventDark detects and prevents problematic usage of smartphones in the dark. [88] Using vibrations instead of notifications to limit app usage has also been found to be effective. [89] Further, researchers have found group-based interventions that rely on users sharing their limiting behaviors with others to be effective. [90] Bans on mobile phone use [ edit ] See also: Mobile phone use in schools In some places in the world the use of mobile phones was banned in classes during instructional time, for example, in France , Ontario .

-
Epileptic Encephalopathy, Early Infantile, 45
OMIM
The patient had onset of seizures at age 12 months and showed developmental regression at age 35 months. ... Lien et al. (2016) reported a 32-month-old boy with severe developmental delay and hypotonia who developed refractory epilepsy at age 3 months. Brain imaging was normal. ... In vitro functional studies in HEK293 cells showed that the mutation altered the kinetic properties of the channel, resulting in the net loss of GABAergic inhibition. In a boy with EIEE45, Lien et al. (2016) identified a de novo heterozygous missense mutation in the GABRB1 gene (T287I; 137190.0002).
-
Estrogen And Neurodegenerative Diseases
Wikipedia
Estrogen has been proposed to act as a neuroprotectant at several levels, and it is probable that deprivation of estrogen as a result of menopause exposes the aging or diseased brain to several insults. In addition, estrogen deprivation is likely to initiate or enhance degenerative changes caused by oxidative stress , and to reduce the brain's ability to maintain synaptic connectivity and cholinergic integrity leading to the cognitive decline seen in aged and disease-afflicted individuals. [5] There is sufficient evidence that estradiol is a powerful neuroprotectant which might have use against AD, stroke and Parkinson's disease both in women and men. [5] Estrogen and Alzheimer's disease [ edit ] This figure shows how APP cleavage produces toxic Abeta in Alzheimer's disease. Amyloid plaques formed by amyloid-β (Aβ) deposition and neurofibrillary tangles formed by tau protein phosphorylation are dominant physiological features of Alzheimer's disease. Amyloid precursor protein (APP) proteolysis is fundamental for production of Aβ peptides implicated in AD pathology. [6] By using a cell line that contains high levels of estrogen receptors, scientists found that treatment with physiological concentrations of 17 beta-estradiol is associated with accumulation in the conditioned medium of an amino-terminal cleavage product of APP (soluble APP or protease nexin-2), indicative of non-amyloidogenic processing. [7] Estrogen and Parkinson's disease [ edit ] Recommendations on the use of postmenopausal hormonal replacement therapy in women with Parkinson's disease or those genetically at risk. [8] But another group of scientists found a positive association between estrogen use and lower symptom severity in women with early PD not yet taking L-dopa . [9] Estrogen and Huntington's disease [ edit ] Huntington's disease (HD) is a polyglutamine disorder based on an expanded CAG triplet repeat [10] leading to cerebral and striatal neurodegeneration. [11] Potential sex differences concerning the age of onset and the course of the disease are poorly defined, as the difficulties of matching female and male HD patients regarding their CAG repeat lengths limit comparability. [12] Estrogen and Amyotrophic lateral sclerosis [ edit ] ALS occurs more commonly in men than in women, and women get the disease later in life compared to men. [13] This suggested the possible protective role of estrogen in ALS. ... The 2002 Women's Health Initiative of the National Institutes of Health found disparate results for all cause mortality with hormone replacement, finding it to be lower when HRT was begun earlier, between age 50-59, but higher when begun after age 60. ... "Octyl Gallate Markedly Promotes Anti-amyloidogenic Processing of APP through Estrogen Receptor-Mediated ADAM10 Activation" .
-
Pick Disease Of Brain
OMIM
The disorder progressed until death at age 62 years. Neuropathologic examination showed severe neuronal loss and ovoid tau-positive argyrophilic intraneuronal inclusions consistent with Pick bodies. Beta-amyloid (see APP; 104760) plaques were not detected. ... None had a family history of the disorder. The age at onset ranged from 42 to 70 years, with a mean of 60. ... Van Leeuwen et al. (2006) postulated that accumulation of APP+1 and UBB+1, which represents defective proteasome function, contributes to various forms of dementia. ... The patient with the G389R mutation showed a decline in intellectual ability with forgetfulness, aggression, and a decline in personal hygiene at age 32, which progressed to death by age 37.
-
Malaria
Mayo Clinic
To reduce malaria infections, world health programs distribute preventive drugs and insecticide-treated bed nets to protect people from mosquito bites. The World Health Organization has recommended a malaria vaccine for use in children who live in countries with high numbers of malaria cases. Protective clothing, bed nets and insecticides can protect you while traveling. ... The World Health Organization estimates that about 94% of all malaria deaths occur in Africa — most commonly in children under the age of 5. Malaria deaths are usually related to one or more serious complications, including: Cerebral malaria. ... Do not use products with oil of lemon eucalyptus (OLE) or p-Menthane-3,8-diol (PMD) on children under age 3. Apply repellent to clothing. Sprays containing permethrin are safe to apply to clothing. Sleep under a net. Bed nets, particularly those treated with insecticides, such as permethrin, help prevent mosquito bites while you are sleeping.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
