Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Joubert Syndrome 32
OMIM
The families were of Italian (family COR369) and Egyptian (family MTI-2023) descent. Brain imaging showed mild cerebellar vermis hypoplasia with elongated superior cerebellar peduncles and a deepened interpeduncular fossa, indicative of the molar tooth sign.
-
Occupational Cancer
Wikipedia
"Night work and breast cancer risk: a systematic review and meta-analysis". Eur. J. Cancer . 41 (13): 2023–32. doi : 10.1016/j.ejca.2005.05.010 .
-
Dementia
Mayo Clinic
The FDA approved this medicine in 2023. A phase 3 clinical trial found that lecanemab slowed cognitive decline in people with early Alzheimer's disease by 27%. ... The FDA may begin the review process of donanemab in 2023. Therapies Several dementia symptoms and behavior problems might be treated initially with therapies other than medicine.TREM2, APP, MAPT, PRNP, NOTCH3, GRN, PSEN2, CST3, DNMT1, CP, HTRA1, FUS, TYROBP, SLC6A3, CSF1R, COL4A1, SLC9A8, C9orf72, ITM2B, APOE, LRRK2, SNCA, PSEN1, VCP, CHMP2B, ATP13A2, SNCB, FMR1, SPAST, GBA, PINK1, SERPINI1, TTR, NOS3, TBP, TMEM106B, ARSA, ATXN2, JPH3, OPA1, SNCAIP, WDR45, ABCD1, NPC2, C19orf12, TRPM7, COX2, SPG21, NPC1, IRF6, NAGLU, AARS2, CLN6, DCTN1, CISD2, MECP2, PPP2R2B, HTT, CERS1, EIF4G1, SDHB, ATN1, ATP7B, EPM2A, HFE, CTSF, MPO, ATXN3, DNAJC5, SMOX, RNF216, GBE1, FTL, ATP6, AMN, VPS13C, APTX, LAMC2, PANK2, GLUD2, IGFALS, VPS35, GM2A, ROGDI, TWNK, HEXA, PRDM8, IL6, HLA-DQB1, IRF2BPL, IL1B, GBA2, TRNC, ADA2, COX1, SDHD, BACE1, PSAP, SMUG1, ATP6V0A2, NECTIN2, ALDH18A1, TARDBP, DNAJC13, ATXN8OS, SDHA, SLC2A3, ATXN10, TREX1, SOD1, PNPLA6, TUBB4A, TNF, DNM1L, MATR3, WFS1, CUBN, XPR1, FBXO7, POLG, COX3, TRNS2, CYTB, ND1, ND5, ND6, TRNE, TRNF, TRNK, TRNL1, TRNQ, TRNS1, TRNV, MMACHC, TRNW, NDP, NEFL, MBTPS2, NR4A2, RRM2B, PRDX1, PDGFB, PLAU, GIGYF2, OSBPL1A, A2M, TYMP, ASAH1, PRICKLE1, ACE, KCTD7, ACHE, MCIDAS, TIMM8A, CRP, BDNF, CYP27A1, DGUOK, SCARB2, CSF2, CSTB, SLC13A5, ATP6V1A, CDR1, ATP6V1E1, ADH1C, ERCC8, SDHAF1, CLN3, ESR1, BCHE, NHLRC1, CHI3L1, COMT, CLU, IGF1, IL1A, SERPINA3, MTHFR, PAEP, RUNX1T1, PON1, ITIH4, NHS, LEP, AKR1C4, UBQLN2, CCL2, ALB, DYRK1A, SLC6A4, CIT, DPP4, IDE, ABCB6, COPD, NRGN, GFAP, ADIPOQ, SORL1, SIRT1, AGER, CHRNA4, ST3GAL4, CHRNA7, SQSTM1, BCL2, APOC1, AGPS, TSHZ1, TGFB1, SCD, MCF2L, LRP2, ADAMTS2, CHAT, PRKN, CYP19A1, NDUFAB1, CETP, ALDH2, TOMM40, CYP2D6, BPIFA4P, FASTK, NGF, MME, MOK, IAPP, IGF1R, DHDDS, SERPINA1, HTR6, STH, ICAM1, S100B, VCAM1, APOA1, WWC1, IL10, SUCLA2, IL1RN, PLA2G7, CPAT1, MIR132, ATM, CCL4, PPARG, CXCL12, PART1, ERVW-1, ATP2B3, EBP, RELN, OPN1SW, REN, SEC14L2, MIR659, TGDS, TXNIP, RAB3A, NF1, APOB, MIR155, AKT1, PDC, SLC18A3, SERPINE1, PAFAH1B1, PDYN, SNAP25, SNCG, CHCHD10, SLC1A3, ABCB1, SLC1A2, NOS2, SHBG, SGCA, SFTPC, PIN1, PARK7, STAR, EDAR, REM1, TBK1, MMP9, SLC30A6, CHRFAM7A, TPI1, GAP43, USO1, GCG, IL18, TRPM2, PLA2G6, SPG11, GLS, SLC17A7, DBN1, ASXL1, PICALM, AKAP6, ACTB, AGTR1, GDF15, HTR2A, ERVK-6, HSD11B1, DRD3, YWHAE, DLG4, ERVK-32, SIGMAR1, INSRR, TIMP1, MTOR, FLNB, MMP2, ESR2, MFGE8, THBS1, TET2, NXF1, TAS2R62P, MAOA, SF3B1, KL, SPTLC1, EPO, ERVK-20, ABCA1, LPA, ABCA7, JAG1, MMP3, CCL4L2, LGI1, AD10, H6PD, H3P28, TRIM2, ASTN2, FAM72A, ATG5, UBR4, TAOK2, FHL5, NCSTN, GRAP2, LIPG, PHLPP1, CYTIP, FPGT-TNNI3K, NBEAL2, RABEPK, MCI2, AD16, MIR2113, LANCL1, ADAMTS13, CNMD, SDS, EBNA1BP2, SPON1, CFDP1, OLFM1, PPARGC1A, NES, FAS-AS1, CRTAP, RIDA, WDHD1, SIRT2, WIF1, ABCG1, FASTKD2, ACOT7, MTCO2P12, RAPGEF5, AD17, PGR-AS1, RBM8A, ZGLP1, CCS, AD11, GNE, WASF2, ERVK-18, MPZL2, LRRC37A, SLCO6A1, FAM72B, ACE2, ANKK1, PCSK9, KIDINS220, PLEKHG5, AICDA, MCOLN1, TIGAR, SLC17A6, PDXP, ACKR3, CHPT1, MRGPRX1, FMN2, RETN, ASAH2, USE1, MBD5, ATF7IP, ADI1, SLC52A1, MARCHF1, HCCAT5, INTS11, DYM, ARHGEF2, ARMH1, NANOS3, ACTBL2, PWAR4, HAMP, LGR6, CABIN1, CLSTN2, MRGPRX3, ESCO1, CSMD2, NLRP3, TMEM54, ATPAF2, GPR151, SHANK3, BPIFA2, CACUL1, COL25A1, TOM1L2, PPP1R2C, NAA50, OXER1, RIN3, EHMT1, DHX40, PTCRA, FTO, UBE2Z, WNK1, GPRC6A, CSMD1, CIAO3, HHIP, ROBO3, FNDC5, ABCG4, GSTK1, SERPINA13P, DNAJC10, CCL4L1, CYFIP2, SLC17A5, SEZ6L2, HSPB8, FBXW8, PTPN22, VN1R17P, LRP10, GPR166P, PLF, SH2B1, MIR384, BACE2, BDNF-AS, PRDX5, TPSG1, MIR193B, GCA, BRI3, QPCT, NPS, POTEM, LOC643387, CD2AP, KLHDC2, DDX58, PADI4, LPAR3, LINC00273, GLS2, POTEKP, SIT1, MIR21, MIRLET7B, MIR106B, MIR107, MIR125A, POLE3, INPP5K, WWOX, UBR5, PLA1A, MIR181C, MIR195, MIR206, CHCHD2, SAR1B, HPGDS, TNNI3K, GEMIN4, MIR210, IL22, DELEC1, NOX4, MIR212, MIR223, KCNIP3, CD274, MIR29A, MRGPRX4, PYCARD, SLC33A1, CCL21, HGS, HHEX, GHR, CBLIF, GCLC, GLP1R, GLRA2, GPR39, GRK5, GPX1, GPX4, GRIN2B, NR3C1, GSK3B, GYPC, HARS1, HDAC2, GH1, GDNF, GDF1, PTK2B, EIF4G2, ELF3, EPHX2, F2RL1, F11, FANCD2, FANCB, GAPDH, FDXR, FOXO3, FLT1, FN1, FOLH1, G6PD, HGF, HLA-C, LRPAP1, HMGB1, IL12B, IL13, IL15, IL17A, CXCL10, INPP5D, INSR, IRS1, ITGB2, KNG1, LAD1, RPSA, LIMK1, LMNA, LPL, IL12A, IL9, CXCL8, IDH2, HMOX1, HP, HSF1, HSPG2, HTR1A, IDH1, IFNG, IL7, IGF2, IGFBP2, IGFBP3, IKBKB, IL4, IL6R, EGFR, EEF1A2, EDN1, ECE1, BAX, BCL2A1, DST, BRCA1, BRCA2, TSPO, SERPING1, CA11, CASP1, CASP2, CASP6, CAV1, CBS, CCT, CD4, BACH1, ATR, ATP7A, AGT, ACTG1, ACTG2, ACY1, ADA, ADAM10, ADSL, AGTR2, APOC3, AHR, AKR1B1, ALK, BIN1, ANGPT1, ANXA1, CD14, CD38, CDKN1A, DIH1, CSF3, CSPG4, CTSB, CTSD, CUX1, DAG1, DLD, CREB1, SARDH, DPP6, DRD2, DSC3, DSPP, E2F1, CRYGD, CPE, CDR2, CHD2, CEACAM5, CEBPB, CECR, CGA, CEACAM3, CEACAM7, CHIT1, COL4A2, CHM, CHRM1, CHRNB2, TPP1, CCR5, CNR1, LRP5, LY6E, F2RL3, TLR2, SST, ST13, STAT1, STC1, STXBP3, SULT1A1, SYP, TAC1, TBCA, TCEA1, TFAM, TFPI, TGFB2, THOP1, TIMP2, SREBF1, SPP1, SORD, SELP, CCL3, CCL11, NAT2, CX3CL1, SEL1L, SELE, SET, SOD2, SFPQ, SRSF5, SLC5A2, SMARCA1, SMPD1, SNTA1, TIMP4, TLR4, MAP2, CLDN5, TFEB, NTT, NUP214, FGF23, CNSN, COLQ, FZD4, SRPX, BHLHE40, STK16, TP63, FADD, NR1I2, SYNJ1, WASF1, DEK, CXCR4, LRP8, TYRP1, TNNI3, TP53, TPO, HSP90B2P, TSC2, TYMS, UBB, VWF, UBE2H, UCHL1, UGCG, NR1H2, UROD, VLDLR, ATXN1, RPL29, BRD2, REG1A, NTRK2, NTS, OXT, P2RY11, P4HB, PEBP1, REG3A, PAWR, SERPINA5, PDE9A, SLC26A4, PENK, PER1, PFN1, PGF, NPTX2, NOTCH4, NOS1, MTR, SMCP, MDM2, MDM4, MEF2C, MSD, MSMB, MYLK, NINJ2, NDUFS3, NFIA, NFATC3, NFIB, NFIC, NFIX, PIK3CA, PIK3CB, PIK3CD, PTGS2, KLK10, PSG2, PSMD2, PSMD7, PSPH, PTH, PTHLH, PSPN, PTK2, PZP, RAP1GDS1, PLAAT4, RBP4, RCN1, KLK7, MAPK3, PIK3CG, PNN, PITX2, PITX3, PKNOX1, PLAUR, PLD1, PLP1, POLE, MAPK1, PON2, PPARA, PPP3CA, PREP, PRKAR1B, PRKCB, H3P11

-
Macrocephaly
Wikipedia
"Genetic disorders associated with macrocephaly" . Am J Med Genet A . 146A (16): 2023–37. doi : 10.1002/ajmg.a.32434 .

-
Mild Cognitive Impairment (Mci)
Mayo Clinic
Another Alzheimer's medicine, lecanemab, has shown promise for people with mild Alzheimer's disease and mild cognitive impairment due to Alzheimer's disease. It could become available in 2023. Not all people with MCI are expected to be eligible for the treatment, since only some have MCI due to Alzheimer's disease.
