Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Alzheimer Disease 8
OMIM
Within this candidate region, 1 gene of particular interest was that encoding cystatin-3 (CST3; 604312), because it is known to be an amyloidogenic protein and is codeposited with the amyloid-beta precursor protein (APP; 104760) in amyloid plaques in the brain of AD patients. Using a covariate-based linkage method, Olson et al. (2001) showed that the APP region on chromosome 21q21 is strongly linked to AD-affected sib pairs of the oldest current age (i.e., age either at last exam or at death) who lacked E4 alleles at the apolipoprotein E (APOE; 107741) locus. ... Two-locus analysis provided evidence of strong epistasis between 20p and the APP region, limited to the oldest age group and to those lacking E4 alleles at the APOE locus.TOMM40, TREM2, ABCA7, APP, APOE, PSEN2, PSEN1, MAPT, SORL1, PRNP, CASP3, BACE1, GSK3B, NCSTN, IDE, IL1B, HFE, A2M, ACE, DHCR24, BIN1, ESR1, ADAM10, ADAMTS1, PGRMC1, VEGFA, ARC, CYP46A1, SLC30A4, VSNL1, PICALM, HMOX1, HLA-DRB5, IGF1R, IGF1, INPP5D, IGF2, MPO, NPY, NOS3, PLAU, PLCG2, PPARG, RELN, MTHFR, PYY, NECTIN2, SLC2A4, IGF2R, SOD2, MAOB, TF, LEP, TFAM, INSR, INS, TNF, TPI1, EPHA1, F2, ENO1, CR1, CASS4, ATP5F1A, CLU, CHRNB2, CHRNA7, MIR766, CD33, IQCK, EIF2S1, MIR505, APOC1, CALM1, MIR100, MIR146A, BDNF, BCL2, MIR375, MIR296, BCHE, MIR708, TPP1, SLC30A6, SNAR-I, DPYSL2, ACHE, CD2AP, GAPDHS, PCDH11X, CYP2D6, MIR4467, CRH, MIR3622B, BAX, AMFR, ABI3, CST3, MS4A4A, WWOX, BRCA2, FANCD2, TFF1, TAS2R64P, CTNNB1, SUCLA2, SNCA, CTSD, RNR2, NEFL, TAS2R62P, SOD1, ITPR3, ITPR2, ITPR1, FLAD1, PSENEN, TP53, CDK5R1, EIF2AK3, UBQLN1, ALG3, PIK3CG, PIK3CA, PIK3CD, SERPINA3, PIK3CB, DOCK3, APLP1, OGDH, CREB1, NOTCH1, CASP6, NGF, CCND1, FOS, DLX4, DLG4, DDIT3, RABGEF1, PEBP1, PYCARD, DAPK2, KCNIP3, CTSB, CSF2, CRMP1, CTSG, EHMT2, ENO2, ERBB4, TMED10, TERF2IP, PTK2B, FCN2, PTGES3, FGF2, ACKR1, DNM1L, SDC3, G6PD, GCHFR, ITM2B, CREBBP, MAP3K8, TRPM7, ADI1, MTCO2P12, UPK3B, ACTB, AKT1, AKT2, ANXA1, APBB1, DNLZ, STS, MIR34A, BRCA1, MIR137, C5AR1, DDR1, CAMK4, TMED10P1, MPEG1, C9orf72, ESCO1, CDCA5, PRRT2, MAP1LC3B, CAT, EHMT1, CNR2, SPPL2B, RAB9A, NRXN3, GFAP, SYNJ1, SERPINB5, CD99, MME, MNAT1, CCL2, RRAS, RPS27, RPS21, RAP1A, PYCR1, COX2, PTS, PTGS2, MTHFD1, MMUT, NCAM1, NFIA, NFIB, MAPK8, MAPK3, PRKCB, PRKCA, PPBP, MED1, NFIC, PPARA, NFIX, PKD1, NOTCH3, NRGN, MEOX2, MEF2A, SPRR2A, TTC3, GRIN2A, DENR, GRIN2B, RAB7A, LRP8, HPRT1, HSP90AA1, VIM, IDUA, UTRN, SUMO1, UBE2I, TTK, TPT1, SULT1E1, IL1A, IL6, IL12A, TSPAN6, TIE1, TGFB1, TG, KNG1, LAMC2, LGALS3, TERT, TERC, STIM1, H3P17
-
Early-Onset Alzheimer's Disease
Wikipedia
This loss of brain volume affects ones ability to live and function properly, ultimately being fatal. [5] Beta-amyloid is a small piece of a larger protein called the amyloid precursor protein (APP). Once APP is activated, it is cut into smaller sections of other proteins. ... Familial Alzheimer disease is caused by a mutation in one of at least three genes, which code for presenilin 1 , presenilin 2 , and amyloid precursor protein (APP). [6] [7] [8] Other gene mutations are in study. ... Alpha-secretase cleavage of APP, which precludes the production of Aβ, is the most common processing event for APP. 21 allelic mutations have been discovered in the APP gene. ... "A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid". ... FEBS Letters . 354 (3): 274–8. doi : 10.1016/0014-5793(94)01137-0 .
-
Estrogen And Neurodegenerative Diseases
Wikipedia
Neurochemistry Research . 28 (7): 1003–8. doi : 10.1023/A:1023246921127 . ^ Zhang, She-Qing; et al. ... "Octyl Gallate Markedly Promotes Anti-amyloidogenic Processing of APP through Estrogen Receptor-Mediated ADAM10 Activation" . PLOS ONE . 8 (8): e71913. Bibcode : 2013PLoSO...871913Z . doi : 10.1371/journal.pone.0071913 . ... "Estrogen and Parkinson's disease". Frontiers in Bioscience . 8 (6): 391–400. doi : 10.2741/1070 . ^ Pullman, R. ... Journal of the Neurological Sciences . 169 (1–2): 126–7. doi : 10.1016/s0022-510x(99)00234-8 . PMID 10540020 . ^ Groenerveld, G.J.; et al.