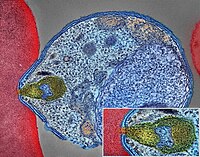
-
Gerontophobia
Wikipedia
Part of a series on Discrimination General forms Age Class ( Caste ) Physical Disability Education Economic Employment Genetics Hair texture Height Housing Language Looks Race / Ethnicity / Nationality Rank Religion Sanity Sex Sexual orientation Size Skin color Specific forms Social Acephobia Adultism Amatonormativity Anti-albinism Anti-autism Anti-homelessness Anti-intellectualism Anti-intersex Anti-left handedness Anti-Masonry Antisemitism (Judeophobia) Aporophobia Audism Biphobia Clannism Cronyism Drug use Elitism Ephebiphobia Fatism Gerontophobia Heteronormativity Heterosexism HIV/AIDS stigma Homophobia Leprosy stigma Lesbophobia Misandry Misogyny Nepotism Pedophobia Perpetual foreigner Pregnancy Reverse Sectarianism Supremacism Black White Transphobia Non-binary Transmisogyny Vegaphobia Xenophobia Religious Ahmadiyya Atheism Baháʼí Faith Buddhism Catholicism Christianity post–Cold War era Druze Falun Gong Hinduism Persecution Islam Persecution Jehovah's Witnesses Judaism Persecution LDS or Mormon Neopaganism Eastern Orthodox Oriental Orthodox Copts Protestantism Rastafarianism Shi'ism Sufism Sunnism Zoroastrianism Ethnic/national African Albanian American Arab Armenian Australian Austrian Azerbaijani British Canadian Catalan Chechen Chilean Chinese Croat Dutch English Estonian European Filipino Finnish French Georgian German Greek Haitian Hazara Hispanic Hungarian Igbo Indian Indonesian Iranian Irish Israeli Italian Japanese Jewish Khmer Korean Kurdish Malay Manchu Mexican Middle Eastern Mongolian Montenegrin Pakistani Pashtun Polish Portuguese Quebec Romani Romanian Russian Scottish Serb Slavic Somali Soviet Tatar Thai Tibetan Turkish Ukrainian Venezuelan Vietnamese Western Manifestations Blood libel Bullying Compulsory sterilization Counter-jihad Cultural genocide Defamation Democide Disability hate crime Dog-whistle politics Eliminationism Ethnic cleansing Ethnic conflict Ethnic hatred Ethnic joke Ethnocide Forced conversion Freak show Gay bashing Gendercide Genital modification and mutilation Genocide examples Glass ceiling Hate crime Hate group Hate speech online Homeless dumping Indian rolling Lavender scare LGBT hate crimes Lynching Mortgage Murder music Occupational segregation Persecution Pogrom Purge Red Scare Religious persecution Religious terrorism Religious violence Religious war Scapegoating Segregation academy Sex-selective abortion Slavery Slut-shaming Trans bashing Victimisation Violence against women White flight White power music Wife selling Witch-hunt Policies Age of candidacy Blood purity Blood quantum Crime of apartheid Disabilities Catholic Jewish Ethnocracy Ethnopluralism Gender pay gap Gender roles Gerontocracy Gerrymandering Ghetto benches Internment Jewish quota Jim Crow laws Law for Protection of the Nation McCarthyism MSM blood donation restrictions Nonpersons Numerus clausus (as religious or racial quota) Nuremberg Laws One-drop rule Racial quota Racial steering Redlining Same-sex marriage (laws and issues prohibiting) Segregation age racial religious sexual Sodomy law State atheism State religion Ugly law Voter suppression Countermeasures Affirmative action Anti-discrimination law Cultural assimilation Cultural pluralism Diversity training Empowerment Feminism Fighting Discrimination Hate speech laws by country Human rights Intersex rights LGBT rights Masculism Multiculturalism Nonviolence Racial integration Reappropriation Self-determination Social integration Toleration Related topics Allophilia Anti-cultural, anti-national, and anti-ethnic terms Bias Christian privilege Civil liberties Cultural assimilation Dehumanization Diversity Ethnic penalty Eugenics Internalized oppression Intersectionality Male privilege Masculism Medical model of disability autism Multiculturalism Net bias Neurodiversity Oikophobia Oppression Police brutality Political correctness Polyculturalism Power distance Prejudice Prisoner abuse Racial bias in criminal news Racism by country Religious intolerance Second-generation gender bias Snobbery Social exclusion Social model of disability Social stigma Stereotype threat The talk White privilege v t e Gerontophobia is the fear of age-related self-degeneration (similar to Gerascophobia ), or a hatred or fear of the elderly due to memento mori . The term comes from the Greek γέρων – gerōn , "old man" [1] and φόβος – phobos , "fear". [2] Contents 1 Ageism 2 See also 3 References 4 External links Ageism [ edit ] Discriminatory aspects of ageism have been strongly linked to gerontophobia . [3] This irrational fear or hatred of the elderly is associated with the fact that someday all young people including oneself will be old inevitably and suffer from the irreversible health decline that comes with old age , which is associated with disability , disease and death . The sight of aged people is a reminder of death ( memento mori ) and inevitable biological vulnerability. ... External links [ edit ] AGEISM AND AGING UP: A Q and A with Mariah MedFriendly Age Wave v t e Discrimination General forms Age Caste Class Disability Education Economic Employment Genetic Hair texture Height Housing Language Looks Race / Ethnicity / Nationality Rank Sanity Sex Sexual orientation Size Skin color Social Acephobia Adultism Amatonormativity Anti-albinism Anti-autism Anti-homelessness Anti-intellectualism Anti-intersex Anti-left handedness Anti-Masonry Antisemitism (Judeophobia) Aporophobia Audism Biphobia Clannism Cronyism Drug use Elitism Ephebiphobia Fatism Gerontophobia Heteronormativity Heterosexism HIV/AIDS stigma Homophobia Leprosy stigma Lesbophobia Misandry Misogyny Nepotism Pedophobia Perpetual foreigner Pregnancy Reverse Sectarianism Supremacism Black White Transphobia Non-binary Transmisogyny Vegaphobia Xenophobia Religious Ahmadiyya Atheism Baháʼí