-
Focal Segmental Glomerulosclerosis
Wikipedia
Pflugers Arch. 2017 Aug;469(7-8):983-988. doi: 10.1007/s00424-017-2023-x. Epub 2017 Jun 29. PMID: 28664408. ^ a b c De Vriese AS, Sethi S, Nath KA, et al.TRPC6, INF2, AGT, NPHS2, ACTN4, CD2AP, APOL1, SERPINE1, PAX2, CRB2, MYO1E, ANLN, MYH9, SGPL1, NPHS1, SYNPO, TGFB1, EDN1, ZMPSTE24, COL4A5, NXF5, LMX1B, ARHGAP24, CCN2, LMNA, MPV17, NOS2, PCNA, FN1, E2F3, ACTA2, GNAQ, HAVCR1, SPP1, MYH10, COL4A1, AGTR1, MMP9, MMP2, LIPC, AGTR2, CASP9, ANGPT2, VLDLR, LPL, ACTR3, PIAS1, WT1, REN, PLCE1, SMARCAL1, COQ6, NUP107, PTPRO, TBC1D8B, ITGA3, COQ8B, SCARB2, CLCNKB, NARS2, NUP160, NUP205, WDR73, SLC12A3, NUP133, COL4A4, NUP93, ACTB, SEC61A1, VPS33A, CLCN5, IGAN1, TRIM8, G6PC, SLC37A4, ACE, NUP85, COL4A3, CD44, VEGFA, TNF, TRPC5, CCL2, POMC, PODXL, TBPL1, TYRP1, ALB, RAC1, NFE2L2, BAX, CAMK4, CCR2, APOE, KRT20, MAPK1, MS4A1, PON1, PLG, CD80, TLR4, MAPK3, IL10, MTOR, MIR193A, CYP11B2, LAMB2, HLA-A, IL1A, IL1B, UBD, HPSE, C1QL1, NES, CHP1, GDF15, POLDIP2, MAFB, AHSA1, SIRT1, YAP1, RAPGEF5, BMS1, RNF19A, PLA2R1, MLYCD, DDN, KEAP1, PTPRU, MMRN1, SERPINA3, SMPDL3B, SIRPA, CNKSR3, RHOV, SDK1, THSD7A, NPNT, NPHP4, LINC01193, MIR106A, MIR150, MIR155, MIR19B1, MIR200C, MIR30A, MIR34C, HNP1, MIR451A, MIR490, MIR663A, KTWS, MIR1225, MIR1915, PLB1, HT, IGKV3-7, PLEKHH2, IL22, DERL2, ATL1, ZDHHC2, WT1-AS, SNX9, NBAS, ADA2, KIRREL1, FBXW7, PLXNA3, RPL23, PDSS2, TMEM63C, VANGL2, RBPJP4, PDLIM2, HHIP, TTC21B, ALG13, STRIP1, GRAP2, CCL19, KLF4, CFH, DMPK, ECT2, EGF, ENPEP, EPHB2, EYA1, F2RL1, GABPA, GH1, CXCL1, HLA-DQA1, MAGI1, HMOX1, HP, HTC2, IL1R1, IL6, IL18, ILK, ITGAE, ITGB1, LAMA5, CTLA4, VCAN, CSF2, MAPK14, ANK3, FASLG, B2M, BAK1, BCL2, BCL2L1, BSG, C3AR1, CALB2, CD9, CD40, CD40LG, CDC42, COL1A1, COL8A1, COL8A2, KLF6, COX10, CP, CRH, CRK, LAMC2, LCN2, LPA, ACACA, SLC5A2, SLC12A1, SNRPB, TAGLN, HNF1B, TGFBR2, TIMP1, TLR2, TYRO3, UBA52, KDM6A, VASP, YWHAZ, PAX8, CXCR4, MANF, AIMP2, CUBN, CASK, NRP2, PDLIM1, SELP, SCN7A, SMAD7, S100A4, MMP14, MTHFR, TRNL1, MUC1, MYH1, NAGLU, RPL10A, NEFH, NOS1, NOTCH1, NPHP1, PKD1, PKD2, PLA2G1B, PLAU, PLCG1, PPARG, PTPRC, RAP1GAP, RARA, RPL17, LOC105374325

- B-Cell Prolymphocytic Leukemia Wikipedia
-
Chromothripsis
Wikipedia
"Chromothripsis: Breakage-fusion-bridge over and over again" . Cell Cycle . 12 (13): 2016–2023. doi : 10.4161/cc.25266 . PMC 3737304 .

-
Autoimmune Hemolytic Anemia
Wikipedia
Br Med J (Clin Res Ed) . 282 (6281): 2023–7. doi : 10.1136/bmj.282.6281.2023 .SLC14A1, TSLP, CTLA4, ADA, RAG2, CASP10, IL2RB, ZAP70, TPP2, STIM1, STAT3, CIITA, STAT1, RFXANK, NBN, PNP, PRKCD, RAG1, IL2RA, RASGRP1, RFXAP, FOXP3, FAS, FASLG, NLRP1, TTC7A, LRBA, RFX5, CD47, IL10, KRT20, CD274, SIRPA, LOC102723407, SOD1, LOC102724971, IGHV3-69-1, IGHV3OR16-7, RN7SL263P, FCGR3B, MS4A1, HP, IGH, FCGR3A, RHD, EPO, IGHV3-21, IGHV3-11, IGHV1-69, MBL3P, CD55, GLS, CRP, IGAN1, CR1, MRGPRF, CD59, MIR150, MIR19A, C5AR1, IGHV4-59, RBFOX2, ANXA5, SYK, RASA1, SMN1, SMN2, PTPN6, MBL2, LTA, LEP, TGFB1, HMOX1, TNF, IL17A, IL6, RNF112, CDR3, NR0B2, BTG3, SLC4A1
-
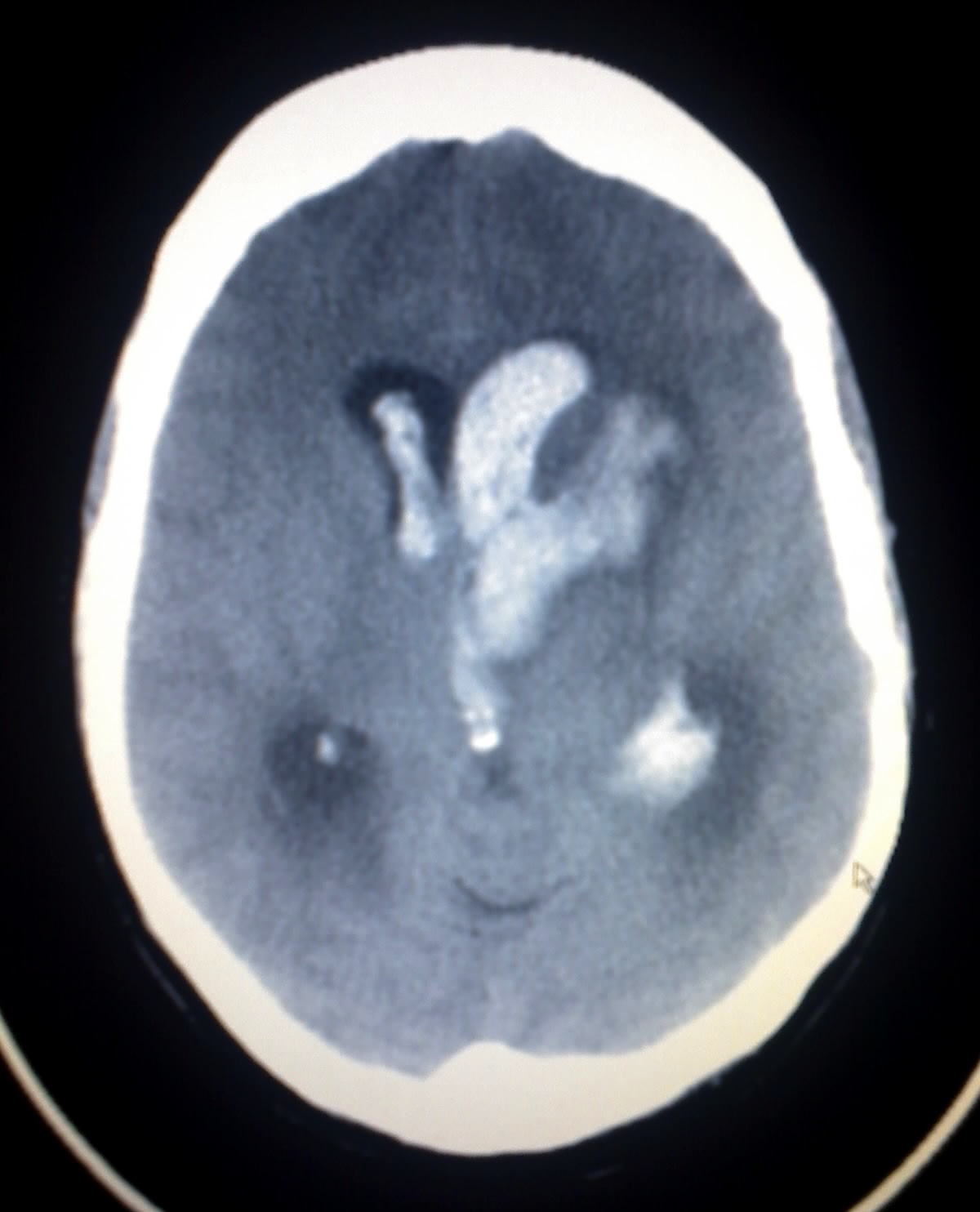
Intracerebral Hemorrhage
Wikipedia
"Guidelines for the Management of Spontaneous Intracerebral Hemorrhage in Adults: 2007 Update: A Guideline From the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists" . Stroke . 38 (6): 2001–2023. doi : 10.1161/strokeaha.107.183689 .CASP8, CASP3, FLT1, PLAT, COL4A1, S100B, ACE, F7, MMP3, COL4A2, MMP9, MMP2, VEGFA, SPP1, ITGB3, ITGAV, ITGA2B, HMOX2, KDR, CNTF, HMOX1, NPPB, BCL2L1, BCL2, BAX, SERPINC1, POMC, PLAU, IL1RN, RELB, RELA, SQSTM1, PTGS2, BECN1, MT2A, NPY, NFKBIA, MT1A, THBS2, THBS1, MMP12, NFKB2, NFKB1, SLC24A1, CXCR4, MTOR, CD14, MAP1LC3A, SLC8B1, C3, DAG1, DCX, NQO1, BNIP3L, AQP4, CASP9, CASP12, APEX1, ANXA1, APP, CST3, SDHD, SMARCB1, CCM2, PMF1-BGLAP, SDHC, SDHB, HELLPAR, FCGR2C, SLC25A44, COLGALT1, TMEM127, VHL, SDHAF2, SUFU, SLC25A11, PARVB, KIF1B, USP18, PMF1, PDCD10, SH2B3, TMEM94, SDHA, LINC02444, RET, GDNF, ABCC6, KRIT1, DLST, ENG, F13A1, F13B, FGA, FGB, FGG, FH, PTCH1, GDF2, FN1, PROS1, CFI, JAK2, SMAD4, MAX, CD46, MDH2, ACVRL1, CFH, NF2, PDGFB, APOE, SERPINA3, AGTR1, APOB, APOH, HCRT, TUBB1, MTHFR, HSPB8, IGFBP3, ESR1
-
Hemolytic Anemia
Wikipedia
Br Med J (Clin Res Ed). 282 (6281): 2023–7. doi :10.1136/bmj.282.6281.2023.G6PD, TPI1, SLC4A1, GCLC, COL4A1, HMOX1, ITPA, SPTB, GPI, EPO, IFNA2, GSR, EIF2AK1, ALAS2, A2M, UROS, ADAMTS13, NT5C3A, SPTA1, HBA2, HBA1, PGK1, CD59, ANK1, GSS, KRAS, CD40LG, RHAG, SLC2A1, HBB, PTPN22, TNFRSF13B, ATP7B, KCNN4, ATP11C, TNFRSF13C, LCAT, NFKB1, NFKB2, BTNL2, NRAS, PFKM, PGM3, PIGA, PRKCD, PIGT, ICOS, ALAD, HLA-DRB1, AK1, KDM6A, WAS, WIPF1, KMT2D, LAT, TNFSF12, TREX1, UROD, CD81, GP1BA, FCGR2B, CTLA4, FECH, GALT, DNASE1, CR2, GATA1, EPB42, CD19, MS4A1, EPB41, FCGR2A, CFH, PKLR, KLF1, HFE, UGT1A1, ADA, PRDX2, CP, HAMP, RN7SL263P, UBL4A, DHS, PIEZO1, H6PD, GCLM, ABCG8, HEPH, MIR451A, MIR144, GSTK1, BTG3, ADD2, IFNL3, IL27, SLEH1, SMUG1, C20orf194, SIRPA, MYDGF, ATN1, SLCO6A1, CRYZ, IL37, HPGDS, DECR1, KRT88P, FOXP3, ACR, RAB14, CD55, MED25, IL17D, ECB2, FUBP1, MTHFR, PTGS2, PSEN1, FOLR1, FOLR2, CD47, ABO, PRDX1, COX2, RPS19, CD247, C3, TNFRSF17, HP, BRCA2, HBG2, HK1, FLNA, S100A8, DMTN, UGT1A, SMARCA5, CDR3, F2, F3, FANCD2, VWF, FBP1, UGCG, SLC6A3, SLC35A2, TRV-AAC1-4, FCGR3A, TFRC, TRIM21, ALDH9A1, ALDOA, MTCO2P12
-
Parkinson's Disease
Mayo Clinic
The test is called an alpha-synuclein seed amplification assay. In a 2023 study, researchers tested the spinal fluid of more than 1,000 people to look for clumps of the protein