-
Angioedema Induced By Ace Inhibitors, Susceptibility To
OMIM
Clinical Features Blais et al. (1999) and Adam et al. (2002) reported significantly lower plasma aminopeptidase P (APP) activities in patients with a history of AEACEI. ... Measured genotype analysis strongly suggested that the linkage signal for APP activity at this locus was accounted for predominantly by the SNP association. ... There was a significant association between the -2399A allele and decreased serum APP activity in both men and women, but the APP activity was lower in men regardless of genotype. ... This haplotype was associated with decreased plasma APP activity and decreased luciferase gene expression compared to other haplotypes of these SNPs. Cilia La Corte et al. (2011) concluded that the ATG haplotype of XPNPEP2 is functional and contributes to the development of ACEi-angioedema through a reduction in APP activity.
-
Party And Play
Wikipedia
For this reason, it is considered "a public health priority." [3] Contents 1 Terminology 2 Participants and drugs 3 Risks 4 Statistics 5 History and cultural significance 6 Criticism 7 See also 8 References 9 Further reading 10 External links Terminology [ edit ] The practice is nicknamed "party 'n' play" ("PNP" or "PnP") by some participants. ... One academic study calls the practice "sexualized drug use" or SDU. [6] The term PnP is commonly used by gay men [1] [ failed verification ] and other men who have sex with men (MSM) in North America and Australia, while the term chemsex is more associated with the gay scene in Europe. [7] Participants and drugs [ edit ] A selection of poppers Methamphetamine is often used recreationally for its effects as a potent aphrodisiac , euphoriant , and stimulant. [8] It has been further described that "an entire subculture known as party and play is based around methamphetamine use." [8] Gay men belonging to this subculture will typically meet up through internet dating sites to have sex. [8] On such sites, men often include notations such as "chems" or "PNP". [8] Since stimulant drugs such as methamphetamine drastically delay the need for sleep , increase sexual arousal , and tend to inhibit ejaculation , PNP sexual encounters can continue for many hours or even days. [8] Methamphetamine taken in excess of amounts prescribed or recommended will prolong symptoms of intoxication for up to eight hours. [9] In some cases, these sexual encounters will sometimes occur continuously for several days along with repeated methamphetamine use. [8] Methamphetamine is used to create euphoria, "heighten sexual appetite", and increase sexual stamina. [10] The crash following the use of methamphetamine in this manner is very often severe, with marked hypersomnia . [8] Ketamine is very different from the main chemsex drugs, as it is a dissociative hallucinogen that distorts perceptions and creates a sense of detachment. Ketamine is used in chemsex encounters to "improve the experience of receptive anal intercourse or fisting". [10] A study of sauna participants in Barcelona, Spain, in 2016, found that the most commonly used drugs in chemsex are "GHB/GBL, cocaine, ecstasy, silver bars ( MDMA ), poppers and Viagra". [11] A 2014 study on chemsex in London, UK, indicated that the drugs associated with chemsex include mephedrone, GHB/GBL, crystal meth, ketamine, and cocaine. [10] Internet posts by men seeking PNP experiences often resort to slang to identify what drug they are partying with. [12] [13] These drugs tend to inhibit penile erection , [8] [9] a phenomenon known by the slang term crystal penis or tweaker dick . ... In some instances, PNP sessions play a part in the formation of loose social networks that are valued and relied upon by participants. [22] For other men, increasing reliance on hookup apps and websites to arrange sex may result in a sense of isolation that may exacerbate the risk of drug dependence, especially in the context of a lack of other venues for gay socializing and sexual community-formation. [23] A 2014 study found that one of the key reasons for taking drugs before and during sex was to boost sexual confidence and reduce feelings of self-doubt, regarding feelings of "internalised homophobia" from society, concerns about an HIV diagnosis, or "guilt related to having or desiring gay sex". A key self-confidence issue for study participants was "body image", a concern that was heightened by the focus on social networking apps on appearance, because on these apps, there is a focus on idealized male bodies that are "toned and muscular".

-
Occupational Hearing Loss
Wikipedia
For example, a person continuously exposed to 85 dB(A) over an 8-hour work shift will reach 100% of their daily noise dose. ... Employers who expose workers to 85 dB or more for 8 hour shifts are required to provide hearing exams and protection, monitor noise levels, and provide training. ... "Evaluation of smartphone sound measurement applications (apps) using external microphones-A follow-up study" . ... "Smartphone-based sound level measurement apps: Evaluation of compliance with international sound level meter standards". ... New York: M. Dekker. ISBN 978-0-8247-8814-8 . ^ Al-Otaibi ST (June 2000). "Occupational hearing loss".