Faith Buddhism Catholicism Christianity post–Cold War era Falun Gong Hinduism Persecution Islam Persecution Jehovah's Witnesses Judaism Persecution LDS or Mormon Neopaganism Eastern Orthodox Oriental Orthodox Protestantism Rastafarianism Shi'ism Sufism Zoroastrianism Ethnic/National African Albanian American Arab Armenian Australian Austrian British Canadian Catalan Chilean Chinese Croat Dutch English Estonian European Filipino Finnish French Georgian German Greek Haitian Hazara Hindu Hispanic Hungarian Igbo Indian Indonesian Iranian Irish Israeli Italian Japanese Jewish Khmer Korean Kurdish Malay Manchu Mexican Middle Eastern Mongolian Pakistani Pashtun Polish Portuguese Quebec Romani Romanian Russian Scottish Serb Slavic Somali Soviet Tatar Thai Turkish Ukrainian Venezuelan Vietnamese Western Manifestations Blood libel Bullying Compulsory sterilization Counter-jihad Cultural genocide Defamation Democide Disability hate crime Dog-whistle politics Eliminationism Enemy of the people Ethnic cleansing Ethnic conflict Ethnic hatred Ethnic joke Ethnocide Forced conversion Freak show Gay bashing Gendercide Genital modification and mutilation Genocide examples Glass ceiling Hate crime Hate group Hate speech Homeless dumping Indian rolling Lavender scare LGBT hate crimes Lynching Mortgage Murder music Native American sports mascots Occupational segregation Persecution Pogrom Purge Red Scare Religious persecution Religious terrorism Religious violence Religious war Scapegoating Segregation academy Sex-selective abortion Slavery Slut-shaming Trans bashing Victimisation Violence against women White flight White power music Wife selling Witch-hunt Discriminatory policies Age of candidacy Blood purity Blood quantum Crime of apartheid Disabilities Catholic Jewish Ethnocracy Ethnopluralism Gender pay gap Gender roles Gerontocracy Gerrymandering Ghetto benches Internment Jewish quota Jim Crow laws Law for Protection of the Nation McCarthyism MSM blood donation restrictions Nonpersons Numerus clausus (as religious or racial quota) Nuremberg Laws One-drop rule Racial quota Racial steering Redlining Same-sex marriage (laws and issues prohibiting) Segregation age racial religious sexual Sodomy law State atheism State religion Ugly law Voter suppression Countermeasures Affirmative action Anti-discrimination law Cultural assimilation Cultural pluralism Diversity training Empowerment Feminism Fighting Discrimination Hate speech laws by country Human rights Intersex rights LGBT rights Masculism Multiculturalism Nonviolence Racial integration Reappropriation Self-determination Social integration Toleration Related topics Allophilia Anti-cultural, anti-national, and anti-ethnic terms Bias Christian privilege Civil liberties Cultural assimilation Dehumanization Diversity Ethnic penalty Eugenics Internalized oppression Intersectionality Male privilege Masculism Medical model of disability autism Multiculturalism Net bias Neurodiversity Oikophobia Oppression Police brutality Political correctness Polyculturalism Power distance Prejudice Prisoner abuse Racial bias in criminal news Racism by country Religious intolerance Second-generation gender bias Snobbery Social exclusion Social model of disability Social stigma Stereotype threat The talk White privilege Category
-
Hearing Loss
Mayo Clinic
Overview Hearing loss that comes on little by little as you age, also known as presbycusis, is common. More than half the people in the United States older than age 75 have some age-related hearing loss. ... Mixed, which is a mix of the two. Aging and being around loud noises both can cause hearing loss. ... Talk to your health care provider if loss of hearing is causing you trouble. Age-related hearing loss happens little by little. ... A whisper test, which involves covering one ear at a time while listening to words spoken at many volumes, can show how you react to other sounds. App-based hearing tests. You can use a mobile app on your tablet to screen yourself for hearing loss.SLC26A4, PCDH15, USH1G, MSRB3, GSDME, DNMT1, TJP2, TECTA, WFS1, GJB6, GJB2, MYO7A, GJB3, OTOF, USH2A, CDH23, EYA4, TMPRSS3, TMC1, PRPS1, ACTG1, WHRN, TRIOBP, USH1C, LRTOMT, MYO15A, MARVELD2, ILDR1, SIX1, POU3F4, PJVK, COL4A5, FGFR3, ADGRV1, CHD7, MYO3A, SALL4, TMIE, FOXC1, GRXCR1, DSPP, FANCC, FGF3, PITX2, COCH, GIPC3, TPRN, EYA1, SLC33A1, CLDN14, FANCG, FANCA, BTD, ARSB, PNPT1, SLC29A3, APOE, CLRN1, GRHL2, SMPX, ESRRB, CIB2, SLC26A5, COL11A2, LOXHD1, CRYM, DIAPH1, SH3PXD2B, TIMM8A, OTOA, RDX, LMX1A, STAG2, CACNA2D2, TLR4, ERCC6, MN1, NTF3, RPS6KA3, TRAPPC4, ABHD5, RPGR, HSD17B4, DIABLO, FOXI1, CCDC50, SOD1, ATP6V1B1, MIR96, ABHD12, BCL2L1, CISD2, BSND, MYO1A, SLC17A8, STAT1, CDK8, SERPINB6, UCP2, BDNF, PDE5A, UCP3, IL10, ARC, KCNQ4, STRC, POU4F3, ELMOD3, OTOGL, SLC26A2, LHFPL5, ESPN, CEACAM16, TBC1D24, SOX10, PAX3, GUSB, TFAP2A, IQGAP2, LMNA, GJB1, GJB4, MANBA, ATP6, KCNE1, MPZ, COL2A1, CDC14A, COL4A4, PMP22, CABP2, RMND1, OTOG, EDN1, TFAP2B, TCF12, HNF1B, UBE2A, FIG4, TWIST1, ALMS1, TELO2, IFT140, PIEZO1, MED12, SEMA3E, SHOC2, CEP57, IQCB1, GDF3, GABBR2, AMMECR1, CREB3L1, SCO2, CERT1, ZMPSTE24, SF3B4, COQ7, KDM6A, BCAP31, SLX4, PIGO, TBX1, NSD2, ZIC1, PQBP1, TBX15, NELFA, LMNB2, WNT5A, ABCB6, XRCC2, PPP1R15B, TULP1, KMT2D, ARL6, TBX4, BMP15, TRRAP, SHANK3, CDC45, BUB3, PLA2G6, AIFM1, SMC3, PLOD3, KYNU, TGFB1, ZNF469, PEX11B, TNFRSF11A, PHF6, OFD1, TP63, TNFSF11, CNTNAP1, TGM1, STUB1, LRAT, TERT, MKKS, PIGL, CHST3, ANTXR1, ITM2B, TERC, RECQL4, COG1, USP9X, ARID1A, RBM10, USP45, TK2, NRXN1, THRB, TRIP13, NAA10, RNF135, AP1S2, CEP290, TCIRG1, TBL1XR1, DARS2, MSTO1, WRAP53, FANCL, CARS2, PHIP, SH3TC2, IMPAD1, MKS1, NSUN2, BCOR, UGT1A1, LZTFL1, NDUFB11, USB1, RTEL1, OTUD6B, LIPT1, MBTPS2, DACT1, WAC, TACO1, TRAPPC12, NDUFA13, WDPCP, GMNN, TPRKB, SOST, TBX22, RFWD3, BBS7, FANCI, HYMAI, PORCN, NMNAT1, EPS8L2, NXN, SLC39A8, PIEZO2, ALOXE3, NDUFAF5, GBA2, FANCM, ARID1B, NUP107, RPGRIP1, COQ8A, NOP10, MRPS22, TWNK, CCDC28B, KLHL7, NDUFA12, VPS11, SPATA7, MCTP2, VAC14, NHP2, PIGV, SLC52A2, FOXRED1, RRM2B, PALB2, NDRG1, NARS2, CTC1, CRB1, SURF1, PLXND1, POGZ, TRIM32, P2RX2, DHX30, MORC2, NTNG1, PUF60, EXOSC8, POLG2, POLR3A, PRDM5, IFT27, SLC19A3, SDCCAG8, CDT1, RAI1, YME1L1, EBP, STAMBP, BRIP1, POMT1, COLEC10, NOP56, SEC23B, MAD2L2, SPIDR, KAT6B, CDK20, IFT172, PYCR2, PSMC3IP, SNX10, ANKRD11, UBE2T, NDUFAF4, BBS10, PCLO, PGAP2, BBS9, INTU, EHMT1, TINF2, ABCA12, WBP2, ZBTB20, CNTNAP2, DNAH1, SIN3A, NDUFAF3, ANAPC15, L2HGDH, RAB3GAP2, AIPL1, KCNE5, DCAF17, ORC6, LEMD3, TAF1, TTR, ABCC8, GCK, GBA, ARID2, FLCN, GALNS, GALC, GAA, FZD2, KDSR, FUCA1, NR5A1, FSHR, FXN, FMR1, CERS3, FLNA, FOXG1, FGFR2, FGFR1, GPC4, FDXR, FANCF, FANCB, ACSL4, FANCE, FANCD2, RNASEH1, NALCN, ERCC5, ERCC4, GCH1, GJA1, STXBP1, GPC3, ITGB6, PDX1, INS, IMPDH1, BEAN1, SIX5, IFRD1, IDUA, IDS, TNC, HSPD1, HNRNPK, HNF4A, HIVEP1, HEXA, HCCS, HBB, HARS1, GUCY2D, C8orf37, BBS12, LCA5, GSN, ASXL1, GNS, GNAS, GNAI3, GLI3, GLB1, ERCC3, ERCC2, EP300, EDNRB, PTPRQ, RUNX2, PCARE, CACNA1D, BUB1B, BUB1, BTK, BRCA2, BRAF, BRCA1, BCS1L, BBS4, BBS2, BBS1, GDF6, ASPA, ARSL, C10orf105, ALG11, APC, SLC25A4, ALOX12B, ABCD1, AKT1, AK2, AHSG, PET100, KLLN, ACVR1, NDUFS7, CDC6, CHD4, CRX, EDN3, ECHS1, DVL3, DVL1, ATN1, SUMF1, DKC1, DHCR7, PNPLA1, SLC26A4-AS1, DDX11, DDX3X, CTNNB1, CREBBP, NIPAL4, COX15, COX7B, RD3, COL10A1, ZFP57, COL7A1, COL4A6, COL4A3, COL1A2, COL1A1, CLCN7, AP1S1, CKMT1B, KARS1, KCNC3, TTC8, RET, SMIM12, COLEC11, RAD51, PTH1R, NLRP3, PTEN, MASP1, GPRASP2, PRKAR1A, PPP1CB, CTSA, POLR2F, POLG, NUS1, PLCB4, PLAGL1, PIK3CA, PIK3C2A, PIGA, TWIST2, PHEX, ATP8B1, PEX13, PEX6, PEX1, PEPD, PDHA1, PDE9A, PDE4D, DPF2, REV3L, PCYT1A, CHST14, CDKL5, STAT3, SRY, SRP72, SOX11, SOX9, SOX4, SOX2, SON, SMARCE1, SMARCC2, SMARCB1, SMARCA4, SNAI2, RFT1, NDUFAF2, COG7, SGSH, SDHD, SDHC, SDHB, SDHA, BBIP1, G6PC3, SC5D, SALL1, DTD1, RPL11, RPE65, PDE1C, RAD51C, KCNJ11, SMAD4, MYCN, TRNL1, MTHFD1, ATP8, BBS5, VPS37A, NDUFAF6, MITF, MGP, HGSNAT, MEOX1, MECP2, MAF, B3GLCT, PAX2, LRP5, LRP4, RDH12, LMX1B, LMNB1, LIG4, LHX1, LETM1, KRAS, KIT, KIF5A, KCNQ1, KCNJ13, MYD88, MYH3, NDUFV1, TNFRSF11B, OAT, ROR2, ORC1, NPHP1, NOTCH2, NFIX, MTFMT, NDUFV2, NDUFS8, NDUFS4, NDUFS3, NDUFS2, NDUFS1, NDUFB8, NDUFA10, ORC4, NDUFA9, NDUFA4, NDUFA2, PARN, NAGLU, NAGA, RNR1, MYO6, MYH9, OPA1, MYH6, NT5E, MYH14, COX1, KCNJ10, NAT2, GSTM1, GATA3, IGF1, ADA, ADCY1, CD226, ACE, UCN, MTHFR, S1PR2, ATP2B2, TRS-AGA2-3, GRM7, CRP, F11, ETS1, TRPV4, EPO, FABP2, SLC26A3, SLC26A11, RSPO1, DFNA47, GATA2, FCGR1A, FOXO3, FLI1, FLNC, DMD, PTCRA, DFNB33, GSTM3, GSTP1, GSTT1, HOXA1, IDH2, SH2D6, CYP2A6, DFNA16, DFNA7, DFNB71, ADCYAP1, DFNB55, ALB, DFNB47, DFNB45, BIRC5, ARL2, MIR34A, CHCHD10, BAK1, BCL2, CALCA, CAPG, CD5L, CD40LG, CD69, GJC3, CKB, COL9A3, COL11A1, DFNB38, DFNA49, KLKB1, DFNX3, IL1B, MET, LRP2, LY6E, UCP1, UROD, BEST1, CD164, ADIPOQ, FHL5, BAG3, SNAP91, PDLIM5, GIPC1, EHD1, SIRT1, BACE1, DFNA24, PDZD7, MTO1, EHF, HPGDS, REM1, EHD4, EHD3, EHD2, TSPEAR, MCPH1, THG1L, FKBP14, POMGNT1, ACTB, TPO, TGFB3, PRKCG, ANKH, ATXN3, MSX1, MTAP, ND4, MTR, TRNS1, NEFL, PAX9, SERPINA1, PLOD1, PLS1, PTGDS, DFNA18, RASA1, ROM1, SERHL, ACSM3, ATXN2, SCD, SKP1, SLC6A11, SLC6A13, SKP1P1, SLC25A21, TGFA, ERICD

-
Maple Syrup Urine Disease
GeneReviews
Acute metabolic decompensation is corrected by treating the precipitating stress while delivering sufficient calories, insulin, free amino acids, isoleucine, and valine to achieve sustained net protein synthesis in tissues. Some centers use hemodialysis/hemofiltration to remove BCAAs from the extracellular compartment, but this intervention does not alone establish net protein accretion. ... Signs of deepening encephalopathy including lethargy, intermittent apnea, opisthotonus, and stereotyped movements such as "fencing" and "bicycling" are evident by age four to five days. Coma and central respiratory failure may occur by age seven to ten days, sometimes before newborn screening results are available. ... Following the neonatal period, acute metabolic intoxication (leucinosis) and neurologic deterioration can develop rapidly at any age as a result of net protein degradation precipitated by infection, surgery, injury, or psychological stress (see Figure 1). ... Plasma leucine levels rise predictably as a result of net protein catabolism provoked by a variety of physiologic stresses, including (more...) ... Coronal T 2 -weighted MRI from a Mennonite boy age five years during an acute metabolic crisis.DBT, BCKDHB, BCKDHA, BCAT2, PPM1K, DLD, ARID4B, BDNF, CTSD, SERPINE1, TNS3, CACNA2D2, MKRN3, UMOD, SPN, NME1, PAH, NBN, MEA1, IL1B, GPR4, GLI2, F2, MECP2

-
Mutyh Polyposis
GeneReviews
Surveillance: Colonoscopy with polypectomy every one to two years beginning at age 25-30 years; upper endoscopy and side viewing duodenoscopy every three months to four years beginning at age 30-35 years with subsequent follow up based on initial findings. ... The risk was found to be higher in one study, with median age at diagnosis 53 years (range 45-76) [Vogt et al 2009]. ... Two of 15 probands with familial NET of the small intestine and four of 215 individuals with nonfamilial NET of the small intestine were heterozygous for MUTYH pathogenic variant p.Gly396Asp [Dumanski et al 2017]. It is unclear if a heterozygous MUTYH pathogenic variant is a risk factor for NET or ACC, as the risk of NET or ACC in individuals with biallelic MUTYH pathogenic variants appears to be quite low. ... NCCN [2019] guidelines propose that MUTYH heterozygotes with a first-degree relative with CRC (who does not have MAP) undergo colonoscopy every five years beginning at age 40 years or ten years prior to the age of the first-degree relative's age at CRC diagnosis.