alpha-synuclein.SNCA, PINK1, DRD2, TH, DDC, MAOB, ATP13A2, PRKN, SLC18A2, PARK7, DRD1, IGF1R, LRRK2, BST1, MAPT, GBA, GAK, HLA-DRA, CYP2D6, CP, PPARGC1A, TNF, SOD2, SOD1, BDNF, ABCB1, VPS35, IL6, NOS1, PARK16, MTHFR, GDNF, GFAP, TMEM230, MAOA, DNM1L, GSTM1, HFE, HMOX1, HSPA9, SLC6A3, DDIT4, GSTP1, CYP2E1, MAP3K5, NGF, AIF1, NQO1, IGF2, GSTA4, TRPM2, IGF2R, INS, HLA-DRB5, GPX1, TCL1B, MIR181C, MTA1, ENO2, BAG5, MAG, FCER2, HGF, HSPA1A, FBP1, INSR, HSPA8, NCAPG2, HBG1, RPL23A, GSK3B, HSPA4, NR4A2, RPS8, COL19A1, TALDO1, SLC30A10, DRAXIN, PITX3, CNTNAP2, NECTIN2, IL1B, ADARB2, CEACAM6, TFAM, MAP2, EDN1, FGB, KCNJ4, RPL6, RPL14, SLC2A14, OPTN, SPR, TWNK, ATG7, GRK5, HTR1A, PENK, GDF5, PPP1R9B, ADH7, DLG4, ABL1, HSF1, LEP, PTN, HSPD1, ND3, GRK6, DLG1, DBN1, ADCY5, HCN3, CCN2, FEZ1, APOE, FASLG, COX10, GJC2, RRN3, RIT2, GCH1, ARPC3, GRK3, GRK2, STK39, LAMP3, SLC41A1, CCDC62, MCCC1, GPNMB, DGKQ, TMEM175, VPS13C, STH, HLA-DRB1, SIPA1L2, CTSB, SREBF1, HIP1R, NUCKS1, MAPT-AS1, RAI1, BCKDK, BIN3, RREB1, TCEANC2, TPM1, PAM, DLG2, ITPKB, MMRN1, KANSL1, FAM47E, ITGA8, LHFPL2, SH3GL2, LMNA, FAM47E-STBD1, NSF, INPP5F, TMC3-AS1, CCDC82, DRD3, SLC50A1, DSCAS, MUL1, RABEP2, NOS2, MANF, ADORA2A, WBP1L, AIMP2, PLPPR1, PTEN, GBF1, EEF1A2, DNAJC1, BAP1, NEFL, SQSTM1, PIP4P2, EIF4G1, PLA2G6, GPR65, MX2, ESR1, PDSS2, NFE2L2, TBC1D5, LINC02224, SMUG1, HTT, SIRT1, TARDBP, SF3B1, GSTT1, LAMC2, GRM5, LRRK2-DT, FRY, FBXO7, GRIN2B, RNF19A, GRN, LINC02451, ACHE, SYNM, RAB29, AHSA1, IL1A, IL10, IGFALS, IGF1, CIT, ITIH1, SIRT2, MCF2L, RET, LOC107987479, TBC1D9, MPHOSPH6, SYT11, TNK2, GPR37, GIGYF2, POLDIP2, MAP4K4, SNCAIP, IP6K2, FYN, MTOR, ATXN3, SYT17, FMR1, FGF20, DCUN1D1, GRAP2, TREM2, MPZ, TET2, CNNM2, GABPA, CHCHD2, GAPDH, ZNF646, GBAP1, GCG, LINC02210-CRHR1, LINC02471, CTNNA3, REM1, HTRA2, HPGDS, GLP1R, CTIF, LY6E, PLEKHM1, LRP2, KCNIP4, ATXN2, ZP3, PARK3, TPTE2P6, PM20D1, LINC02210, MAPK1, TP53, TPO, LINC01121, SLCO6A1, DPY19L2P3, CA8, CDNF, TYW1B, APP, POLG, CNTN1, COL5A2, COL13A1, KLHDC1, COMT, BORCS7, AGAP1, CRHR1, CRK, PIK3CD, CRP, RAB39B, GSTK1, TLR4, CHM, CASP3, PIK3CG, PRSS53, PIK3CB, PIK3CA, TMC3, ALDH1A1, C9orf72, NAT2, ASXL1, PARK10, SPTSSB, SPPL2C, CDH8, CDK5, PRNP, MCIDAS, MDGA2, TMEM229B, PTGS2, CBLL2, THY1, CNKSR3, CASC16, TMPRSS9, SNCA-AS1, NLRP3, RPA2P1, MAPK14, NDUFAF2, UCHL1, SMPD1, YWHAE, BCHE, ALDH2, BCL2, SCN2A, CCL2, AKT1, LINGO1, WNT3, VDR, SLC45A3, CYP17A1, MAPT-IT1, UNC13C, SNCG, CCNT2-AS1, ADAMTSL1, CTSD, TPTE2, BORCS7-ASMT, ZNF165, PREP, SLC2A13, PON1, SNCB, BRINP1, CSF2, ACE, PAFAH1B1, ZSCAN16-AS1, NRTN, CAB39L, DBH, VEGFA, HCRT, GAD1, FUS, SLC11A2, CXCL8, SLC6A4, PARP1, CRMP1, DENR, PPARG, ULK1, DNAJC13, P2RX7, REG1A, IFNG, UTRN, MIR133B, DAPK2, LAMP2, APLN, CTNNB1, TYR, HIF1A, ESR2, PAEP, HNMT, CYP1A2, MC1R, EHMT1, LRRK1, ACMSD, CALB1, USP24, NUP62, CASP9, GRIN2A, TSPO, S100B, CAT, LMX1A, TAC1, GTF2H1, UBE2K, SIRT3, TLR2, LINC01672, DYRK1A, CNR2, ADRB2, CNR1, PDE10A, SYNJ1, HSP90AA1, KHDRBS1, SLC1A2, TPPP, ANG, DNAJB1, ZGLP1, CHRNA4, RMDN3, SCARB2, GIP, LILRB1, DCTN4, PSPN, ROS1, TGFB1, ACTB, SERPINA3, DECR1, IL17A, GRM4, PSPH, CRYZ, STX1B, HTR2A, SLC6A2, BPIFA2, STXBP3, MSMB, MPO, AGER, MAPK8, MEF2D, LMX1B, RIDA, HMGB1, CAV1, CXCR4, CYBB, TOMM40, POTEKP, CCR2, REST, MIR433, AQP4, AGFG1, POTEM, SIGMAR1, ACOT7, ACTG1, MIR34B, ZFPM1, RPS27A, RMDN2, CLOCK, FHL5, NMS, COX2, GSTO2, SUCLA2, SYBU, UBC, BECN1, CST3, CYP1A1, NTS, NNMT, NTF3, DRD4, TFEB, PPP1R2C, RMDN1, MMP9, NQO2, MIR221, TBP, ACTG2, ACTBL2, STAT3, CHMP2B, MFN2, CHCHD10, GSTM2, DNAJC6, GLA, CCK, PLK2, NEDD4, NPC1, VCP, PICALM, NGFR, SYP, SEPTIN4, TRPV1, PTPA, NPY, XBP1, LCN2, TFRC, MAPK3, SKP1, AKR1A1, PRDX2, GDF15, GSTO1, MMP3, LAG3, KLK6, OGA, PSEN1, DNAJB6, PHOX2B, CDK5R1, CXCL12, TCF3, CX3CL1, A2M, REN, CREB1, SNHG1, MIR34A, MIR205, DNM3, MALAT1, NEAT1, GLRX, RLS1, DNAJB1P1, LINGO2, ZNF746, CUX1, SV2C, CYP2D7, DMRT1, NIF3L1, ELAVL4, FKBP1A, ELK3, EPHB1, SLC2A9, FGF2, PTK2B, FABP3, ATF4, RHOT1, MIR4697, HP, HSPA5, ADCYAP1, ICAM1, APOA1, IAPP, CX3CR1, MAP1LC3B, WNT1, AGT, CYP2B6, VIPR2, OGDH, OGG1, VDAC1, APRT, NRGN, P2RX3, VCAM1, AGTR1, UCP2, CTSL, PEBP1, UBB, PINK1-AS, PROKR2, TMEM163, RAB10, NRF1, ADH1B, MOK, RAC1, NR1I2, PARL, F2, ACP3, RAB1A, EPHB2, MARK2, NDUFV2, TRPC1, NEDD9, PVALB, RTN4R, RIC3, HSD3B7, DNMT1, ADH1C, DNAJC5, YWHAZ, TSC2, CLU, PCNA, PACRG, PSMC4, PON2, PPARA, PPARD, MIR29C, PRKAA1, HOTAIR, PRKAA2, PRKAB1, AVP, SMN2, SMN1, PRKAR2A, PRKD1, SGCA, MIR135B, ASAH1, ARNTL, WASH6P, FAS, SKIL, APEX1, MIR200A, SEPTIN5, ATXN8OS, SIAH1, PDE4A, CHRM3, PDYN, TIMP2, ALB, CFP, CDKN2D, CDKN2A, CD38, MIR19B1, WASHC1, USP40, APAF1, PIN1, CASP1, CALCA, SST, PNOC, MIR195, TFR2, IL4, DCTN1, DCDC2, FKBP1AP2, FOXA2, TRAP1, ATG12, GAP43, GRIA2, FKBP1AP3, FKBP1AP4, PRRX2, HMOX2, FTL, MSC, MEFV, FKBP4, ISYNA1, GPX4, NCS1, PPIG, FOXO1, LSAMP, TLR9, LGALS4, MACF1, LAMP1, LRPPRC, ATG5, GRIN2D, MFAP1, BAG3, LDLR, LRP10, GRM2, IREB2, KEAP1, COQ2, FAF1, SBNO2, VPS41, SGSM3, NOX1, SEMA5A, IL1RN, PSIP1, FKBP1AP1, TOMM20, CYP4F3, SLC40A1, PTPN22, GLUD2, SBNO1, IL18, MIR27A, MIR185, MIR137, SNAP25, MIR132, MIR126, TAL1, MIR29A, SP1, SRY, PPIF, MIR204, SPG7, SPP1, FBXO8, MIR21, HDAC6, MIR212, MIR223, USP15, MIR22, RNF11, SOAT1, SNRPF, SNRNP70, SOD3, BDNF-AS, BACE1, FST, ARL6IP5, NLRP1, P2RX5-TAX1BP3, SCD, TXNIP, MGLL, CHP1, MIR4519, HRH3, NES, OPN1MW3, RIPK3, RAB32, TMED10, RNASE4, RNASE1, MTCO2P12, PRSS21, TRIM27, RFC1, LINC02605, H3P13, RENBP, CCL5, P2RX2, ATP6AP2, ABCA7, SMARCA1, CD2AP, SLC22A1, SLC18A3, SRRM2, MIR326, SLC16A1, SLC8A1, MIR494, MIR505, ARC, UCA1, RNF41, MPRIP, HMGXB3, ST3GAL2, MIR563, SI, SHMT2, TRIM32, SHMT1, OPN1MW2, SGTA, SIGLEC7, SPHK1, ERCC6L2, TET1, P2RX6, AXIN2, NEUROG2, NOD2, FBRS, HS1BP3, DDRGK1, SLC52A2, PANK2, MAP3K13, NELFE, H3C1, RAB7A, XPR1, COASY, ZNF184, NDFIP1, EGLN1, MFHAS1, GPR55, KLF4, RNF146, HIVEP3, H3C4, XIST, TMED9, TRPM7, CISD1, RIPK1, DNAJC12, B3GALT4, H3C7, PDE8B, KHSRP, SLC17A6, MGAM, STBD1, H3C3, NOL3, SEMA6A, H3C2, H3C10, H3C12, TMEM106B, H3C8, H3C11, H3C6, TRIB3, C19orf12, SESN2, TCOF1, TIMP3, TRAF2, MZB1, SESN3, GAL, TNFRSF1B, PWAR1, TRAT1, PLB1, TLN1, HDAC4, ATG13, CD200R1, TAS2R13, BMS1, RMC1, EIF2B3, MCAT, MTIF3, ANKK1, PGP, RAB7B, HCAR2, TRH, TTR, PPP1R1B, IL23A, SYVN1, GPR88, HOMER1, SHC3, VIM, ATAD1, MIDN, SELENOT, ROCK2, SLC25A27, DNER, TPH2, USP1, SCLY, UCHL3, FTMT, OPN4, UBE2L3, TXNRD3, VPS26A, CARD16, TXN, TGM2, TGFB2, IDUA, CHRNA5, HK2, LGALS1, BAG1, PLA2G1B, F9, HLA-C, LBP, E2F1, BAX, CLK1, NCL, HCN2, F2R, NR4A1, FOXA1, NDUFA1, NDUFA2, NDUFS3, PLD2, HK1, HOXD13, TOR1A, COL17A1, COL11A2, PPID, FCGR3A, POU2F1, POMC, POLB, ARSA, ASCL1, LGALS3, HCLS1, PLXNA2, LIFR, FAAH, ATM, ATOX1, NRG1, ATP1A3, BNIP3L, NDUFS4, PDIA3, CBS, P2RY2, IL7, KCNJ6, P2RY1, P2RX5, P2RX4, P2RX1, OPRL1, CCKAR, KDR, GPR143, NUCB2, IL13RA1, ISG20, ITGAM, NTRK2, NT5E, CD19, IL2RB, IL2RA, EGF, PCP4, HRES1, NEFM, CHAT, CTSC, ERG, CEBPB, PDC, NFKB1, EGR1, CAST, HTC2, CACNA1A, NHS, HTR1B, KRT7, IDE, CAPN1, NOS3, COX8A, ELAVL2, FRAXE, CBLIF, RBM3, PSMB8, DDIT3, CD200, DAPK1, CSF3, PSEN2, APOD, PSAP, CYTB, GPX7, APOB, DNMT3B, MFGE8, ADCYAP1R1, FXN, GLB1, PTBP1, GLUL, GRP, FTH1, PTPRC, ALOX5, AKR1B1, MSRA, ADH4, MEF2C, GAD2, AHR, GALC, MS, DPEP1, FOSL2, ND1, ATF2, CYP3A4, DRD5, MIF, ACACA, MTRR, NR3C1, MTR, DPYSL2, MAPK10, DPP4, KLK3, ND5, MRC1, FOXO3, OPN1MW, GC, CD55, ND2, CRY1, SLC39A8, FSD1L, SNX6, CYP27B1, KREMEN1, TNMD, DLGAP3, EHHADH, ANO3, GBA2, CYP3A5, TINAGL1, MAK16, DTNBP1, EIF2B1, NEUROD6, LSM2, ATP13A4, MINDY4, DAPK3, TLCD3B, FKBP10, EIF4E, PROK2, EIF4EBP1, HAPLN2, HAMP, LGR6, ACE2, EIF4A2, LIN28A, APH1B, PAGR1, DOCK3, SLC8B1, ASPSCR1, ELOVL7, FA2H, DHRS11, BRCC3, FSD1, CWC22, DHDDS, XYLT2, DVL1, DUSP8, TNFAIP8L2, MCPH1, PEAK1, NEIL1, RTL10, ATN1, DUSP2, DUSP26, DMPK, DLD, TIMM8A, SLC25A28, TFB2M, MS4A6A, EGFR, MEGF8, HIF3A, RAB1B, CSMD1, DAXX, DBI, DCC, DCN, TAS1R2, ZNF436, PDIA2, EEF2, RAPH1, TNKS2, EEF1A1, FYCO1, REL, USP37, FBXO42, FPR2, WWOX, INPP5K, IL17D, PANK1, FN1, PPIL3, IMMTP1, AFF2, FLNB, H2BS1, FKBP5, TREM1, WNT4, FKBP3, ATG16L1, FKBP2, UGT1A9, FKBP1B, FGG, MARCHF5, FGF13, FGF9, FGF8, NDE1, FGF1, GPC4, FDXR, ANKHD1, FCGR3B, VPS29, TRAPPC2L, NUB1, FKBP7, GDF1, DPP7, PSAT1, UBQLN2, UBQLN1, SLC39A2, OBP2A, VSX1, NOX4, ASAP1, GATA2, SOST, TMED7, ST8SIA3, NMD3, POLR1D, GALNS, TFB1M, B4GALNT1, TRIM17, GABBR1, TRAPPC4, G6PD, GAST, HSD17B7, FRAXA, SIRT7, RAB6B, PPIL1, HEATR3, OXR1, SH3RF1, PELI1, EPRS1, NPDC1, TCIM, PNO1, EPO, PAK6, ARNTL2, ZGPAT, RGMA, EPHX2, CASS4, TIGAR, EPHX1, PGLYRP4, ENDOG, THAP1, ANKRD50, VN1R1, MCOLN1, CYP20A1, NDRG2, PPM1H, ZNF512B, NLN, XPO5, GPR158, NUFIP2, NCEH1, ELF4, ELAVL1, ERBB2, ERBB3, CTNNBL1, ZNF253, MTPAP, FCGR2B, MAP1S, FASN, RCBTB1, ADI1, QRSL1, F12, FBXW7, CHDH, YOD1, TRPV6, GPRC5D, SAGE1, SMPD3, F2RL1, SOX6, AMBRA1, EZH2, MECOM, ARFGAP1, MBD5, VPS11, ESRRG, PSENEN, GPRC5C, ERN1, MYO5C, MYDGF, MAP1LC3A, CRY2, EBPL, GET3, MIR342, MIR335, MIR331, ASIP, MIR324, MIR148B, GPR166P, SPAG11A, VN1R17P, ATP5F1B, ATP7A, ATR, MIR96, MIR34C, PLF, MIR425, FBXO48, MIR410, ARRB1, MIR520D, ARR3, MIR486-1, ARG1, POU5F1P3, ARF3, AR, POU5F1P4, CCDC154, NORAD, LINC00273, AQP9, ADGRB1, MIR320A, MIR30E, MIR19A, MIR107, CA12, CA2, MIR134, C4B, MIR142, MIR144, MIR15B, MIR181A2, C4A, MIR183, C1R, MIR190A, KLF9, BRAF, MIR30C2, DST, BMX, BMP2, BMI1, MIR214, MIR217, BGLAP, CFB, MIR222, BCR, MIR27B, BCL2L1, MIR30A, MIR30C1, CXADRP1, AQP1, MIRLET7D, PGR-AS1, LOC102724334, MIR8061, LOC101928100, ADORA1, UCHL1-AS1, TLE5, APLNR, OCM, ERVW-4, MIR4639, ALAD, MICOS10-NBL1, AMFR, BIN1, CBSL, HAGLROS, TP53COR1, APOA1-AS, SNHG14, ADAM10, ADA, ACO2, SIRT1-AS, BUB1B-PAK6, ACO1, ASIC2, LOC110366354, LOC110806262, AOC1, ABCA1, H3P17, MIR3143, ANGPT1, TMED7-TICAM2, PLIN4, MIR545, MIR542, PARK12, MIR561, MIR579, MIR590, MIR599, MIR613, MIR625, MIR626, MIR634, ATXN8, POTEF, APOC3, MIR671, C4B_2, XIAP, VTRNA2-1, MIR543, APC, MUPP, APBB1, ANXA5, ANK2, ANK1, CD24, MIR1224, MFT2, ANGPT2, TIMM23, MIR100, CALB2, SPZ1, KLF6, CCR7, ABCC2, CNGA1, CNTF, GPR139, MTFMT, CPOX, CHI3L1, CRAT, EXOSC6, MRGPRX4, MRGPRX3, RAB3IP, NUS1, GNPDA2, CCR5, GPR151, CCR3, PXDNL, CMA1, SIRPA, STK35, CACUL1, CLN3, CHRNB4, ZFP90, CHRNB3, ZNF569, GLIS1, PDIK1L, FBXO41, PGLYRP3, PGLYRP2, EEF2K, PLXNA4, CYP2C9, USP30, PTPN5, ORAI1, CYP2C19, CYP1B1, PPIL4, DNAJC14, CYLD, CHRFAM7A, CYC1, MCU, IL33, TRIM41, DEPDC7, CRYAB, CXADR, MTDH, CTSZ, CTSK, PRDM6, CTRL, CADPS2, CTNS, CSNK1D, TGS1, NACC1, TRIM9, CSF1R, CSF1, CHIT1, CEL, HSP90AB2P, TMED10P1, CASR, GOLGA6A, CPP, RSPO2, CAV2, RUNX1, PPIL6, SHOC1, FBXW12, ZNF763, LYPD5, CCKBR, CCND3, OR10A4, NANOS3, HSD17B13, PFN3, CASP8, CASP7, SEPTIN14, AMIGO2, HCN1, TICAM2, CASP6, NDUFS7, CALR, CALD1, CALCR, TMEM189, TMEM189-UBE2V1, UOX, CD14, SELENOH, MRGPRX1, ARID2, CDKN1B, CDKN1A, FSIP1, CDK6, CDC42, TMPRSS6, OXER1, CDK1, DDX53, CDA, CD69, RHOV, CD59, CD47, MLKL, GPX6, CDCP2, PRSS55, CD44, CD36, TPCN2, ZUP1, GPRC6A, CD34, IL27, LCLAT1, EBF3, PHYHD1, CD28, GK5, HOOK2, GPR3, USP25, UMOD, VHL, VGF, OPA1, OPRM1, UQCRH, UQCRFS1, UCP1, NPPC, UCN, P2RY6, P4HB, UBTF, UBE3A, UBE2V1, OMP, VIP, VIPR1, VSNL1, WAS, WFS1, NUCB1, WNT5A, NTRK1, NTF4, XK, XRCC1, XRCC3, YWHAG, YWHAH, NPTX2, CNBP, PRDX1, PAH, SERPINE1, TRAF6, PECAM1, TKT, PDR, PDPK1, PDHA1, TM7SF2, PDE1B, PDE7A, PDCD2, PDB1, TPT1, CRISP2, NR2C1, HSP90B1, PCNT, TYROBP, PCMT1, TRPC6, SERPINA5, PAX4, PHLDA2, TST, PAWR, TUBA4A, TUFM, TNFRSF4, REG3A, TXNRD1, TYMS, PAK1, ZSCAN21, NPPB, EIF2S2, MYOC, PEA15, MYL4, EIF3A, HSD17B6, TP63, PSMG1, USO1, NPPA, PRKRA, CASK, NAGLU, PDXK, NBL1, LGR5, MYD88, SNX3, MYCN, MYC, TNFRSF14, FADD, SNAP23, RGS9, MTX1, FGF18, CCN6, TRNL1, ASAP2, MTTP, MTNR1B, FUBP1, EIF2B4, CHST1, DGKD, NUBP1, SLC14A2, MAP3K12, PRDM2, LRP8, TUBA1A, NPM1, NPAS2, NOTCH4, ST8SIA4, NOTCH2, LAP, NKTR, CSRP3, FGF23, SLC7A5, MLRL, CUL1, SYN3, GTPBP6, NFKBIA, AXIN1, NFKB2, FZD4, NFE2L1, NFE2, NEK1, NEFH, NEDD8, NDN, NR0B2, NCK2, SERPINF1, TIMP1, TIAL1, PSMC1, PSMA2, SGK1, SELENOP, SELE, SEL1L, PSMB9, PSMD11, PER1, CCL3, SCN9A, SCN8A, TAS2R38, SCN1A, PTGER2, PSG5, SHH, HTRA1, KLK7, PRSS2, LGMN, PROS1, SLC1A1, PROP1, SLC1A6, SLC2A1, SLC2A4, PROC, PRL, MAP2K7, SLC6A8, SLC6A11, PTGER4, PTH, SATB1, ROCK1, RB1, RAP2B, RAP1B, RAN, RGS2, RGS4, RGS10, RHEB, RAD51B, RING1, RAD51, RAB6A, RNH1, RNY1, RAB3B, SAT1, QDPR, PTX3, RPL32, RPS6, RPS6KB1, PTPRA, RRAS, PTPN6, RTN1, RXRA, S100A9, S100A10, QSOX1, SALL1, SLC6A13, SLC7A1, MAP2K6, DYNLT3, CDKL5, STXBP1, SERPINF2, ABCC8, PLEK, PLG, ADAM17, TACR1, TAF1, PLD1, TAP1, CNTN2, PLAG1, PKD1, PITX2, STAT4, TEF, TMBIM6, TF, PIK3C2B, TFDP2, PIGC, SERPINA1, PHF1, SLC25A3, PHB, PGK1, THAS, THBS1, THOP1, STIM1, PLK1, SLC8A3, PPP3R1, SLC11A1, MAP2K2, PRKCG, SLC19A1, SLC20A2, PRKCD, SLC22A5, PRKAR1B, PRKAR1A, PRKACB, PRKACA, SMPD2, SMS, PPY, PPP4C, STAT1, PPP2R2B, PPP1R1A, PPBP, MED1, POU5F1, SORL1, SOX2, SPN, PODXL, PNMT, SPTBN1, PRRX1, FXYD1, ST13, EIF2B2, ENDOU, GFPT1, SARM1, HLA-A, HLA-B, ACSBG1, HLA-DQB1, KDM6B, CUL9, SMG1, GSR, TNIK, FAIM2, HLF, HNF4A, HNRNPA1, SEPHS1, SATB2, NEDD4L, HEXA, ZNF629, NCSTN, HDAC2, DICER1, SIRT5, HDAC1, HCRTR1, TPSD1, HAS2, ATP1B4, HSD17B10, ABCA5, TRAM1, GTF2H4, TLX2, RPH3A, HPD, RER1, MRPS30, JTB, PNPLA6, SUGT1, PAPOLA, RALBP1, PRDX3, IKBKB, IGHG3, COPS5, IMMT, RAB35, IFNAR1, STMN2, HTR6, DLGAP4, SLC2A6, PTGDR2, HSPB2, HSPB1, COPE, ECD, FKBP9, HSPA1L, PDAP1, TUSC2, GABARAP, HSPA1B, GABARAPL2, RRAS2, ZFYVE26, SEC14L2, MBD2, PDE7B, GLI1, RBMS3, SIT1, GLI2, GPR78, AGO2, CACYBP, LPAR3, CACNG5, B3GAT1, GLS, GLUD1, GNAL, ACAD8, TOX3, GJB2, GJB1, MAT2B, TOR2A, GHSR, GIT1, ACAD9, FLVCR1, MCTS1, DBNL, HIPK2, GHR, SETD2, SCG3, PYCARD, TBK1, ATP2C1, GNAQ, SEC22A, ARIH1, GSPT1, PDSS1, OPN3, MKRN2, GRM8, GRM3, LDOC1, PPP1R15A, SLC7A11, PPIL2, MAFF, GRIN2C, ATXN10, NOCT, TPSG1, CHORDC1, PRDX5, BACE2, PART1, GRIN1, GRIK3, GORASP2, FFAR1, GPER1, MCHR1, GPR17, UTS2R, FBXO2, FBXO9, GPR6, IL9, PPP1R13L, IL13, MICD, MAP3K3, RAB8A, PTGES, MGMT, TMEM59, NPEPPS, TBX4, PPP1R17, MAPK8IP1, MIP, MAP3K11, SH3BP5, KMT2A, AFF1, DNAJB9, ISG15, MCL1, HERPUD1, MBP, SART3, HDAC9, SETD1A, CLSTN3, PHACTR2, MATN3, MAS1, MARK1, PIEZO1, MAP6, MAP1B, CD180, AFDN, NR3C2, MME, MSX1, MTHFD1, NUDT1, USP13, MT3, HSPB3, PGLYRP1, MT2A, SOCS3, MT1E, ERDA1, MT1B, MT1A, PIAS2, USP10, AIFM1, COX5A, OSMR, SLC16A7, SLC33A1, VAPB, MOG, MMP8, GPR37L1, MMP7, MMP2, TAOK2, MMP1, LONP1, KL, ADIPOQ, LTF, LTA, LRPAP1, NXF1, SPRY1, STUB1, DNAJA2, KLC1, DLEU1, KNG1, CCL26, KLK2, NPM3, NDRG1, SPAG11B, UBAC1, KIF2A, KCNMA1, CIB1, CALCOCO2, PROCR, KCNA5, ARFGEF1, NPC2, SCGN, JUP, TRIM3, ITPR3, YKT6, ITPR1, ITGB2, PTGES3, GJB6, CPLX1, KRT10, STX6, HEPH, LPA, PSMD6, SV2B, SRGAP3, RBM19, NCAPD2, SEC16A, LRP6, LRP1, USP3, MVP, NR1I3, SRA1, LPO, BCL2L11, ABCB6, EIF1, LIG4, PTPRU, PQBP1, LIF, TRIM10, LCN1, G3BP1, RPSA, WASF2, LAMB2, LAD1, L1CAM, KRT31, TRIM13, H3P42