-
Al Amyloidosis
Wikipedia
Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Treatment 5 Prognosis 6 Epidemiology 7 See also 8 References 9 External links Signs and symptoms [ edit ] AL amyloidosis can affect a wide range of organs, and consequently present with a range of symptoms. ... However many patients are too weak to tolerate this approach. [8] [9] Other treatments can involve application of chemotherapy similar to that used in multiple myeloma. [9] A combination of melphalan and dexamethasone has been found effective in those who are ineligible for stem cell transplantation, [8] and a combination of bortezomib and dexamethasone is now in widespread clinical use. [10] [11] Prognosis [ edit ] Median survival for patients diagnosed with AL amyloidosis was 13 months in the early 1990s, but had improved to c. 40 months a decade later. [12] Epidemiology [ edit ] AL amyloidosis is a rare disease; only 1200 to 3200 new cases are reported each year in the United States. ... "Association of melphalan and high-dose dexamethasone is effective and well tolerated in patients with AL (primary) amyloidosis who are ineligible for stem cell transplantation" . Blood . 103 (8): 2936–8. doi : 10.1182/blood-2003-08-2788 . ... "Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone" . Haematologica . 92 (10): 1351–8. doi : 10.3324/haematol.11325 . PMID 18024372 . ^ Ashutosh D. ... External links [ edit ] Classification D ICD - 10 : E85 ICD - 9-CM : 277.3 OMIM : 254500 MeSH : C531616 DiseasesDB : 315 External resources MedlinePlus : 000533 eMedicine : med/3363 v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2 v t e Immunoproliferative immunoglobulin disorders PCDs / PP Plasmacytoma Multiple myeloma ( Plasma cell leukemia ) MGUS IgM ( Macroglobulinemia / Waldenström's macroglobulinemia ) heavy chain ( Heavy chain disease ) light chain ( Primary amyloidosis ) Other hypergammaglobulinemia CryoglobulinemiaCCND1, LINC02343, CBX7, LINC00457, SMARCD3, TTR, DNAH11, CD38, SCT, GDF15, NPPB, SPP1, IGKV2-29, MIR34A, SMN1, SMN2, ST2, TNF, BMS1P20, KRT20, IGKV1-8, BABAM2, GLIPR1, WDHD1, MAGEC2, SLC1A4, PLXNB2, IGKV3D-15, IGKV1D-8, IGKV3-20, PRAME, ALB, SDC1, FGA, BCR, PRDM1, CAT, MS4A1, CDA, CDH1, CRP, CSF3, CYP1B1, DDT, ETFA, GSN, FAS, IGKV@, LGALS3, LGALS4, MDK, MMP1, MMP2, MUC1, MYD88, NCAM1, PFDN5, RPS27A, SAA1
-
Alzheimer's Disease
Wikipedia
The hypothesis holds that an amyloid-related mechanism that prunes neuronal connections in the brain in the fast-growth phase of early life may be triggered by ageing-related processes in later life to cause the neuronal withering of Alzheimer's disease. [64] N-APP, a fragment of APP from the peptide's N-terminus , is adjacent to beta-amyloid and is cleaved from APP by one of the same enzymes. N-APP triggers the self-destruct pathway by binding to a neuronal receptor called death receptor 6 (DR6, also known as TNFRSF21 ). [64] DR6 is highly expressed in the human brain regions most affected by Alzheimer's, so it is possible that the N-APP/DR6 pathway might be hijacked in the ageing brain to cause damage. ... Osaka mutation A Japanese pedigree of familial Alzheimer's disease was found to be associated with a deletion mutation of codon 693 of APP. [65] This mutation and its association