-
Rahman Syndrome
OMIM
Clinical Features Tatton-Brown et al. (2017) reported 5 unrelated patients, ranging in age from 1.9 to 16 years, with mild to severe intellectual disability associated with variable somatic overgrowth, including height, weight, and/or head circumference. ... Additional features, each found only in 1 or 2 patients, included kyphoscoliosis, camptodactyly, talipes equinovarus, advanced bone age, dental anomalies, skin nevi, strabismus, astigmatism, and amblyopia. ... The truncated proteins were predicted to have a reduced net charge compared to the wildtype protein, rendering them likely to be less effective in neutralizing negatively charged linker DNA. ... INHERITANCE - Autosomal dominant GROWTH Height - Increased birth length - Increased height Weight - Increased birth weight - Increased weight HEAD & NECK Head - Large head circumference Face - Full cheeks Eyes - Telecanthus - Strabismus - Amblyopia - Astigmatism ABDOMEN Gastrointestinal - Poor feeding in the neonatal period SKELETAL - Advanced bone age Spine - Kyphoscoliosis Hands - Camptodactyly Feet - Talipes equinovarus SKIN, NAILS, & HAIR Skin - Nevi MUSCLE, SOFT TISSUES - Hypotonia, neonatal - Hypertonia, neonatal NEUROLOGIC Central Nervous System - Delayed development - Intellectual disability, mild to severe MISCELLANEOUS - Highly variable features - De novo mutation MOLECULAR BASIS - Caused by mutation in the histone gene cluster 1, H1 histone family, member E gene (HIST1H1E, 142220.0001 ) ▲ Close
-
Epilepsy, Progressive Myoclonic, 10
OMIM
The proband, who was the most severely affected, developed school difficulties, dysarthria, and myoclonus at age 5 years. She then developed seizures, ataxia, bladder incontinence; she became mute by age 12 and was wheelchair-bound by age 14. At age 34, she was bedridden and unresponsive, with spastic tetraplegia. ... Studies in patient tissues showed no net change in glycogen synthase (see, e.g., GYS1, 138570) activity, indicating that polyglucosan formation was not related to glycogen synthase.
-
Supranuclear Palsy, Progressive, 1
OMIM
Three family members were affected. At 37 years of age, the proband developed an akinetic-rigid syndrome, gait disturbance, frequent falls, micrographia, dysarthria, eyelid apraxia, abolition of upgaze, and hyperreflexia with unilateral extensor plantar response. ... Other variable features included tremor, retrocollis, limb dystonia, and favorable response to levodopa. Average age at onset was 66 years, and the mean survival was 5 to 6 years. ... Pathogenesis Van Leeuwen et al. (2006) detected aberrant frameshifted proteins, APP+1 (APP; 104760) and UBB+1 (UBB; 191339), within the neuropathologic hallmarks of Alzheimer disease (AD; 104300) and other MAPT-related dementias, including Pick disease, progressive supranuclear palsy, and less commonly frontotemporal dementia. Van Leeuwen et al. (2006) postulated that accumulation of APP+1 and UBB+1, which represents defective proteasome function, contributes to various forms of dementia. ... There was no difference between PSP cases with one H1 or two H1 alleles in the age of onset, severity, or survival of patients, thus showing that tau genotyping does not predict the prognosis of PSP.MAPT, STX6, MOBP, EIF2AK3, SRSF2, TRA2B, SLCO1A2, TRIM11, SP1, PSPH, REG1A, SNCA, RIDA, STXBP3, MSMB, PSPN, TPO, CD8B, ASAP1, RUNX2, BPIFA2, PIK3C2G, IRF4, APOE, SLC6A3, TARDBP, LRRK2, NEFL, SOD1, CIT, C9orf72, CSF2, MAOB, GRN, LAMC2, DCTN1, STH, SMUG1, PRKN, UBB, PYCARD, NPC1, APP, TYMS, CRHR1, TH, TGM2, SLC25A38, ATXN2, GFAP, IGLON5, CST3, NPEPPS, VEGFA, RAB35, YWHAE, OGA, CXCR4, PICALM, NPC2, SNCAIP, BSN, MAP3K14, OPN1MW3, DUSP10, ARL17B, ROCK2, SCRN1, MAP4K4, NF1P1, UNC13A, DNAJB1P1, FLAD1, UBASH3B, SPECC1, FOXP2, RMDN2, ASXL1, MCIDAS, SETX, MIR132, MIR518E, GGTLC5P, GGTLC3, GGT2, OPN1MW2, CTNNBL1, SYBU, PSPC1, RMDN3, LRRC37A4P, TET2, TMEM106B, TREM2, LCMT1, PPME1, RMDN1, GGTLC4P, PSAT1, TBK1, CSDC2, LMOD1, SF3B1, MINK1, NAT1, TPI1, OPN1MW, FMR1, MTOR, FUS, GABPA, GABRG2, GBA, GGT1, EGFR, GLDC, GSTM1, NRG1, HSPA4, DNAJB1, IFNG, ERBB4, DLX1, IGFALS, CASP3, AP2A2, ANXA6, KLK3, BDNF, BNIP1, BRCA1, CBS, ACE, CDK5, CHI3L1, CLU, CRP, CTSS, CYP2D6, IGF1, IL2, TP53BP1, MAP2K4, PSEN2, PTEN, PTPRC, RAPSN, ROCK1, ATXN8OS, NAT2, PROS1, SPOCK1, SPP1, TCOF1, TGFB1, TGM1, TNF, PSEN1, PRNP, IL6, NR4A2, IRS1, MUSK, NFE2L2, NGF, NOS1, NSF, PAEP, PTPA, PAFAH1B1, PDK1, PIN1, PLAG1, PLCG2, PLXNA2, ATXN2-AS
-