-
Alzheimer's Disease
Mayo Clinic
Another Alzheimer's medicine, lecanemab (Leqembi), has shown promise for people with mild Alzheimer's disease and mild cognitive impairment due to Alzheimer's disease. The FDA approved the medicine in 2023. A phase 3 clinical trial found that the medicine slowed cognitive decline in people with early Alzheimer's disease by 27%.APP, ACE, TREM2, ADAM10, APOE, PSEN1, GSK3B, HFE, MAPT, PLAU, NPY, BCL2, CASP3, BDNF, IDE, INSR, IL1B, LEP, BACE1, IGF2, IGF1R, ATP5F1A, INS, BAX, CR1, A2M, ABCA7, TOMM40, CD2AP, BIN1, EPHA1, CLU, PICALM, NOS3, PSEN2, APOC1, MPO, SORL1, VSNL1, INPP5D, NECTIN2, MS4A4A, PCDH11X, CASS4, BCHE, MIR146A, CYP46A1, DHCR24, CHRNA7, NCSTN, VEGFA, DPYSL2, PRNP, ESR1, PPARG, RELN, HMOX1, ACHE, CST3, MAOB, TNF, MTHFR, IGF1, CD33, TFAM, IL6, CYP2D6, CRH, SOD2, UNC5C, PLCG2, TF, ABI3, WWOX, SLC30A6, CHRNB2, ARC, PGRMC1, F2, CALM1, EIF2S1, HLA-DRB5, ENO1, TPI1, IGF2R, SLC30A4, MIR296, SLC2A4, MIR100, IQCK, MIR375, AMFR, SNAR-I, ADAMTS1, MAPK14, PIN1, PYY, PTGS2, S100B, PPARGC1A, NOS2, NGFR, NGF, NFE2L2, SOD1, SYP, CDK5, NGB, MIR505, GAPDHS, MME, MAP2, CTNNB1, TPP1, LRP1, IRS1, CHAT, GAPDH, MIR4467, MIR3622B, AGER, MIR766, MIR708, CAV1, NTRK2, PTGS1, APLP2, ADAM17, MFN2, DNM1, HSF1, GSR, IL33, CCR5, HSPD1, HSPB1, CIB1, CASP8, IKBKB, SERPINF1, ATP7A, MT2A, ADAM9, INS-IGF2, BCL2L2, CASP9, GAB2, PTK2B, PLCB1, ABCA1, GRN, CASP12, SQSTM1, FERMT2, HLA-DRB1, NFIC, CSF1R, APOB, MARK4, HSPG2, MS4A6A, CELF1, VCP, SYNJ1, ZCWPW1, MS4A4E, APH1B, APOC2, F13A1, EXOC3L2, PLXNA4, ADAMTS4, AKAP9, MADD, DST, PILRA, FRMD4A, LAMP1, SLC24A4, GLIS3, SPON1, CADPS2, IL34, COL18A1, TRIP4, SPI1, TGFB2, BCL3, MTHFD1L, AICDA, IL6R, DCHS2, MEGF10, SLC16A7, EPHX2, NDUFAF6, DSG2, OSBPL6, CELF2, UBE2L3, SPPL2A, MAPK7, CDH13, LAMA1, SGK1, SUCLG2, LUZP2, PTPRG, ST6GAL1, AP2A2, RBFOX1, SORCS3, TSPOAP1-AS1, TLN2, ZAP70, ALDH1A2, TCF7L2, FMN2, OTOF, EXOC4, HSD17B10, DNM1L, ALOX5, GULOP, HTR2A, AHCYL1, SDR42E2, HTR6, GTF2H1, GRM5, IAPP, AHSA1, STAG3, ALB, FARP1, TSHZ1, HRES1, AGFG2, DCAF7, SIGMAR1, BCKDK, RTN3, TPPP, HSPA4, HCLS1, HSP90AA1, G3BP1, PGAM5P1, BACE1-AS, KHDRBS1, ALDH2, CFH, HCRT, GPC6, ABCA8, GLP1R, NR3C1, TNRC6A, IL19, PARVB, DDX25, BZW2, FCN2, FGF10, GOLIM4, LINC00476, HPGDS, FANCD2, PDE7B, SIGLEC7, TSPAN16, TRPC4AP, POLDIP2, CNTNAP2, RNF19A, BACE2, UBQLN1, ITSN2, GRIN2B, ZGLP1, BCAS3, EPDR1, TMED9, ENO2, CYCS, ANXA1, EPHB2, EPO, RAPGEF6, APH1A, LARS1, EYA4, SNX9, ESR2, WAC, SLC8A1-AS1, DCTN4, RMDN1, QPCT, MTOR, SGK3, FBXL7, SZT2, GIP, ACSL6, WWC1, CLEC16A, KAZN, LINC01672, PRRC2C, COLGALT2, PLD3, HECW1, ZNF292, MYO16, DKK1, SIRT2, ACOT7, PSIP1, CLASRP, LINC00271, SIRT3, SIRT1, GFAP, NUP62, FYN, CBLC, INHCAP, TRIM51CP, GABPA, GABRA2, GABRG3, DAPK2, SMUG1, GAP43, GATA1, GCG, GCHFR, TARDBP, NCS1, GDNF, HDAC6, RAB3D, MVP, OGG1, LINC02268, LINC02325, SOAT1, ACTG2, ACTG1, SNCG, SNCA, SNCB, SNAP25, NOS1, ACTB, MEF2C-AS1, SLC6A4, NPC1, SLC6A3, GEMIN7-AS1, SLC1A2, LINC01508, LINC01725, NEFL, COX2, TNFRSF1B, STAG3L5P, TLR4, TLR2, MSH2, THY1, MT3, TH, SST, STAG3L5P-PVRIG2P-PILRB, TGFB1, TFF1, RNR2, LINC02653, LINC01712, TCF3, NRGN, CX3CL1, ELMO1, CCL2, PIK3CG, ABCA2, PLA2G1B, EIF2AK2, SERPINA3, PLG, MAPK8, MAPK1, PRKCB, PRKCA, PRKAB1, PRKAA2, PRKAA1, PMS2P1, POLD1, PTPA, PON1, PIK3CD, PIK3CB, PIK3CA, MOK, UPK3B, SORT1, ROS1, SERPINE1, REST, REN, RELB, RAC1, LINC01965, PVR, PVALB, PTPRA, LINC00972, ABCB1, LINC02008, MTCO2P12, TP53, OVCH1-AS1, MOBP, MNAT1, SLC4A8, AGT, INSIG1, AZIN1-AS1, EIF3E, FHL5, IRF2, GSTO1, ITM2B, GRAP2, LIPG, ADIPOQ, KL, MSC, LRAT, AP4M1, CCRL2, IL18, IL17A, IL12A, ST18, IFNG, ARL17B, AKT1, AIF1, KRBOX1, IGFALS, HDAC9, PHF14, IL10, IL1A, ALOX12-AS1, MICAL2, IL2RB, IL4, CLOCK, CXCL8, KCNN2, MPZL1, HERC2, VDR, TFEB, MAOA, COX10-AS1, ZNF232, YWHAZ, VLDLR, PARP1, UTRN, BCAM, UCHL1, UBB, TYROBP, AFF1, TTR, TRPM1, MMP9, AIMP2, LTBP2, KNG1, CRADD, CACNA1G, LAMC2, RPSA, CDK5R1, SUCLA2, LCN2, LDLR, BECN1, LPL, PDE5A, ABCB11, APOC4-APOC2, KHSRP, DENR, AGPS, LPA, CDKAL1, PPARA, MEIKIN, COL4A4, CRK, CEACAM22P, SCIMP, CREB1, CR1L, ZNF862, SH2D4B, CP, PNPLA7, SIMC1, GGACT, COMT, COL12A1, FAM181A, BRCA2, PPP1R37, GPR141, TENM3-AS1, CNR2, L3MBTL4, UBXN11, ACKR2, TMEM132C, CASTOR3, CLPTM1, STRADA, FNIP1, CDCA5, NLRP3, APOA1, CRMP1, KAT8, CSMD1, CLMN, PINK1, CYP8B1, AHNAK, MIR132, MIR107, CUX1, POTEM, PPP1R3B, FAS, SAP30L, ANKRD55, CTSD, CTSB, GEMIN7, EHMT1, LINC01184, CTNNA2, LINC01185, TMC5, THSD4, CCDC134, SP6, LINC01567, PDCD1LG2, SETD7, APOD, BHMG1, CSF2, HYI, BLOC1S3, TSPO, CHRNA4, CHRNA2, TAS2R62P, C3orf67, C9orf72, CCDC83, CCDC89, KDM1B, CD14, TGM6, ATXN7L1, RSPO4, ADGRF2, STH, TAS2R64P, CALHM1, RUNX1T1, PPP1R42, ALPK2, PCSK9, CAT, ANKRD31, CASP6, NKAIN3, TRIQK, CALB1, STEAP1B, CASP1, EPHA1-AS1, CAPN1, APOC4, FAM181A-AS1, NKPD1, SPRED2, CD36, SCARB1, PLPP4, MED12L, ARAP2, CHN2, CHI3L1, ACTBL2, C10orf71, MCIDAS, LRRK2, AKR1C4, ANO4, AGBL1, CEACAM20, ZNF813, RMDN3, CETP, CDR1, LINC00343, TCAM1P, APOC1P1, IGSF23, RMDN2, CDK1, SLC25A48, NKAIN2, FSIP1, CD68, BMPER, C3, CD40, CYP19A1, CRP, NIT2, ANO3, DLG4, ARHGAP20, RCAN1, WDR41, NDUFA12, STK32B, EDEM2, DSCAML1, RNF165, SH3RF1, DYRK1A, MIR29A, SYBU, AQP4, APBB1, DLX5, DBN1, PALM2AKAP2, CEACAM19, DPP4, IL6-AS1, ARVCF, CDC42SE2, DMXL1, TULP4, DAPK1, PMS2CL, POTEKP, MIR34A, VAT1L, OLR1, HDAC2, LRP8, GSN, CCL11, S100A9, COL25A1, POTEF, KLK6, BLMH, HSD17B7, P2RX7, COX8A, ABCB6, PRRT2, IL2, SORCS1, NR1I2, MAPK3, ITGAM, CASR, ATP7B, VDAC1, EGR1, PDE4A, RAB5A, SUMO1, NRG1, OXER1, NTRK1, TFCP2, ANK1, CSNK1D, DLST, APLP1, BLVRA, NFIB, IL1RN, HTT, ACAT1, PLA2G4A, NFIX, NLRP1, GPRC6A, HMGCR, PPID, LPAR3, FZD4, REG1A, MRGPRX1, LRP2, DBH, PSENEN, VPS35, ESCO1, HSD11B1, VN1R17P, SOX2, AGTR1, XBP1, MIR155, MRGPRX4, MRGPRX3, GAL, GPR151, IL13, PAEP, OGDH, GPR166P, STAT3, SET, NFIA, PLB1, AR, LGR6, DHRS11, ABCG2, C4A, KCNIP3, HSD17B13, ABCG1, TTBK1, NOTCH1, EIF2AK3, SLCO6A1, CHMP2B, RBM45, CD44, RIPK1, APBA1, GSTK1, ADNP, ICAM1, BRCA1, APCS, TNFRSF1A, NFKB1, CNTF, MMP3, KLC1, LBP, CTNNA3, SGSM3, FGF2, C4B, HIF1A, CREBBP, SERPINA1, TMEM106B, GRIA1, GRIA2, ECE1, C4B_2, GSAP, OGA, TFRC, PLA2G6, ST3GAL4, PAWR, MFAP1, KAT5, GSTM1, APRT, COX1, HP, NTF3, MIR206, FPR2, CDC42, FUS, MARK1, FGF1, PREP, C5AR1, PON2, MIR29C, CALB2, PDIK1L, SYK, S100A1, CH25H, SREBF2, COX5A, GRIN2A, VCAM1, TMED10, GSTP1, KLK8, PHF1, CXCL10, MEFV, SP1, GJA1, IGFBP3, SLC17A7, CYP3A4, FOXO3, HMGA1, SLC11A2, XPR1, MARK2, PPIF, CRHR1, SHANK3, MYC, CD40LG, CPOX, FKBP5, ANPEP, CAST, C1D, FKBP4, HSPA1A, FLT1, MIF, PLA2G2A, CX3CR1, CSF3, IFNB1, KALRN, PLTP, STXBP3, DDR1, PWAR1, PRDX2, TP63, VIM, IL23A, F2RL3, MMP14, MEF2C, TREM1, TMED10P1, NAT2, MIR342, SAMD9, RAB7A, PGR-AS1, TRPM2, EGFR, ADRB2, CLDN5, ETS2, SYT1, TIMP1, NME8, ELANE, F2R, CD59, EPHA4, CBS, MSMB, APLN, MMP2, MYCL, CALML5, SYN1, XRCC1, TGM2, EEF2, PLA2G7, ELAVL2, EDN1, TMEM97, HMGB1, MIR455, HTRA1, BPIFA2, SLC52A2, NQO1, TUBA1B, FOS, CRYAB, SLC2A1, GPR3, LGMN, SLC2A3, RIDA, FN1, ABCA4, HSPA1B, PECAM1, PTEN, HSPA8, HLA-A, PTPN1, HAMP, TXNIP, GRM2, P4HB, LIN28A, PSPH, GSTT1, CCN2, DECR1, CPLX1, BCYRN1, NES, POU5F1P4, MIR21, GRK5, POU5F1, NTSR1, MIR212, PRKN, LINC02210-CRHR1, HSPA5, DLD, DAB1, HTRA2, POU5F1P3, MIR137, DNAH8, MAPK10, GH1, SERPING1, ADAMTS2, EEF2K, GSTO2, ROCK2, NEDD9, SPTBN1, NTN1, CEBPD, GDF2, CEBPB, PWAR4, SYNM, IGFBP2, GLUL, ABCC9, ATM, PSPN, PTPRC, MIR29B1, RANBP9, NDRG2, CNR1, RTN4R, PTBP1, AQP1, PDK1, MIR106B, PDE4D, ARNTL, PRDX1, ADM, RENBP, POMC, PTPN4, MS, PDGFRB, P2RY2, MIR142, PDE9A, SSTR4, KLK3, BCL2A1, C2, SRPK2, NFATC2, ADCYAP1, LRRTM3, PLD1, NUBP1, MIR424, ATF4, BSG, MIR29B2, PPY, BMP4, TBP, SLC18A3, POLB, NOTCH3, SLC18A2, NPTX2, MTR, SI, MIR222, SH3GL2, ND2, NR4A2, SELENOP, CXCL12, CCL5, ATXN1, CALM3, ITGAX, IFNA13, DISC1, OPTN, HTR1F, HTR4, WNT3A, COL11A2, C20orf181, IFNA1, KEAP1, HDAC4, KLK4, SEMA6A, LRRC4, CRTC1, IL9, DAO, ALOX15, AGTR2, IDO1, SLC25A27, ABCG4, CD55, APOA4, MCOLN1, REM1, ATCAY, EBPL, HSPB2, HSPA9, PLK2, GAD1, NANOG, DCX, COASY, UBE2K, DDIT3, TREML2, APBB2, MAP1LC3B, SRRM2, GZMB, FXN, HNRNPA1, HPSE, RAB10, CIP2A, FOLH1, STIM2, DIO2, MMP24, CEBPZ, GBA, CDR2, ITPR3, CDKN2A, MELTF, SLC30A3, ADRA2B, FTO, FNDC5, GGA3, XPNPEP1, VGF, NR1H2, UGCG, MFGE8, MGAT3, CXCR4, TLR9, APOC3, GPT, ELK3, NEAT1, ADORA2A, MMEL1, TRPC6, EIF4E, CAMK2A, MS4A6E, SRR, HSPA14, IRS2, MGAM, C1orf52, HDAC3, PABPC4, ACKR3, LGALS3, FAM20C, WNK1, DRD4, CYP2C9, MBTPS1, DRD1, LRP6, GRK2, CYP2B6, OGT, LIPA, AD11, GORASP1, PTGDS, SPEN, MIR200B, NPTXR, DNMBP, MIR200A, NCOA6, MIR181C, RBP4, RELA, OPN1LW, EFHD2, MIR188, TPH1, HNRNPA1P10, CTNNBL1, SLC40A1, PNO1, CHCHD2, SDF4, RETN, GOLM1, PPP3R1, PYCARD, PAG1, CCR2, DDIT4, RCBTB1, SBNO1, PPARD, CD274, PCBP4, ACE2, PROS1, PRND, PPP1R15A, CIZ1, MIR26B, TPSG1, GGA1, CFAP97, MAP2K1, AATF, SHANK2, PRL, RBMS3, LOC107987479, MAP2K2, PAXIP1, CHCHD10, SBNO2, PTGES, SPHK1, LPAR2, PPIG, NRXN3, MED23, SPP1, MAPK8IP1, BAG3, APBA3, TAP2, PRDX6, CARTPT, SNAP91, SV2A, MALAT1, MAK16, SYVN1, GDF11, TAC1, TAT, SLC6A2, TRPV1, CISD3, TPT1, TSC2, TM7SF2, TXN, UBE2I, TLE1, TIMP2, XK, CNTN2, TGFBR2, YY1, GOLGA6A, ANP32A, TAM, TERT, DCP1B, TNFSF10, PPP1R1B, GPHN, RGS2, ADAM30, SAA1, METAP2, IMMT, SDS, ADAP1, RYR3, ECHDC3, RYR2, RXRA, SCD, RPS6KB1, CIT, RPS6, CTXN3, LMTK2, NLGN1, ROCK1, RGS4, ATXN2, SRL, STUB1, SHBG, LILRB2, AKR1A1, OLFM1, SKIL, SLC9A6, CREB3, PITRM1, SCGN, CXCR6, OCM, RIN3, SGCA, SFPQ, NCKAP1, CPLX2, TP73, EDAR, CCL3, NPS, BEST1, HPS1, CXCL1, GLO1, LIF, CDH1, LHCGR, CDK4, PCNA, PCK1, L1CAM, GPR42, GPX1, GRB2, ANGPT1, ANGPT2, CETN1, MYD88, GLB1, AKT2, LMNA, DNMT3B, TSC22D3, CD38, PER1, CD69, DRD3, LOX, CD74, LMNB1, DNM2, GAS6, AVP, GC, DMRT1, SARDH, GRIN1, ANXA5, BACH1, P2RY1, INPPL1, CSF1, CS, NEFM, NTS, APEX1, STS, HTC2, NEUROD1, NPTX1, NPPA, ATF2, HTR2C, ARRB2, IGFBP7, HMGCS2, CLK1, CKB, APBA2, CYP17A1, GSTM3, CHGA, CYBB, JUN, CTSS, ORM1, ITPR1, CHRM1, CHRM2, OPRK1, OPRD1, CTRL, HK1, ADRB1, LAMP2, CASP2, EDNRA, PLD2, CAPN2, F2RL1, ACO1, ERBB4, FAT1, FGFR3, CALM2, BRS3, CALCA, CAD, EGF, CASP4, FCGR3B, FDPS, LYZ, FCGR3A, FLNA, MECP2, FABP3, ADRA1A, PLXNA2, MBP, FAAH, ENPEP, F12, BAG1, MEOX2, HOMER1, ITGB2, DBA2, ITGAL, ITGB1, AIM2, AZIN2, CD80, ITIH4, CD46, VIP, CHRNA3, ATG5, TREML1, MCL1, GPRASP2, VWF, APOA5, TMEM119, KLF4, SOCS6, WNT1, XBP1P1, FOXQ1, C3AR1, OPN4, USF1, C1QA, CXCR2, VPS26A, MOGAT3, CCR3, IL6ST, IL5, IL1RAP, LMF2, IL1R1, CREB3L1, UNG, MCU, CLSTN3, SNPH, IL9R, NPEPPS, NAPSA, USF2, MEF2A, ING1, CGB8, FTMT, CHM, VAV1, IMPA1, C1R, ILK, ADAMTS3, IL16, MAP3K5, IL15, GDF15, CGB5, CA2, KCNB1, TSPOAP1, SMAD2, DPPA2, SGO1, LIG3, IFNL3, TNK1, CP20, APCDD1, HSD17B6, CDKN1A, TTBK2, CDKN1B, SLC2A14, CFLAR, STMN1, LIPC, TAB3, CHIT1, BRAP, SPARCL1, MLKL, PTCRA, CD47, LGR5, CD8A, CCT, NR4A3, USP9X, MSRB3, CDR3, CCK, LNPEP, CASP7, CHRFAM7A, CAMP, PER3, YES1, CGA, MARK3, CGB3, SYNGR1, CALCR, IL1RL1, ARHGEF2, PER2, SLC33A1, TPH2, CHEK1, RAB7B, NOG, MBL2, CFL2, HSPB6, AHSA2P, SLC30A1, CD200R1, SOCS3, KDR, KIF5A, HAP1, CALR, CES1, TRPA1, HSPB3, WASF1, SLC32A1, ARHGEF7, CAMK4, COL3A1, H3P40, SCRN1, PTCD1, SPHK2, ATN1, PPIL2, POU2F1, FOSB, GCA, FLT4, SH2B1, APPL1, DNMT1, FLG, FOXO1, DUSP1, DUSP6, HHAT, HSPB8, E2F1, PLXNA3, DOCK2, PNPLA2, ADI1, GMFB, GPI, GPC1, KIF21B, NMNAT2, TRIB3, ALS2, DLG2, DLG3, GLS, PDSS2, ASTN2, MCF2L, GLI2, KIDINS220, CBLIF, SYNE1, DMD, GGT1, FKBP1A, SIT1, SV2C, GDE1, FOXP3, ASCC1, TMED7, FIS1, PRLH, CRYL1, ADIPOR1, LSR, F11, MBL3P, SIRT6, TRMO, NRN1, LCMT1, PRRX2, ERN1, BIN2, UBR5, HEBP1, GEMIN4, PDCD4, TBK1, SLC25A38, FGF14, PCSK1N, TRPM7, DLL1, FLVCR1, AHI1, SETD2, ELK1, IL22, NCAPH2, ELN, PADI1, BPTF, NRBF2, FABP5, EP300, PLA2G3, GRHL3, CXCR3, NAV3, SIRPB1, FLOT1, MET, KCNMB2, CRISPLD2, ARHGAP24, HNMT, SNX27, NPL, BHLHB9, TRIM13, HMOX2, KLF2, CSNK1E, LPAL2, CPQ, PPP1R2C, RAPGEF3, TET1, CRYZ, SORBS3, CTBP1, BCL2L11, COL17A1, NCAPD2, COX10, UBASH3B, NR1I3, ACOT8, PTPN5, PPP1R9B, TOM1, MINDY4, CPE, HTR7, NR1H3, HTR1B, HTR1A, LRPPRC, PDIA6, RHBDD1, NAA25, HLA-C, TPX2, BCAN, ADAMTS13, MOAP1, TNMD, GRIA3, NEUROD6, CHEK2, PADI2, HRH3, PHB2, SIL1, MGLL, FFAR1, GADD45A, MMRN1, DEFA1, IL21, NLRC4, GPR17, AZI2, HHIP, CTF1, HCRTR2, HLA-B, CTNND2, HHEX, HGF, CTSG, HDAC1, CTCF, DHX40, PTGES3, STIP1, CTSZ, CXADR, CARD14, CYP1A