with Alzheimer's disease was first reported in 2008. [66] This mutation is known as the Osaka mutation. ... A β is a fragment from the larger amyloid precursor protein (APP). APP is a transmembrane protein that penetrates through the neuron's membrane. APP is critical to neuron growth, survival, and post-injury repair. [103] [104] In Alzheimer's disease, gamma secretase and beta secretase act together in a proteolytic process which causes APP to be divided into smaller fragments. [105] One of these fragments gives rise to fibrils of amyloid beta, which then form clumps that deposit outside neurons in dense formations known as senile plaques . [98] [106] AD is also considered a tauopathy due to abnormal aggregation of the tau protein .APP, ACE, TREM2, ADAM10, APOE, PSEN1, GSK3B, HFE, MAPT, PLAU, NPY, BCL2, CASP3, BDNF, IDE, INSR, IL1B, LEP, BACE1, IGF2, IGF1R, ATP5F1A, INS, BAX, CR1, A2M, ABCA7, TOMM40, CD2AP, BIN1, EPHA1, CLU, PICALM, NOS3, PSEN2, APOC1, MPO, SORL1, VSNL1, INPP5D, NECTIN2, MS4A4A, PCDH11X, CASS4, BCHE, MIR146A, CYP46A1, DHCR24, CHRNA7, NCSTN, VEGFA, DPYSL2, PRNP, ESR1, PPARG, RELN, HMOX1, ACHE, CST3, MAOB, TNF, MTHFR, IGF1, CD33, TFAM, IL6, CYP2D6, CRH, SOD2, UNC5C, PLCG2, TF, ABI3, WWOX, SLC30A6, CHRNB2, ARC, PGRMC1, F2, CALM1, EIF2S1, HLA-DRB5, ENO1, TPI1, IGF2R, SLC30A4, MIR296, SLC2A4, MIR100, IQCK, MIR375, AMFR, SNAR-I, ADAMTS1, MAPK14, PIN1, PYY, PTGS2, S100B, PPARGC1A, NOS2, NGFR, NGF, NFE2L2, SOD1, SYP, CDK5, NGB, MIR505, GAPDHS, MME, MAP2, CTNNB1, TPP1, LRP1, IRS1, CHAT, GAPDH, MIR4467, MIR3622B, AGER, MIR766, MIR708, CAV1, NTRK2, PTGS1, APLP2, ADAM17, MFN2, DNM1, HSF1, GSR, IL33, CCR5, HSPD1, HSPB1, CIB1, CASP8, IKBKB, SERPINF1, ATP7A, MT2A, ADAM9, INS-IGF2, BCL2L2, CASP9, GAB2, PTK2B, PLCB1, ABCA1, GRN, CASP12, SQSTM1, FERMT2, HLA-DRB1, NFIC, CSF1R, APOB, MARK4, HSPG2, MS4A6A, CELF1, VCP, SYNJ1, ZCWPW1, MS4A4E, APH1B, APOC2, F13A1, EXOC3L2, PLXNA4, ADAMTS4, AKAP9, MADD, DST, PILRA, FRMD4A, LAMP1, SLC24A4, GLIS3, SPON1, CADPS2, IL34, COL18A1, TRIP4, SPI1, TGFB2, BCL3, MTHFD1L, AICDA, IL6R, DCHS2, MEGF10, SLC16A7, EPHX2, NDUFAF6, DSG2, OSBPL6, CELF2, UBE2L3, SPPL2A, MAPK7, CDH13, LAMA1, SGK1, SUCLG2, LUZP2, PTPRG, ST6GAL1, AP2A2, RBFOX1, SORCS3, TSPOAP1-AS1, TLN2, ZAP70, ALDH1A2, TCF7L2, FMN2, OTOF, EXOC4, HSD17B10, DNM1L, ALOX5, GULOP, HTR2A, AHCYL1, SDR42E2, HTR6, GTF2H1, GRM5, IAPP, AHSA1, STAG3, ALB, FARP1, TSHZ1, HRES1, AGFG2, DCAF7, SIGMAR1, BCKDK, RTN3, TPPP, HSPA4, HCLS1, HSP90AA1, G3BP1, PGAM5P1, BACE1-AS, KHDRBS1, ALDH2, CFH, HCRT, GPC6, ABCA8, GLP1R, NR3C1, TNRC6A, IL19, PARVB, DDX25, BZW2, FCN2, FGF10, GOLIM4, LINC00476, HPGDS, FANCD2, PDE7B, SIGLEC7, TSPAN16, TRPC4AP, POLDIP2, CNTNAP2, RNF19A, BACE2, UBQLN1, ITSN2, GRIN2B, ZGLP1, BCAS3, EPDR1, TMED9, ENO2, CYCS, ANXA1, EPHB2, EPO, RAPGEF6, APH1A, LARS1, EYA4, SNX9, ESR2, WAC, SLC8A1-AS1, DCTN4, RMDN1, QPCT, MTOR, SGK3, FBXL7, SZT2, GIP, ACSL6, WWC1, CLEC16A, KAZN, LINC01672, PRRC2C, COLGALT2, PLD3, HECW1, ZNF292, MYO16, DKK1, SIRT2, ACOT7, PSIP1, CLASRP, LINC00271, SIRT3, SIRT1, GFAP, NUP62, FYN, CBLC, INHCAP, TRIM51CP, GABPA, GABRA2, GABRG3, DAPK2, SMUG1, GAP43, GATA1, GCG, GCHFR, TARDBP, NCS1, GDNF, HDAC6, RAB3D, MVP, OGG1, LINC02268, LINC02325, SOAT1, ACTG2, ACTG1, SNCG, SNCA, SNCB, SNAP25, NOS1, ACTB, MEF2C-AS1, SLC6A4, NPC1, SLC6A3, GEMIN7-AS1, SLC1A2, LINC01508, LINC01725, NEFL, COX2, TNFRSF1B, STAG3L5P, TLR4, TLR2, MSH2, THY1, MT3, TH, SST, STAG3L5P-PVRIG2P-PILRB, TGFB1, TFF1, RNR2, LINC02653, LINC01712, TCF3, NRGN, CX3CL1, ELMO1, CCL2, PIK3CG, ABCA2, PLA2G1B, EIF2AK2, SERPINA3, PLG, MAPK8, MAPK1, PRKCB, PRKCA, PRKAB1, PRKAA2, PRKAA1, PMS2P1, POLD1, PTPA, PON1, PIK3CD, PIK3CB, PIK3CA, MOK, UPK3B, SORT1, ROS1, SERPINE1, REST, REN, RELB, RAC1, LINC01965, PVR, PVALB, PTPRA, LINC00972, ABCB1, LINC02008, MTCO2P12, TP53, OVCH1-AS1, MOBP, MNAT1, SLC4A8, AGT, INSIG1, AZIN1-AS1, EIF3E, FHL5, IRF2, GSTO1, ITM2B, GRAP2, LIPG, ADIPOQ, KL, MSC, LRAT, AP4M1, CCRL2, IL18, IL17A, IL12A, ST18, IFNG, ARL17B, AKT1, AIF1, KRBOX1, IGFALS, HDAC9, PHF14, IL10, IL1A, ALOX12-AS1, MICAL2, IL2RB, IL4, CLOCK, CXCL8, KCNN2, MPZL1, HERC2, VDR, TFEB, MAOA, COX10-AS1, ZNF232, YWHAZ, VLDLR, PARP1, UTRN, BCAM, UCHL1, UBB, TYROBP, AFF1, TTR, TRPM1, MMP9, AIMP2, LTBP2, KNG1, CRADD, CACNA1G, LAMC2, RPSA, CDK5R1, SUCLA2, LCN2, LDLR, BECN1, LPL, PDE5A, ABCB11, APOC4-APOC2, KHSRP, DENR, AGPS, LPA, CDKAL1, PPARA, MEIKIN, COL4A4, CRK, CEACAM22P, SCIMP, CREB1, CR1L, ZNF862, SH2D4B, CP, PNPLA7, SIMC1, GGACT, COMT, COL12A1, FAM181A, BRCA2, PPP1R37, GPR141, TENM3-AS1, CNR2, L3MBTL4, UBXN11, ACKR2, TMEM132C, CASTOR3, CLPTM1, STRADA, FNIP1, CDCA5, NLRP3, APOA1, CRMP1, KAT8, CSMD1, CLMN, PINK1, CYP8B1, AHNAK, MIR132, MIR107, CUX1, POTEM, PPP1R3B, FAS, SAP30L, ANKRD55, CTSD, CTSB, GEMIN7, EHMT1, LINC01184, CTNNA2, LINC01185, TMC5, THSD4, CCDC134, SP6, LINC01567, PDCD1LG2, SETD7, APOD, BHMG1, CSF2, HYI, BLOC1S3, TSPO, CHRNA4, CHRNA2, TAS2R62P, C3orf67, C9orf72, CCDC83, CCDC89, KDM1B, CD14, TGM6, ATXN7L1, RSPO4, ADGRF2, STH, TAS2R64P, CALHM1, RUNX1T1, PPP1R42, ALPK2, PCSK9, CAT, ANKRD31, CASP6, NKAIN3, TRIQK, CALB1, STEAP1B, CASP1, EPHA1-AS1, CAPN1, APOC4, FAM181A-AS1, NKPD1, SPRED2, CD36, SCARB1, PLPP4, MED12L, ARAP2, CHN2, CHI3L1, ACTBL2, C10orf71, MCIDAS, LRRK2, AKR1C4, ANO4, AGBL1, CEACAM20, ZNF813, RMDN3, CETP, CDR1, LINC00343, TCAM1P, APOC1P1, IGSF23, RMDN2, CDK1, SLC25A48, NKAIN2, FSIP1, CD68, BMPER, C3, CD40, CYP19A1, CRP, NIT2, ANO3, DLG4, ARHGAP20, RCAN1, WDR41, NDUFA12, STK32B, EDEM2, DSCAML1, RNF165, SH3RF1, DYRK1A, MIR29A, SYBU, AQP4, APBB1, DLX5, DBN1, PALM2AKAP2, CEACAM19, DPP4, IL6-AS1, ARVCF, CDC42SE2, DMXL1, TULP4, DAPK1, PMS2CL, POTEKP, MIR34A, VAT1L, OLR1, HDAC2, LRP8, GSN, CCL11, S100A9, COL25A1, POTEF, KLK6, BLMH, HSD17B7, P2RX7, COX8A, ABCB6, PRRT2, IL2, SORCS1, NR1I2, MAPK3, ITGAM, CASR, ATP7B, VDAC1, EGR1, PDE4A, RAB5A, SUMO1, NRG1, OXER1, NTRK1, TFCP2, ANK1, CSNK1D, DLST, APLP1, BLVRA, NFIB, IL1RN, HTT, ACAT1, PLA2G4A, NFIX, NLRP1, GPRC6A, HMGCR, PPID, LPAR3, FZD4, REG1A, MRGPRX1, LRP2, DBH, PSENEN, VPS35, ESCO1, HSD11B1, VN1R17P, SOX2, AGTR1, XBP1, MIR155, MRGPRX4, MRGPRX3, GAL, GPR151, IL13, PAEP, OGDH, GPR166P, STAT3, SET, NFIA, PLB1, AR, LGR6, DHRS11, ABCG2, C4A, KCNIP3, HSD17B13, ABCG1, TTBK1, NOTCH1, EIF2AK3, SLCO6A1, CHMP2B, RBM45, CD44, RIPK1, APBA1, GSTK1, ADNP, ICAM1, BRCA1, APCS, TNFRSF1A, NFKB1, CNTF, MMP3, KLC1, LBP, CTNNA3, SGSM3, FGF2, C4B, HIF1A, CREBBP, SERPINA1, TMEM106B, GRIA1, GRIA2, ECE1, C4B_2, GSAP, OGA, TFRC, PLA2G6, ST3GAL4, PAWR, MFAP1, KAT5, GSTM1, APRT, COX1, HP, NTF3, MIR206, FPR2, CDC42, FUS, MARK1, FGF1, PREP, C5AR1, PON