Neurodegeneration
Wikipedia
Beta-amyloid is a fragment from a larger protein called amyloid precursor protein (APP), a transmembrane protein that penetrates through the neuron's membrane. ... Mitochondrial DNA mutations as well as oxidative stress both contribute to aging. [41] Many of these diseases are late-onset, meaning there is some factor that changes as a person ages for each disease. [2] One constant factor is that in each disease, neurons gradually lose function as the disease progresses with age. ... The Alzheimer conundrum : entanglements of dementia and aging . Princeton. ISBN 978-1-4008-4846-1 . ... "Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression" . Neurobiology of Aging . 37 : 209.e1-209.e7. doi : 10.1016/j.neurobiolaging.2015.09.014 . ... S2CID 4421515 . ^ Bernstein C, Bernstein H (1991). Aging, Sex, and DNA Repair . San Diego: Academic Press. pp. 121–139.NGF, SNCA, APP, EPO, SIRT1, ATXN1, MAPT, HMOX1, PSEN1, PANK2, GDNF, HCRT, IL6, CRYAB, GSTO1, IDO1, TBCD, TTC19, SERPINA1, SOD2, GSTM1, GSTM2, SELENOP, APOD, VIM, KYNU, FTH1, PDE8B, AGPAT3, GSR, NGFR, GPX3, SPTAN1, PKD2, MGST1, GSTM4, GSTM5, SEPTIN5, LRRK2, INA, CAT, FTL, AIMP1, CLEC16A, APLP2, GOT2, ATG7, NQO1, TNF, PRNP, OPA1, RMDN2, CLN3, SACS, MTOR, NPC1, FXN, ATXN7, TREM2, NFE2L2, NEFL, P2RX7, TGM2, ATXN2, FMR1, PPARGC1A, RMDN1, SNCG, TPP1, SETX, TSPO, SNCB, CDK5, PIK3CG, PIK3CD, SMN2, SMN1, SIRT2, PPARG, SNRPN, PIK3CB, C9orf72, PIK3CA, NLRP3, PTPA, PPT1, PRKN, CLU, SOD1, FUS, CSF2, AR, STMN1, APOE, LAMC2, SIGMAR1, GRN, PLA2G6, TTR, DNM1L, IL1B, ABCD1, CDK5R1, HDAC6, SQSTM1, IGFALS, SNURF, MFN2, PARP1, PINK1, IGF1, HTT, HFE, ACTB, HSP90AA1, ACHE, HSPA4, LCN2, OPTN, TARDBP, SYBU, GABPA, BDNF, MAOB, GCG, UCHL1, BCHE, RMDN3, ATXN3, GFAP, VEGFA, VCP, ATM, GRIN2B, EIF2B2, TLR4, GAPDH, GBA, KHDRBS1, GTF2H1, EIF2S2, ANG, CP, EIF2B4, REN, DCTN4, NUP62, EIF2B1, PIN1, TPPP, GSK3B, HSF1, ITM2B, HRES1, S100B, TP53, BACE1, CTSD, MAOA, TMED9, TMEM106B, CASP6, TFEB, CASP3, PTBP1, NGB, MAPK8, MAPK1, DAPK2, MAK16, RBMS3, PNO1, SRRM2, GLP1R, CTNNB1, MGLL, SIRT3, DENR, CNR2, KEAP1, SLC6A4, UTRN, CNTF, CRMP1, OGA, PNPLA6, EPM2A, SLC1A2, LEP, SLC6A3, PREP, AKT1, CLN5, DYRK1A, ITPR1, STH, LY6E, ARSA, HTRA2, DNAJC5, MPO, P4HB, GALC, TRPM2, CHCHD10, AGER, NCL, PTGS2, CHMP2B, ADIPOQ, HDAC9, SGSH, ABHD12, CNR1, TXN, SPP1, WFS1, NRGN, STAT3, TRPV1, EIF2AK3, CX3CL1, TDP1, ATP13A2, VDR, EPHB2, UBB, KL, MS, TAC1, APTX, TTPA, HSP90B2P, DNMT1, PRKCG, DCTN1, SUCLA2, PQBP1, SORT1, MAPK10, VPS13A, CHI3L1, SMUG1, HSPB8, CASP8, ADAM10, KLK6, TRPM7, HMGB1, CST3, REST, STUB1, MIR29A, MIR132, GCHFR, CLN6, HSPD1, TXNIP, ANXA1, SV2A, IL10, UPK3B, IL12A, IREB2, CAMK4, ELP1, SUGP1, BCL2, HAMP, YWHAZ, DNAJB1, MBP, NR1H2, MNAT1, UCP2, CSF1R, AFG3L2, PARK7, MCIDAS, SURF1, UBQLN2, ABCB1, CDCA5, ACE, SST, DKK1, KIF1B, PLA2G1B, TLR2, PLP1, DISC1, SARM1, ATN1, PPARA, EWSR1, DPP4, ATL1, CSTB, CREBBP, FCN2, NPY, HNRNPA1, PTEN, HOXD13, PRKAB1, RHO, REG1A, RIPK1, PRKAA2, IL1A, HDAC3, SI, HSPA1A, HNRNPA2B1, APLN, HSPB1, SYNJ1, TECPR2, IAPP, PSMD2, PTN, SOCS3, MARK4, LGMN, KMO, MAPK3, EPG5, IFNG, PTPN1, EIF2AK2, CCL2, MGAM, NAGLU, TAF15, PRKAA1, HSPA14, CHCHD2, WDR45, COX2, TAT, TYROBP, MTHFR, TYR, GEMIN4, TBP, NOS3, TDO2, TGFB1, S100A1, PDE10A, MSI1, TBK1, POLG, SPTBN2, MCOLN1, KCNC3, CFDP1, MANF, MAP3K12, SPAST, TMEM97, PDYN, VIP, SGSM3, MEFV, MFGE8, PFN1, AHSA1, CD200, PICK1, ALDH2, KIF1A, GLB1, CBLL2, CRH, TAAR1, MUL1, CRP, ATP7A, GPER1, CDNF, ACO2, SESN2, ATF4, PDIA3, GRM5, GRIA2, MIR34A, CBS, HNRNPA1P10, EIF4G2, CX3CR1, CYBB, NR3C1, FOLR2, ALOX5, ADCYAP1, DECR1, ABCA1, AQP4, HDAC2, DYNC1H1, LYST, APEX1, CACNA1A, CASP1, NIPA1, TTBK1, HIF1A, NRG1, DPYSL2, ASPA, DNAJB1P1, FOLR1, HEXA, CYP46A1, MRGPRX3, PLCG2, PLG, PLA2G4A, MRGPRX4, GSTO2, YME1L1, CALB1, TMSB4X, C5, C5AR1, BSCL2, MIR137, DRD2, PON1, TNFRSF1B, TPO, A2M, MIR183, EGFR, CXCR4, GPNMB, SLC11A2, PTK2B, NRTN, NTRK2, WNT1, SLC25A46, SEPSECS, ASAH1, F2R, MTDH, NPC2, GPR166P, VN1R17P, VDAC1, UBQLN1, ERBB2, PDC, PYCARD, PDE4A, DNAH8, PDE7A, SUMO1, CLN8, TYMS, BRAF, PHPT1, POLDIP2, CANT1, RRAS, OXER1, ATXN8OS, EXOSC8, CRK, STIM1, MAPK14, PNKP, CCL11, ZFYVE26, ROS1, PLB1, LPAR3, TACR1, RGS10, HEXD, SGCG, MIR107, SOX3, PCSK9, KDM1A, SNAP25, CETP, GPRC6A, BRAT1, SLC18A2, SGK1, SLC6A2, ACSBG1, SLC2A1, MLKL, CDC42, SPTBN1, PDIK1L, PADI4, RBM3, DBN1, RELN, CASP2, DBH, CD200R1, MAP2K7, LINGO1, TIMM8A, CUX1, CAST, FAM168B, PRKCD, DHCR24, DIO2, DLG4, DAPK1, STIP1, DAG1, CASP9, CYP27A1, PARS2, TMED