2, PDE10A, LILRB1, EHMT2, PDIA2, UMOD, ANGPT4, MIR339, SYN2, MSD, ACP3, APEH, ST8SIA1, AZU1, PI4KA, NCAM1, MIR144, SMIM10L2B, MSI1, NAP1L1, PRSS3, PEBP1, MASP1, SIM2, TIA1, MIR15B, ATD, RPL29, ABCC1, NCAM2, MIR125A, TLR5, SERPINF2, TNFAIP1, CXADRP1, MAPK9, ZFHX3, GGTLC4P, AD10, SGCG, MIR451A, CDR1-AS, TPTEP2-CSNK1E, MIR384, ITSN1, CBSL, MPZ, NFATC4, PKM, NCL, NTF4, LOC643387, SLC1A3, APOA2, THAS, PSMB6, SERPINB6, PSMB9, RHOA, ARMCX5-GPRASP2, SMPD1, REG3A, ATP4A, MIR193B, RFC1, NOTCH4, SLPI, PGF, AEBP1, MIR214, MIR219A1, SLC19A1, MIR22, PCSK1, ALPP, AMD1, MTNR1A, TGFBR1, COX3, ATP12A, NM, ADD3, PSD, TGM1, ARR3, NPM1, PAK1, TMED7-TICAM2, PNP, MIR195, MTHFD1, ADH1B, AMPH, ND4, AMD1P2, ARG1, SULT2A1, RRAS, PDE7A, TTPA, TYK2, TXNRD1, PPP1R1A, PPP1R10, SPG7, SPAST, RAB4A, PPP2CA, OTC, PPP2R2B, MMP1, ARMS2, RAB3A, GGTLC3, RTL1, P2RX4, ST13, SPARC, ALAS1, PPP3CA, NEFH, SEL1L, TYR, RAB6A, MICB, PPIA, CCL4, BST1, TICAM2, BNIP3, MIR326, OPRL1, PON3, BMP6, OPRM1, AHSG, H3P17, SDC2, AGRN, TYRP1, PPP1CA, BMI1, TYRO3, PNMT, DEFA1B, GGT2, ORI6, SMIM10L2A, CISD2, ARSA, EIF2AK4, PRKAR1A, LRP1-AS, SRSF2, MIR98, MIRLET7B, CD200, PDCD1, RAP1A, GGTLC5P, CCND1, ANXA6, FXYD1, S100A6, NFATC3, PLK1, ABO, PTGER3, APC, S100A12, ASL, HSP90B2P, SETMAR, PRRX1, PZP, STAT1, ODC1, CFB, CDNF, ZBTB4, PARK16, SUGP1, DIO1, SORCS2, MAGEE1, ALDH1A1, MIR1306, DES, DIAPH1, LSM2, MIR1229, XPO5, HCN3, CFD, MIR664A, KIF17, WDR48, MTRNR2L12, PRX, DHFR, EPG5, SEPTIN1, FAS-AS1, RNF213, MIR320E, MIR1908, HECW2, LINC00672, NUFIP2, ABCD1, DLG1, QRFP, ZNF410, AOC2, NBEAL1, CYP11A1, OPN1MW2, AD6, P2RY12, NMNAT1, DEPTOR, TNS3, CYP2D7, FAM72A, NUCKS1, CLEC7A, CYP26A1, ARAP3, GREM2, CDKN2B-AS1, CYP2J2, UBE2Z, MIR1246, TSPY3, MIR632, CTSK, GTDC1, CTSL, MIR650, MIR660, SNORD118, TNFAIP8L2, LYNX1, MUL1, PAGR1, CYLD, MAPKAP1, APOF, PDCL3, CYP2C19, NOC3L, CYP27A1, DPEP2, MFT2, PROK2, HPSE2, AD14, AKR1C2, ALPI, TRPV4, NTN4, PRM3, PDF, JAM2, ALOX5AP, TSPY10, DEFB4A, DEFB4B, ALOX12, PTBP2, DCN, NECAB3, FKBPL, NEUROG2, DGKQ, SLC25A4, MIR873, MIR301B, CENPK, DAXX, GFRA4, GOLPH3, ERVK-6, MTUS1, DBI, MIR937, ANG, ACE3P, SOD2-OT1, ANK3, DRD2, ZNF608, NAT10, DYM, LOC102724334, TRIT1, EIF4G2, TET2, EIF5, SERPINB1, ELAVL4, PDP1, ACO2, THRA1/BTR, UGT1A1, CCHCR1, CPVL, SMOX, TOLLIP, TERF2IP, SNTG1, EMP1, LOC102723407, EIF4EBP1, MIR6845, EIF2S3, ADCY2, NUDT11, EGR2, MSTO1, ADARB1, SLC6A15, ADA, TAPBPL, TESC, MIR6840, ACVRL1, FOCAD, EIF4A1, CASZ1, QRICH1, PGPEP1, EIF4A2, NDE1, ASIC2, CTTN, ACADVL, GSKIP, LNCRNA-ATB, ATP6V1H, H3P7, TDP2, ERBB2, CINP, ZCCHC17, H3P13, DTL, GPRC5B, ERCC1, DCDC2, NAT8B, GULP1, H3P23, ERG, H3P28, H3P11, PPIL1, STIN2-VNTR, NANS, EPHA8, H2BS1, POLE3, ACACA, SLC29A1, FXYD6, LRP1B, CST12P, SIRT1-AS, INPP5K, MSRB1, ARID4B, EPOR, ABL1, AAVS1, NR2F6, ERVK-32, LOC110366354, MNS16A, EFNA5, SLC47A1, ALAD, EEF1A1, SNHG19, MICA, DNTT, SOX21-AS1, DOCK3, DPYSL3, MIR626, XAB2, MFF, DUSP22, ARNTL2, SPPL2B, MCCC1, TMX2-CTNND1, ANKS1B, DPYSL5, FXYD6-FXYD2, BARHL1, DSC1, TWSG1, TLE5, DNASE1, DNA2, OCLN, NLN, AMIGO1, AHR, PLEKHG5, SLC24A3, SPC25, TTC7A, PELI1, JAG1, TMEM159, RTN4, APMAP, CD177, CAMK1D, PLAAT1, NR0B1, TIGAR, P2RX5-TAX1BP3, PARD3, GKN1, ADH6, INAVA, CDK5RAP2, OGDHL, LINC01080, ATF7IP, IPO9, VAC14, DVL1, PPP4R3A, OPN1MW3, EBM, OTUB1, SOX6, SLC30A10, SMPD3, MEG3, PLIN2, FBXW7, TDP1, ADORA1, DSC3, ACSS2, BTNL2, KIAA1217, ZNF253, CFC1, MIR4668, DSG1, APOM, MYO5C, MIR4487, NOTCH2NLC, USE1, SELENOS, GDNF-AS1, DSPP, ADCY10, ADRA2A, ZNF415, LINC-ROR, NARS2, CSF2RB, MIR616, MIR20A, CDC25C, PROM2, ATP6V1E1, IL23R, GLIS1, PM20D1, PHF13, CDH2, ZNF569, MIR191, CDK9, MIR192, PRIMA1, CDKN2D, MIR196A1, OR2AG1, LAYN, PIWIL4, MIR19B1, GPBAR1, CDC25B, GDF7, ZDHHC15, MIR139, CD63, SGMS2, MIR140, TMPRSS6, RHBDL3, AVPR2, MIR15A, CBLL2, MIR186, PRUNE2, AMOTL1, CD81, MIR18A, SLC2A12, CDA, MIR181A2, ATP5PO, SESN3, ATP6V1B2, UBR1, ATP5PF, PPME1, MIR224, LYZL4, KCNH8, MTERF4, MIR23A, CPO, ACMSD, MIR23B, BHLHE23, MIR25, CFL1, OSCAR, SPNS2, SEZ6, SLC38A10, MSI2, CFTR, MIR27A, MIR223, ALDH7A1, MIR221, SELENOM, ATP5MC2, CACUL1, HECTD2, SREK1, CTCFL, CBLN4, CDSN, ATP5MC1, DEFB104A, OCIAD2, MAGEC3, MIR210, CEACAM5, CECR, PPARGC1B, ATP5F1B, IL31RA, GNPDA2, SCARB2, NSMCE1, SOCS4, UBE2L1, BNC1, CACNA1C, SLC25A20, SERPINA13P, BLM, SREK1IP1, MIF-AS1, C20orf203, SYPL2, ZNF763, CCL4L1, BID, BGN, ZSCAN1, ZADH2, SMIM20, MILR1, PGP, GOLGA6L2, TMEM189-UBE2V1, TMEM189, IL31, C4BPA, BTK, AMIGO2, HCN1, NHLRC2, ATP9B, SBSN, BMP1, OSTN, C5, CFAP410, BARHL2, NANOS3, C9, STING1, GADL1, ARMH1, VPS51, HCAR2, CAPG, LINC00639, TMEM201, LIN28B, CD5L, MIR127, CD19, KIF6, MS4A1, MS4A3, HYLS1, STOX1, FOLH1B, OR8J1, TRIML2, CD28, KHDRBS2, GAPT, CENPV, KLHDC8B, MIR134, CD86, PIKFYVE, SLC29A4, CCNC, KRIT1, PTF1A, DAOA-AS1, BDKRB2, HCA1, BRD3OS, ASPM, BCS1L, SGMS1, BCR, MIRLET7D, MIR122, RUNX1, ANKK1, BCL6, PHYHD1, BAK1, MIR10A, EBF3, CCKAR, MIR28, FRMD6, MIR613, ADAMTS10, TMEM175, XIAP, MAF1, BIRC3, SLA2, ANTXR1, ASCC2, CRHBP, EVA1A, QRFPR, MAGT1, NCALD, LOC646506, ROPN1L, L3MBTL2, GMNC, SCFV, CSE1L, RNF146, PHF6, HOOK3, BRSK1, MBOAT4, COX15, MIR497, MFSD2A, MIR501, ACCS, ARG2, FAM126A, CPB1, CPN1, ECSCR, MAP1LC3A, MIR484, AQP9, CPS1, SNORD35B, CPT1A, ABLIM2, FASLG, NETO1, DOCK8, RNFT2, C1QBP, TM2D3, ASRGL1, PTGES2, PANK2, MIR590, SCD5, CSNK2A1, VCAN, ZC3H14, CSPG4, CTBS, MIR592, MIR598, CAMKMT, CTNS, SLTM, PTCD2, CTNND1, MIR603, NUBPL, WDR26, SPHKAP, GSTT2B, TMEM163, NCF1, LBH, SPAG11A, SFTPA1, CSF3R, ZNF436, CSN2, NDFIP1, MIR545, SLC44A4, SLC19A3, FAM72B, AIRE, IQCJ, CSNK1G2, DNAJC5, CSNK1G3, LINGO1, ATG4C, ATP2B4, MIR346, SERPINC1, CISH, ASPA, CLC, UCN3, TMEM54, ASS1P1, CLCN3, NACC1, ASIP, STX1B, IFT43, MIR133B, MIR151A, CLK2, TP53INP1, MIR330, MIR335, MIR338, MIR93, SLC26A7, OMA1, CHGB, PLD4, TDRD9, CHD1, MIR299, ATIC, ATHS, H4-16, LRIG3, EXOSC6, CHRM3, MIR30B, MIR30E, AGAP2, MIR31, MIR34C, MIR9-1, GRIN3B, GRIN3A, ASAH1, MIR369, H2BC12, KPRP, LRSAM1, MIR429, H4C15, SHF, GADD45GIP1, COL11A1, ZNF628, MIR431, HNP1, NAV2, MIR409, SLC31A1, RPPH1, SNORD14E, SNORD14D, SNORD14C, SNORD14B, COX6B1, MIR485, CNTN1, DNM1P33, CNTFR, CHRDL1, CLN3, MIR361, MYOCD, PRDM6, DNER, SPECC1, MIR377, CLN5, EXOC3L4, CNP, MIR425, ARRB1, ZNF804A, BDNF-AS, NLRP12, CCR6, ABCC2, DEFB104B, LRRC15, POU3F4, HOOK1, TERC, NAT1, MCM2, EZR, MDH1, VEGFC, MDH2, MDM4, MEF2D, UVRAG, UROD, UQCRC1, UGT1A, SLC35A2, UCP2, UBTF, UBP1, MID1, UBE3A, UBE2V1, CXCL9, ATXN3, UBE2D2, UBE2A, UBC, MAP3K10, MC1R, WARS1, WAS, ZMYM2, MANF, SCG2, FZD5, SMAD7, MAG, MAP3K12, MAP1A, MAP1B, ZNF236, ZNF224, ZNF217, RNF112, WEE1, MZF1, ZIC1, MARS1, MAS1, MAT1A, MAT2A, MAZ, XIST, WT1, WNT2B, WNT5A, UBA52, KMT2A, MLLT3, THBS1, TLR3, TLE3, MSH3, TKT, TIMP4, TIMP3, MSR1, MSRA, THRA, THOP1, THBS4, CYTB, MRE11, NUDT1, TGFBI, TGFB3, ND1, TFF3, TFDP1, MTNR1B, MTRR, TRNL1, MUC1, TERF2, TSPAN7, MRC1, TWIST1, TRAF2, TUBA4A, NR3C2, TTN, TSPY1, TSHR, TSG101, TSC1, MMP7, MMP8, TRPC1, TRH, NR2C2, TNFAIP6, MMP13, TPM1, MOG, MOV10, TP53BP2, TP53BP1, MPG, TNR, TNNI3, MPI, MPST, SLBP, REEP5, DEK, SNX3, TNFRSF6B, RAB11A, LDHA, LEPR, LGALS4, LGALS9, GPAA1, RNMT, GBF1, ADAM19, URI1, TRADD, LCK, B3GALT4, LIFR, LIG1, SOCS1, NUMB, LIMS1, AOC3, PDE8B, USO1, TNFSF11, STK16, LCT, CES2, KMO, KLRC1, BRSK2, KCNQ1, KIR2DL2, NOL3, ATP6V0E1, SELENBP1, USP13, KLKB1, CDK5R2, RAB29, MBD2, KIF11, TMEM11, ENDOU, EIF2S2, TAX1BP1, NAE1, KRT14, KRT18, LAMC1, LBR, PROM1, LCAT, SOCS2, PRKRA, DEGS1, DDX39B, PABPN1, EOMES, BAP1, LTF, H4C9, COLQ, DYSF, CHAF1B, LYN, NRIP1, COIL, SLC7A5, AD5, H4C1, FGF23, ADAM12, BRD3, PSCA, ARHGEF5, TFPI2, FZD3, GHS, M6PR, MARCKS, SMAD1, LTC4S, H4C4, BHLHE40, IRS4, MAPKAPK5, LMO4, LOXL1, CST7, DDO, DGKZ, GAS7, PIK3R3, PKP4, PPFIA1, LRPAP1, SORBS2, H4C6, CUL4A, GNPAT, LTB, H4C14, H4C13, H4C5, H4C2, H4C8, H4C3, H4C11, H4C12, TERF1, MUC4, KCNMA1, TDO2, PCP4, CDK18, RBM3, RBL2, RBBP6, RB1, RASGRF1, RASA1, RARRES2, RAN, RAF1, RAD52, PCYT1A, RAD23B, RAC2, PDB1, RAB27B, RAB27A, PDC, PDE2A, PURA, PDGFB, ENPP2, PDYN, PTPN13, PC, PAX6, PARN, RPL15, P2RX1, S100A8, P2RX3, P2RX5, P2RY4, RREB1, RPS23, RPS21, RPS6KB2, P2RY6, RPS3A, RPL13, RET, RPA1, PAFAH1B2, RORA, ROM1, SNORD15A, BRD2, RNASE1, RHO, RHD, PAK3, TRIM27, PTPN11, PENK, PTN, PMM2, PLAUR, PLCL1, PLEK, PRKCE, PRKCD, PLP1, PRKAR1B, PRKACB, PRKACA, PLXNB1, PML, PRG2, PLAT, PMP22, PRB1, PPT1, PPP2R5E, POLG, PPP2R1A, PPP1CB, PPL, PPIC, PPIB, POR, MAP2K3, PLAG1, PFDN5, PSMD2, PFKFB3, PTGER2, PTGER1, PGD, PTGDR, PTCH1, PGR, PHB, PSMD9, PSMD7, PSMD3, PSMB2, PITX2, SERPINE2, SERPINI1, KLK10, PIK3C3, PIK3R1, KLK7, PIK3R2, PRS, PROS2P, PIN4, PROC, S100A10, OXT, OXA1L, SQLE, STAR, ST14, ST2, NDUFA6, SSTR3, SSTR2, NDUFA9, NDUFB8, SRM, SRF, NEDD4, SEPTIN2, STC1, SP4, NEU1, SOX5, SOX3, SOS2, SOS1, SOD3, NFE2L1, NFKB2, SNRPG, SNRNP70, NDUFA5, STIM1, NME1, MYH9, TRBV20OR9-2, TCP1, TCN2, MMUT, MUTYH, TCF4, ELOC, TBX2, MX1, MYH6, TARBP2, MAP3K7, STK11, MYO6, TACR2, NACA, VAMP2, VAMP1, SURF1, ABCC8, SUOX, NDP, STXBP1, STX1A, NINJ2, NME2, SAA2, OMP, SFTPC, TRA2B, SRSF6, SRSF5, SRSF3, SRSF1, SFRP1, OCA2, MAP2K4, ODF1, SELE, OPA1, OAS3, CXCL11, CCL21, CCL20, CCL19, CCL8, CCL1, SCP2, SCN1A, ORM2, OSM, TSPAN31, OAT, SGSH, SUMO2, SLC8A3, SMPD2, SLN, SLIT3, SLC22A5, SLC22A2, NQO2, SLC18A1, SLC16A1, SLC12A3, SLC11A1, SLC10A2, SLC8A1, NUP98, SLC6A12, NPHP1, SLC6A1, SLC5A2, NRCAM, NRDC, SLC1A1, NRF1, PMEL, YBX1, NT5E, NAT8, SEMA5A, ESRRA, RAB31, MACF1, HEY2, BRD4, TRAM1, CBX5, ANGPTL2, OPN1MW, GRIP1, KCNH4, MSTN, GFER, GFRA1, SIRT5, COTL1, GFRA3, GHR, ZNF629, UBR4, GHSR, WASHC4, GLI1, NUP160, CLUH, GLI3, SCFD1, KCTD2, CLCF1, SEC14L2, NR5A1, SLC24A2, PRPF6, TFIP11, MAFF, EID1, RAB38, FSHR, TMEFF2, PLA2G15, SLC7A11, FTH1, SNHG1, TSPAN15, GAST, ACKR1, G6PD, GAB1, NTSR2, GABBR1, GAD2, GALNS, DDAH1, PADI4, GART, NBEAL2, GMPR, PLEKHM2, WDHD1, GPR39, CARD8, ARHGEF15, AAK1, SYNPO, GPX4, ECD, PARK7, KLF8, TREX1, WIF1, WDR45, ATF6, CORO1A, TBC1D8, SLC7A9, GRIA4, FAF1, GRIK4, RER1, GRM1, STMN2, RAPGEF4, ADRM1, MSRB2, RAB3GAP1, GNA12, ZNF423, ATG4B, UBXN4, RCOR1, SEPTIN8, STAB1, GNAI1, MAPK8IP3, GRAMD4, GNB3, NFASC, KIF1B, GOLGA2, GPER1, SETX, GOLGA4, PDZD2, SAMD4A, KDM1A, GPM6A, RAB21, GPR6, P2RX2, GPR20, SNW1, FRK, FBXO7, BRI3, SNX12, SOCS7, CD209, FABP6, TBX21, NOP53, FABP7, SLC2A8, UBQLN2, A1CF, PSAT1, FANCG, HOOK2, DELEC1, FASN, SNX8, NPC1L1, BLNK, MS4A2, FCGR1A, FCGR2A, MYLIP, SCG3, DROSHA, TMEM230, NOX4, PCA3, SPCS1, VRK3, ETFA, SLC25A37, TLR8, SLC22A17, ECSIT, CD320, EZH2, DNAJC27, CLEC1B, NT5C3A, MZB1, F2RL2, HP1BP3, F3, F9, IRAK4, SAR1B, UTP11, F13B, SH3GLB1, SIDT2, SHANK1, TRAT1, F11R, RGCC, TMEM176B, NOCT, FBXO2, NPTN, TPK1, VCX, GREM1, FKBP1AP2, FKBP1AP3, AGO1, SEZ6L2, FKBP1AP4, FGF21, CLDN17, NOC2L, SND1, FOXM1, EPC2, ATRNL1, LRP10, FLNB, FMR1, POU2F3, TXN2, FBXL2, FOLR1, FOLR2, FKBP1AP1, B3GAT1, IGHV1-68, TNFRSF21, FEB1, FES, FGF9, RABGEF1, PRPF19, FGF13, FGFR1, FGFR4, PDLIM3, RND1, KLHL20, COQ2, CACYBP, HCAR1, FHL2, VPS4A, IL37, GLS2, NAAA, CYTH4, DKK2, DKK3, BBC3, SDCBP2, GRM3, IL24, SPAG9, CXCL2, PCLAF, IGFBP1, ACAP1, IGFBP5, SART3, KDM4A, IGHG3, SDC3, SH3PXD2A, RGS6, SNCAIP, TCL1B, IL4R, IL7, BCAR1, CCL4L2, CXCR1, BAG2, IL12B, BAG5, TMEM59, TBPL1, GAL3ST1, AKAP5, STX8, PIEZO1, BMS1, PTDSS1, IDH1, SH2D3C, GNE, SH2B3, IRF8, SRA1, TANK, KCNE3, HNRNPDL, CCS, NUP153, MED12, HS3ST1, TOMM20, RBM8A, IDH2, CFI, SV2B, TECPR2, IFI27, TOMM70, KIAA0319, IFIT3, IFNAR1, IGBP1, INSRR, PCYT1B, IL27RA, CCNE2, NOLC1, TIAF1, JUND, ZMYM3, KCNC4, KCNJ13, HGS, SYNGR3, ATG12, CBFA2T2, RABEP1, P2RX6, RAB11B, SLC16A3, SLC16A4, ATP6V0D1, RGN, USP10, USP2, USP14, DNAJA3, CLDN1, CLDN8, ARTN, JUNB, GPR50, PICK1, ITPKB, IRAK1, HOMER2, IREB2, IRF3, IRF6, IRF7, ITGAV, CYP7B1, ITGB3, NRXN1, SLC22A8, ITPR2, AIMP1, JAG2, ITGBL1, LHX2, SLIT2, TAOK2, CD163, JAK2, GPR37L1, PIWIL1, MAPKAPK2, PDLIM7, IARS1, TNC, IL18BP, CCT2, HCK, NRG3, USP39, DCTN6, CD226, HDC, HDLBP, CAMKK2, HIP1, TXNRD2, NPC2, SLC35A1, HBG2, PRDX4, ZNRD2, HYOU1, HLA-DQA1, SEMA4D, HLA-DQB1, NXF1, HLA-DRA, ATP5PD, COG5, GPNMB, CHL1, HAS3, FAM3C, GYPA, GSK3A, COPS5, GSM1, GSTM2, EBNA1BP2, PRSS21, PRDX3, GSTZ1, GTS, GUSB, C1QL1, GYPB, HAS1, GYPC, CCL27, ALDH1L1, GYPE, HAGH, WASF3, HSPH1, GJB6, HARS1, ARPP19, DHS, HLA-DRB4, HLA-G, PQBP1, MPHOSPH6, STAM2, GLYAT, HOXA@, RABEPK, HPCA, CALCOCO2, HRC, OLIG2, DDX39A, TOPORS, EIF1, HSD17B1, RAMP2, WASF2, HSD17B4, HSPA2, HSP90AB1, DNAJB1, NAMPT, BCAP31, CTDSP2, TSPAN3, ACTR2, CERT1, ABCC4, HNRNPK, HMBS, NPM3, CFDP1, PRMT5, HMGB2, YAP1, IFITM3, SPAG11B, PEMT, RACK1, SYCP2, TUBB4B, SEMA3A, WARS2, HNRNPC, FOXA1, CCL26, TLR6, FOXA2, LAMC3, LANCL1, HNF4A, TCIRG1, APBB3, APC2, HNRNPA2B1, MTCH2

-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Neuroendocrine Tumor
Wikipedia
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... According to the WHO and UNICEF, deaths attributable to malaria in 2015 were reduced by 60% [77] from a 2000 estimate of 985,000, largely due to the widespread use of insecticide-treated nets and artemisinin-based combination therapies. [74] In 2012, there were 207 million cases of malaria.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5