2, MIR29C, CALB2, PDIK1L, SYK, S100A1, CH25H, SREBF2, COX5A, GRIN2A, VCAM1, TMED10, GSTP1, KLK8, PHF1, CXCL10, MEFV, SP1, GJA1, IGFBP3, SLC17A7, CYP3A4, FOXO3, HMGA1, SLC11A2, XPR1, MARK2, PPIF, CRHR1, SHANK3, MYC, CD40LG, CPOX, FKBP5, ANPEP, CAST, C1D, FKBP4, HSPA1A, FLT1, MIF, PLA2G2A, CX3CR1, CSF3, IFNB1, KALRN, PLTP, STXBP3, DDR1, PWAR1, PRDX2, TP63, VIM, IL23A, F2RL3, MMP14, MEF2C, TREM1, TMED10P1, NAT2, MIR342, SAMD9, RAB7A, PGR-AS1, TRPM2, EGFR, ADRB2, CLDN5, ETS2, SYT1, TIMP1, NME8, ELANE, F2R, CD59, EPHA4, CBS, MSMB, APLN, MMP2, MYCL, CALML5, SYN1, XRCC1, TGM2, EEF2, PLA2G7, ELAVL2, EDN1, TMEM97, HMGB1, MIR455, HTRA1, BPIFA2, SLC52A2, NQO1, TUBA1B, FOS, CRYAB, SLC2A1, GPR3, LGMN, SLC2A3, RIDA, FN1, ABCA4, HSPA1B, PECAM1, PTEN, HSPA8, HLA-A, PTPN1, HAMP, TXNIP, GRM2, P4HB, LIN28A, PSPH, GSTT1, CCN2, DECR1, CPLX1, BCYRN1, NES, POU5F1P4, MIR21, GRK5, POU5F1, NTSR1, MIR212, PRKN, LINC02210-CRHR1, HSPA5, DLD, DAB1, HTRA2, POU5F1P3, MIR137, DNAH8, MAPK10, GH1, SERPING1, ADAMTS2, EEF2K, GSTO2, ROCK2, NEDD9, SPTBN1, NTN1, CEBPD, GDF2, CEBPB, PWAR4, SYNM, IGFBP2, GLUL, ABCC9, ATM, PSPN, PTPRC, MIR29B1, RANBP9, NDRG2, CNR1, RTN4R, PTBP1, AQP1, PDK1, MIR106B, PDE4D, ARNTL, PRDX1, ADM, RENBP, POMC, PTPN4, MS, PDGFRB, P2RY2, MIR142, PDE9A, SSTR4, KLK3, BCL2A1, C2, SRPK2, NFATC2, ADCYAP1, LRRTM3, PLD1, NUBP1, MIR424, ATF4, BSG, MIR29B2, PPY, BMP4, TBP, SLC18A3, POLB, NOTCH3, SLC18A2, NPTX2, MTR, SI, MIR222, SH3GL2, ND2, NR4A2, SELENOP, CXCL12, CCL5, ATXN1, CALM3, ITGAX, IFNA13, DISC1, OPTN, HTR1F, HTR4, WNT3A, COL11A2, C20orf181, IFNA1, KEAP1, HDAC4, KLK4, SEMA6A, LRRC4, CRTC1, IL9, DAO, ALOX15, AGTR2, IDO1, SLC25A27, ABCG4, CD55, APOA4, MCOLN1, REM1, ATCAY, EBPL, HSPB2, HSPA9, PLK2, GAD1, NANOG, DCX, COASY, UBE2K, DDIT3, TREML2, APBB2, MAP1LC3B, SRRM2, GZMB, FXN, HNRNPA1, HPSE, RAB10, CIP2A, FOLH1, STIM2, DIO2, MMP24, CEBPZ, GBA, CDR2, ITPR3, CDKN2A, MELTF, SLC30A3, ADRA2B, FTO, FNDC5, GGA3, XPNPEP1, VGF, NR1H2, UGCG, MFGE8, MGAT3, CXCR4, TLR9, APOC3, GPT, ELK3, NEAT1, ADORA2A, MMEL1, TRPC6, EIF4E, CAMK2A, MS4A6E, SRR, HSPA14, IRS2, MGAM, C1orf52, HDAC3, PABPC4, ACKR3, LGALS3, FAM20C, WNK1, DRD4, CYP2C9, MBTPS1, DRD1, LRP6, GRK2, CYP2B6, OGT, LIPA, AD11, GORASP1, PTGDS, SPEN, MIR200B, NPTXR, DNMBP, MIR200A, NCOA6, MIR181C, RBP4, RELA, OPN1LW, EFHD2, MIR188, TPH1, HNRNPA1P10, CTNNBL1, SLC40A1, PNO1, CHCHD2, SDF4, RETN, GOLM1, PPP3R1, PYCARD, PAG1, CCR2, DDIT4, RCBTB1, SBNO1, PPARD, CD274, PCBP4, ACE2, PROS1, PRND, PPP1R15A, CIZ1, MIR26B, TPSG1, GGA1, CFAP97, MAP2K1, AATF, SHANK2, PRL, RBMS3, LOC107987479, MAP2K2, PAXIP1, CHCHD10, SBNO2, PTGES, SPHK1, LPAR2, PPIG, NRXN3, MED23, SPP1, MAPK8IP1, BAG3, APBA3, TAP2, PRDX6, CARTPT, SNAP91, SV2A, MALAT1, MAK16, SYVN1, GDF11, TAC1, TAT, SLC6A2, TRPV1, CISD3, TPT1, TSC2, TM7SF2, TXN, UBE2I, TLE1, TIMP2, XK, CNTN2, TGFBR2, YY1, GOLGA6A, ANP32A, TAM, TERT, DCP1B, TNFSF10, PPP1R1B, GPHN, RGS2, ADAM30, SAA1, METAP2, IMMT, SDS, ADAP1, RYR3, ECHDC3, RYR2, RXRA, SCD, RPS6KB1, CIT, RPS6, CTXN3, LMTK2, NLGN1, ROCK1, RGS4, ATXN2, SRL, STUB1, SHBG, LILRB2, AKR1A1, OLFM1, SKIL, SLC9A6, CREB3, PITRM1, SCGN, CXCR6, OCM, RIN3, SGCA, SFPQ, NCKAP1, CPLX2, TP73, EDAR, CCL3, NPS, BEST1, HPS1, CXCL1, GLO1, LIF, CDH1, LHCGR, CDK4, PCNA, PCK1, L1CAM, GPR42, GPX1, GRB2, ANGPT1, ANGPT2, CETN1, MYD