10, CYP19A1, CLIC4, GPR151, RNF19A, PTPN11, PRDX5, TERT, RIPK3, RAC1, MOK, BDNF-AS, P2RY2, MRGPRX1, MSC, IL4, VPS35, SPG11, IL3, GLE1, MID1, GJA1, MBTPS1, PEA15, BECN1, IL2, ABCD2, MMP9, MMP14, GIP, MOG, SNCAIP, MAD2L1BP, LRPPRC, MTPAP, GH1, IGF2, DNAJB6, MDK, ALS2, NDRG2, HAP1, ADM, ACP3, INSR, RARS2, MTCO2P12, GRIN1, AIFM1, RPSA, SLC52A2, LDLR, GLUL, AGTR1, TUBB4A, CISD1, LPL, LRP1, SLC33A1, VAPB, NARS2, ALB, MAS1, HSD17B10, CCR2, NDUFA1, ABCG2, RNR2, GRAP2, CFH, HGF, SPOAN, PRUNE1, APC, NEDD4, NEFH, HSPA9, HSPA5, ANPEP, ANP32A, HK1, HSPA1B, DNAJB2, ABCA7, AAVS1, SLC25A27, LGR6, AIMP2, HMBS, FZD4, JPH3, BAG3, SNX27, ABL1, BLZF1, ATP6, SLC25A38, OXR1, POLR3A, TMEM189, CIT, LZTS3, RAB21, NDUFS7, CLOCK, UNC13A, PTGES, KIF20B, HES3, TMEM189-UBE2V1, CDC37, SCRN1, TMED10P1, RUFY3, NLRP1, ADAP1, H3P13, DNAJC6, KIF21B, SIRT1-AS, P2RX2, TMEM59, GEN1, DDX19B, NANOS3, PGP, PADI2, GOSR1, RGS6, SLC2A6, C14orf177, NEAT1, NPAS4, MMRN1, STXBP5L, ZNF763, TMEM119, PWAR4, ISG15, IMMT, PDAP1, H3P14, TUSC2, HDAC4, SBNO2, COPD, PSIP1, H3P7, RACK1, SETDB1, CXCL13, RAMP2, COQ7, GDF11, SCGN, ATP6AP2, CEBPZ, NAMPT, TMEM41B, SCA26, SH2B2, SFTPA1, MIR30B, MIR25, CXCR6, MIR21, MIR200A, MIR184, LOC643387, DNAJA2, FIG4, HSPA12A, CLN9, MIR504, CISD2, TUBA1B, CACNG2, KLF2, CLEC10A, NXF1, TFG, KAT5, NOP56, SULT1A4, XRCC6P5, BATF, AKR1A1, SPTLC1, RTN3, PARK12, CTCF, MIR603, MIR155, SLC12A6, SLX1A-SULT1A3, PAPOLA, ARMCX5-GPRASP2, P2RX5-TAX1BP3, TCERG1, NR1H4, ERP29, MVP, SCGB1D4, RBM8A, GDNF-AS1, PGR-AS1, ATXN2-AS, CRYAA2, CLP1, COPS5, LINC01672, RAB10, MRPS30, CYSLTR1, FARS2, MIR146A, MIR144, PPIF, PTGES3, C20orf181, ARPP19, PTPRU, AAA1, CCL27, HPSE, PGRMC1, HBD, ABCB6, AOS, KCNE3, NUP153, NECAB1, MFSD8, SYT14, SYVN1, CCDC115, MINDY4, PPP1R1B, RNF146, ADPRS, OCIAD1, TXNDC5, TESC, GDPD5, DARS2, BHLHB9, SBNO1, ZNF436, ROCK2, NAT10, UBA6, SPTLC3, YOD1, FOXRED1, AMBRA1, PPP4R3A, OPA3, NAXD, IFT122, DNAJC10, TERF2IP, ABLIM2, ADO, NAGPA, RPPH1, CRBN, ABI3, VRK3, BFAR, DNAJC14, NRN1, DCDC2, IL23A, TDP2, ATP6V1H, GDAP1, CIAO2B, RASD1, TPPP3, ORAI1, PARP10, ATP8A2, PTPN5, PANK1, DUOX1, TLR9, TREM1, ASRGL1, ZNF415, SLC8B1, REEP1, ZNF512B, LYNX1, NLN, KIDINS220, TAOK1, SEMA6A, CFAP97, FUNDC2, WNK1, STIM2, DPP10, BCL11B, BIRC6, PORCN, SPG16, MTHFSD, SMURF2, LSM2, PDIA2, SENP2, GORASP1, INF2, SIL1, PROK2, MYORG, SCYL1, L2HGDH, SPHK2, PAG1, CCDC51, SELENOS, HDAC8, ZNF253, PRMT8, SLC2A9, TMPRSS4, TWNK, EFHD2, RETN, FA2H, COLEC11, ACKR3, PCBP4, SLC17A6, RPGRIP1, TIGAR, GGCT, THAP11, GJD2, AICDA, METRN, ABHD6, CHRFAM7A, SDF4, MCU, PLXNB2, CABIN1, SEC14L2, GAREM2, MANEAL, ZNF569, TTBK2, PWAR1, CD2AP, PIWIL4, PRND, HSPBP1, GABARAPL1, MACF1, FBXO7, QPCT, SEMA4A, POLR1A, PPARGC1B, ATL3, SH2B1, ACOT11, ATRNL1, CHD5, OCIAD2, SLC39A14, QPRT, APPL1, MGRN1, FNDC5, NMNAT2, OARD1, FOLH1B, MAST2, RTL3, UBR1, PAOX, RLS1, WWC1, AGTPBP1, ARX, SLC44A1, IDO2, SPATA5, DNAJC13, ZNF629, TOR1AIP2, KCTD7, SIRT5, SLC2A12, NCS1, KCNH4, GPBAR1, GIGYF2, KCNH8, LRSAM1, PADI1, SLC52A3, SLC46A1, PRRT2, PCLO, OPN4, REM1, OSTM1, FLVCR1, FTMT, HIPK2, TMEM230, TBCK, CYGB, PILRB, DNER, NOP53, ERVW-1, DUOX2, MTG1, ASAP1, HDGFL3, GMNN, PLXNA4, ADIPOR1, CYP2U1, GPRASP2, AHSA2P, BBC3, PTPN22, FBXL5, HIBCH, BHLHE23, FGF20, GNL3, SLC17A5, RNF11, FETUB, SIGLEC7, B3GAT1, DKK3, UHRF2, EXOSC6, AGO2, IP6K3, COQ2, TNFRSF21, PDLIM3, APEX2, HPGDS, RAB39B, NXNL1, RABGEF1, ATXN10, SORL1, SH3BP5, GRM4, GLRX, GM2A, GMFB, CXCR3, GPR17, GPR18, MCHR1, GPR26, GPR42, GRK5, GRIK3, GRIN2D, GRM2, GRM3, GSN, HSPA6, GSS, GSTP1, GUSB, HBB, HCLS1, HDAC1, HEXB, UBE2K, HK2, HLA-C, HLA-G, HMGA1, NR4A1, HPX, GLO1, GIPR, CBLIF, GHRH, EPRS1, EREG, ERN1, ESR2, EZH2, F2RL1, F3, F5, F9, FAAH, FABP3, BPTF, FDPS, FGF1, FGF9, FOXO1, FOXO3, FLNB, FOLH1, FOS, FOSB, FPR2, FRAXE, G6PD, GAD1, GAP43, GBAP1, KAT2A, GFER, AGFG1, HSPB2, ENO2, MECP2, KCNN1, KIT, LGALS1, LGALS3, LIFR, LIPC, LMNB1, LMX1B, LOX, LPA, NBR1, MARK1, MCL1, MDM2, MAP3K5, HSP90AB1, KITLG, MGST3, MIF, MLF1, MAP3K11, KMT2A, MMP1, MMP2, MPP1, MPST, MPZ, MRC1, MSH2, MSN, KCNMA1, KCND3, KCNB1, JUND, HTR2A, HTR2C, ICAM1, IRF8, IDE, IDH2, IFIT3, RBPJ, IKBKB, IL1RN, IL2RA, IL2RB, IL7R, CXCL8, IL13, IL13RA1, IL15, IL17A, IL18, INPPL1, IRF4, ISG20, ITGAM, ITGB2, ITIH4, ITPR2, ITPR3, JUN, JUNB, EPHA1, ENG, MST1, C9, ATR, AVP, BAX, BCL2L1, BCL6, BCYRN1, BGN, BLMH, BLVRA, BNIP3, BRCA2, BRS3, BTK, CAPN5, CAD, SEPTIN7, CAPN2, CASP7, CASR, CAV1, CAV2, CD14, CD33, SCARB2, CD38, CD40, CD40LG, CD63, CD68, CDK1, ATP5MC1, ATHS, ARNTL, RHOA, SERPINA3, ACADM, ACOX1, ACP1, ACYP2, ADCYAP1R1, ADORA1, ADORA2A, ADRA1A, ADRA2B, ADRB2, AHR, AHSG, AIF1, AK4, AKT2, ALOX12, ALOX15, ALPP, AMD1, AMD1P2, AMPD2, ANK1, APAF1, APBB1, APLP1, APOA1, KLK3, AQP1, CDK11B, CDC27, MARK2, DMPK, CTBP2, CCN2, CTSB, CTSK, CTSZ, CYP2B6, CYP2D7, CYP2D6, DARS1, DBI, DDX3X, DES, COCH, DLST, DOCK2, CDH1, SLC26A3, DRD1, DUSP2, DUSP6, E2F1, EDN1, EDNRA, EEF1A1, EGF, EIF4E, EIF4G1, ELANE, ELAVL2, ELK1, CST6, SLC25A10, VCAN, CSNK1D, CDH15, CDK2, CDK6, CDK9, CDKN2A, CDKN2D, CEBPD, CETN1, CFL2, CHAT, CHD2, CHGA, CHIT1, CHRM1, CHRNA4, CHRNA7, CISH, CLK1, CCR5, COL11A2, COL17A1, COMT, COX8A, CPN1, CPOX, ATF2, CRHR1, CRYAA, CRYZ, MSRA, MT3, AIM2, TNFRSF1A, TF, TFAM, TFCP2, TFRC, THBS1, THOP1, THY1, TIA1, TIMP1, TIMP2, TIMP3, TKT, TLR1, TLR3, TNR, VEGFB, TPP2, TPR, TPT1, TRAF6, TRPC3, TRPC5, TSC1, TSC2, TUBA4A, UBC, UBE2V1, UBE3A, UCHL3, UNG, TMBIM6, TEAD1, PRDX2, TCF3, SLC9A5, SLC18A1, SLC18A3, SLC20A2, SLPI, SON, NAT2, SOS1, SP100, SPARC, SPG7, SPR, SRF, TRIM21, SSTR4, STAT1, STC1, ELOVL4, STX5, SULT1A3, SUPT4H1, SUPT5H, SYK, SYN1, SYT1, TAF1, TAF2, TAP1, CNTN2, VARS1, VGF, SKIL, USP14, RAB11A, ASAP2, NR1I2, SPHK1, SGPL1, MTMR2, ENDOU, CACNA1G, BSN, MBD2, HSPB3, KALRN, F2RL3, SPAG9, P2RX6, VRK2, SYNGR3, LPAR2, XPR1, NOG, TSPOAP1, PIWIL1, GPR55, KLF4, SLIT2, PPIG, PPT2, COX5A, SELENOF, CYP7B1, PABPC4, RNMT, EIF3A, USO1, VSNL1, WNT2, XBP1, XK, XPNPEP1, YY1, SLC30A3, RAB7A, BAG6, SLC39A7, TFPI2, ARHGEF5, NCOA4, AAAS, CLLS2, GAN, GDF5, TAM, USP9X, EPX, TRRAP, PICALM, BAP1, NR0B2, SUPT3H, OGT, KHSRP, AKR7A2, PRKRA, SLC5A2, SHH, NUDT1, PDE2A, P2RX3, P2RX4, P2RX5, P2RY1, PAEP, PRDX1, PAK1, PAK3, REG3A, PAWR, PAX6, PCBP1, PCBP2, PCSK1, PDE4D, PML, PDE9A, PDK1, SERPINF1, PEX6, PFDN5, SLC25A3, PHEX, PHF1, SERPINI1, PITX2, PLA2G2A, PLA2G5, PLAT, PLS3, P2RX1, OXT, OPRD1, OGG1, MTM1, ND3, ND5, MUTYH, MYOC, PPP1R12A, NACA, NAP1L2, NDN, NDUFAB1, NDUFS8, NEK1, NF2, NFKB1, NNAT, NME3, NQO2, NOS1, NOTCH1, NOTCH3, NOVA1, PNP, NPPA, NPY2R, NTF3, NTRK1, NTS, NR4A2, OGDH, PLXNA2, PMP22, SH3GL3, RPE, RAN, RAP1A, RARB, RARS1, RASA1, RASGRF1, RBBP6, OPN1LW, RELA, RENBP, RNASE4, BRD2, RORC, RPGR, RPS4X, PRRX1, RPS6KB1, RPS25, RPS27A, RREB1, RRM2, RXRA, S100A6, S100A9, S100A10, SCN8A, SCP2, SCT, SRSF7, ITSN1, RAB1A, QARS1, PTPRA, PTPN13, SEPTIN4, POLD1, POU3F2, POU3F4, PPARD, PPIA, PPID, PPP1CB, PPP2CA, PPP2R2B, PPP5C, PRKAR1B, PRKCA, PRKCB, PRKD1, MAPK9, MAP2K5, PRL, PROC, HTRA1, PSD, PSEN2, PSG5, PSMC1, PSMD1, PSMD8, PSMD12, PTK2, PTPN6, H3P17

-
Yellow Fever
Orphanet
Management and treatment As there is presently no antiviral drug available for YF, treatment is supportive, following the guidelines for treatment of severe septicemia. Insecticide-treated bed nets and/or room screens should be used in open-air settings to prevent further transmission. ... YF-17D is indicated for persons over 9 months of age who are traveling to or living in YF endemic areas. Vaccination is contraindicated in children <4 months old and pregnant women and caution is advised in persons with egg allergy, the immunocompromised, children 4-9 months of age and elderly persons, especially if the risk is minimal, such as trips restricted to attending conferences in modern urban hotels with no rural exposure.ERVK-32, ERVK-6, DDOST, PARTICL, TRIM56, CYP20A1, EMC3, TRAV12-2, BACE1, AMACR, TRPA1, TFPI, RPL19, RAF1, CRP, MRC1, IL10, IL6, IL1RN, IFNG, IFNAR1, GYPC, GPT, GLS, FCGR3B, FCGR3A, F10, MAP2K1

-
Epithelial Recurrent Erosion Dystrophy
OMIM
The disorder became manifest between 4 and 6 years of age. Recurring ulcerations are also seen in macular and lattice types of classic dystrophy. ... Affected individuals presented within the first decade of life with corneal erosions that occurred approximately every 2 to 3 months. With increasing age, the erosions became much less frequent, and the episodes appeared to cease in the third decade of life. ... Most patients had onset of symptoms between 6 and 7 years of age, although 3 patients reported late onset and 2 family members were asymptomatic despite characteristic corneal changes. ... Examination revealed characteristic diffuse subepithelial opacities in the paracentral cornea, sometimes showing a net-like pattern with raised areas of Salzmann degeneration, and the opacities tended to increase with age. ... Patients presented between 5 and 7 years of age, and episodes of erosions decreased in frequency over time, subsiding in the third to fourth decade.