88, GLB1, AKT2, LMNA, DNMT3B, TSC22D3, CD38, PER1, CD69, DRD3, LOX, CD74, LMNB1, DNM2, GAS6, AVP, GC, DMRT1, SARDH, GRIN1, ANXA5, BACH1, P2RY1, INPPL1, CSF1, CS, NEFM, NTS, APEX1, STS, HTC2, NEUROD1, NPTX1, NPPA, ATF2, HTR2C, ARRB2, IGFBP7, HMGCS2, CLK1, CKB, APBA2, CYP17A1, GSTM3, CHGA, CYBB, JUN, CTSS, ORM1, ITPR1, CHRM1, CHRM2, OPRK1, OPRD1, CTRL, HK1, ADRB1, LAMP2, CASP2, EDNRA, PLD2, CAPN2, F2RL1, ACO1, ERBB4, FAT1, FGFR3, CALM2, BRS3, CALCA, CAD, EGF, CASP4, FCGR3B, FDPS, LYZ, FCGR3A, FLNA, MECP2, FABP3, ADRA1A, PLXNA2, MBP, FAAH, ENPEP, F12, BAG1, MEOX2, HOMER1, ITGB2, DBA2, ITGAL, ITGB1, AIM2, AZIN2, CD80, ITIH4, CD46, VIP, CHRNA3, ATG5, TREML1, MCL1, GPRASP2, VWF, APOA5, TMEM119, KLF4, SOCS6, WNT1, XBP1P1, FOXQ1, C3AR1, OPN4, USF1, C1QA, CXCR2, VPS26A, MOGAT3, CCR3, IL6ST, IL5, IL1RAP, LMF2, IL1R1, CREB3L1, UNG, MCU, CLSTN3, SNPH, IL9R, NPEPPS, NAPSA, USF2, MEF2A, ING1, CGB8, FTMT, CHM, VAV1, IMPA1, C1R, ILK, ADAMTS3, IL16, MAP3K5, IL15, GDF15, CGB5, CA2, KCNB1, TSPOAP1, SMAD2, DPPA2, SGO1, LIG3, IFNL3, TNK1, CP20, APCDD1, HSD17B6, CDKN1A, TTBK2, CDKN1B, SLC2A14, CFLAR, STMN1, LIPC, TAB3, CHIT1, BRAP, SPARCL1, MLKL, PTCRA, CD47, LGR5, CD8A, CCT, NR4A3, USP9X, MSRB3, CDR3, CCK, LNPEP, CASP7, CHRFAM7A, CAMP, PER3, YES1, CGA, MARK3, CGB3, SYNGR1, CALCR, IL1RL1, ARHGEF2, PER2, SLC33A1, TPH2, CHEK1, RAB7B, NOG, MBL2, CFL2, HSPB6, AHSA2P, SLC30A1, CD200R1, SOCS3, KDR, KIF5A, HAP1, CALR, CES1, TRPA1, HSPB3, WASF1, SLC32A1, ARHGEF7, CAMK4, COL3A1, H3P40, SCRN1, PTCD1, SPHK2, ATN1, PPIL2, POU2F1, FOSB, GCA, FLT4, SH2B1, APPL1, DNMT1, FLG, FOXO1, DUSP1, DUSP6, HHAT, HSPB8, E2F1, PLXNA3, DOCK2, PNPLA2, ADI1, GMFB, GPI, GPC1, KIF21B, NMNAT2, TRIB3, ALS2, DLG2, DLG3, GLS, PDSS2, ASTN2, MCF2L, GLI2, KIDINS220, CBLIF, SYNE1, DMD, GGT1, FKBP1A, SIT1, SV2C, GDE1, FOXP3, ASCC1, TMED7, FIS1, PRLH, CRYL1, ADIPOR1, LSR, F11, MBL3P, SIRT6, TRMO, NRN1, LCMT1, PRRX2, ERN1, BIN2, UBR5, HEBP1, GEMIN4, PDCD4, TBK1, SLC25A38, FGF14, PCSK1N, TRPM7, DLL1, FLVCR1, AHI1, SETD2, ELK1, IL22, NCAPH2, ELN, PADI1, BPTF, NRBF2, FABP5, EP300, PLA2G3, GRHL3, CXCR3, NAV3, SIRPB1, FLOT1, MET, KCNMB2, CRISPLD2, ARHGAP24, HNMT, SNX27, NPL, BHLHB9, TRIM13, HMOX2, KLF2, CSNK1E, LPAL2, CPQ, PPP1R2C, RAPGEF3, TET1, CRYZ, SORBS3, CTBP1, BCL2L11, COL17A1, NCAPD2, COX10, UBASH3B, NR1I3, ACOT8, PTPN5, PPP1R9B, TOM1, MINDY4, CPE, HTR7, NR1H3, HTR1B, HTR1A, LRPPRC, PDIA6, RHBDD1, NAA25, HLA-C, TPX2, BCAN, ADAMTS13, MOAP1, TNMD, GRIA3, NEUROD6, CHEK2, PADI2, HRH3, PHB2, SIL1, MGLL, FFAR1, GADD45A, MMRN1, DEFA1, IL21, NLRC4, GPR17, AZI2, HHIP, CTF1, HCRTR2, HLA-B, CTNND2, HHEX, HGF, CTSG, HDAC1, CTCF, DHX40, PTGES3, STIP1, CTSZ, CXADR, CARD14, CYP1A2, PDE10A, LILRB1, EHMT2, PDIA2, UMOD, ANGPT4, MIR339, SYN2, MSD, ACP3, APEH, ST8SIA1, AZU1, PI4KA, NCAM1, MIR144, SMIM10L2B, MSI1, NAP1L1, PRSS3, PEBP1, MASP1, SIM2, TIA1, MIR15B, ATD, RPL29, ABCC1, NCAM2, MIR125A, TLR5, SERPINF2, TNFAIP1, CXADRP1, MAPK9, ZFHX3, GGTLC4P, AD10, SGCG, MIR451A, CDR1-AS, TPTEP2-CSNK1E, MIR384, ITSN1, CBSL, MPZ, NFATC4, PKM, NCL, NTF4, LOC643387, SLC1A3, APOA2, THAS, PSMB6, SERPINB6, PSMB9, RHOA, ARMCX5-GPRASP2, SMPD1, REG3A, ATP4A, MIR193B, RFC1, NOTCH4, SLPI, PGF, AEBP1, MIR214, MIR219A1, SLC19A1, MIR22, PCSK1, ALPP, AMD1, MTNR1A, TGFBR1, COX3, ATP12A, NM, ADD3, PSD, TGM1, ARR3, NPM1, PAK1, TMED7-TICAM2, PNP, MIR195, MTHFD1, ADH1B, AMPH, ND4, AMD1P2, ARG1, SULT2A1, RRAS, PDE7A, TTPA, TYK2, TXNRD1, PPP1R1A, PPP1R10, SPG7, SPAST, RAB4A, PPP2CA, OTC, PPP2R2B, MMP1, ARMS2, RAB3A, GGTLC3, RTL1, P2RX4, ST13, SPARC, ALAS1, PPP3CA, NEFH, SEL1L, TYR, RAB6A, MICB, PPIA, CCL4, BST1, TICAM2, BNIP3, MIR326, OPRL1, PON3, BMP6, OPRM1, AHSG, H3P17, SDC2, AGRN, TYRP1, PPP1CA, BMI1, TYRO3, PNMT, DEFA1B, GGT2, ORI6, SMIM10L2A, CISD2, ARSA, EIF2AK4, PRKAR1A, LRP1-AS, SRSF2, MIR98, MIRLET7B, CD200, PDCD1, RAP1A, GGTLC5P, CCND1, ANXA6, FXYD1, S100A6, NFATC3, PLK1, ABO, PTGER3, APC, S100A12, ASL, HSP90B2P, SETMAR, PRRX1, PZP, STAT1, ODC1, CFB, CDNF, ZBTB4, PARK16, SUGP1, DIO1, SORCS2, MAGEE1, ALDH1A1, MIR1306, DES, DIAPH1, LSM2, MIR1229, XPO5, HCN3, CFD, MIR664A, KIF17, WDR48, MTRNR2L12, PRX, DHFR, EPG5, SEPTIN1, FAS-AS1, RNF213, MIR320E, MIR1908, HECW2, LINC00672, NUFIP2, ABCD1, DLG1, QRFP, ZNF410, AOC2, NBEAL1, CYP11A1, OPN1MW2, AD6, P2RY12, NMNAT1, DEPTOR, TNS3, CYP2D7, FAM72A, NUCKS1, CLEC7A, CYP26A1, ARAP3, GREM2, CDKN2B-AS1, CYP2J2, UBE2Z, MIR1246, TSPY3, MIR632, CTSK, GTDC1, CTSL, MIR650, MIR660, SNORD118, TNFAIP8L2, LYNX1, MUL1, PAGR1, CYLD, MAPKAP1, APOF, PDCL3, CYP2C19, NOC3L, CYP27A1, DPEP2, MFT2, PROK2, HPSE2, AD14, AKR1C2, ALPI, TRPV4, NTN4, PRM3, PDF, JAM2, ALOX5AP, TSPY10, DEFB4A, DEFB4B, ALOX12, PTBP2, DCN, NECAB3, FKBPL, NEUROG2, DGKQ, SLC25A4, MIR873, MIR301B, CENPK, DAXX, GFRA4, GOLPH3, ERVK-6, MTUS1, DBI, MIR937, ANG, ACE3P, SOD2-OT1, ANK3, DRD2, ZNF608, NAT10, DYM, LOC102724334, TRIT1, EIF4G2, TET2, EIF5, SERPINB1, ELAVL4, PDP1, ACO2, THRA1/BTR, UGT1A1, CCHCR1, CPVL, SMOX, TOLLIP, TERF2IP, SNTG1, EMP1, LOC102723407, EIF4EBP1, MIR6845, EIF2S3, ADCY2, NUDT11, EGR2, MSTO1, ADARB1, SLC6A15, ADA, TAPBPL, TESC, MIR6840, ACVRL1, FOCAD, EIF4A1, CASZ1, QRICH1, PGPEP1, EIF4A2, NDE1, ASIC2, CTTN, ACADVL, GSKIP, LNCRNA-ATB, ATP6V1H, H3P7, TDP2, ERBB2, CINP, ZCCHC17, H3P13, DTL, GPRC5B, ERCC1, DCDC2, NAT8B, GULP1, H3P23, ERG, H3P28, H3P11, PPIL1, STIN2-VNTR, NANS, EPHA8, H2BS1, POLE3, ACACA, SLC29A1, FXYD6, LRP1B, CST12P, SIRT1-AS, INPP5K, MSRB1, ARID4B, EPOR, ABL1, AAVS1, NR2F6, ERVK-32, LOC110366354, MNS16A, EFNA5, SLC47A1, ALAD, EEF1A1, SNHG19, MICA, DNTT, SOX21-AS1, DOCK3, DPYSL3, MIR626, XAB2, MFF, DUSP22, ARNTL2, SPPL2B, MCCC1, TMX2-CTNND1, ANKS1B, DPYSL5, FXYD6-FXYD2, BARHL1, DSC1, TWSG1, TLE5, DNASE1, DNA2, OCLN, NLN, AMIGO1, AHR, PLEKHG5, SLC24A3, SPC25, TTC7A, PELI1, JAG1, TMEM159, RTN4, APMAP, CD177, CAMK1D, PLAAT1, NR0B1, TIGAR, P2RX5-TAX1BP3, PARD3, GKN1, ADH6, INAVA, CDK5RAP2, OGDHL, LINC01080, ATF7IP, IPO9, VAC14, DVL1, PPP4R3A, OPN1MW3, EBM, OTUB1, SOX6, SLC30A10, SMPD3, MEG3, PLIN2, FBXW7, TDP1, ADORA1, DSC3, ACSS2, BTNL2, KIAA1217, ZNF253, CFC1, MIR4668, DSG1, APOM, MYO5C, MIR4487, NOTCH2NLC, USE1, SELENOS, GDNF-AS1, DSPP, ADCY10, ADRA2A, ZNF415, LINC-ROR, NARS2, CSF2RB, MIR616, MIR20A, CDC25C, PROM2, ATP6V1E1, IL23R, GLIS1, PM20D1, PHF13, CDH2, ZNF569, MIR191, CDK9, MIR192, PRIMA1, CDKN2D, MIR196A1, OR2AG1, LAYN, PIWIL4, MIR19B1, GPBAR1, CDC25B, GDF7, ZDHHC15, MIR139, CD63, SGMS2, MIR140, TMPRSS6, RHBDL3, AVPR2, MIR15A, CBLL2, MIR186, PRUNE2, AMOTL1, CD81, MIR18A, SLC2A12, CDA, MIR181A2, ATP5PO, SESN3, ATP6V1B2, UBR1, ATP5PF, PPME1, MIR224, LYZL4, KCNH8, MTERF4, MIR23A, CPO, ACMSD, MIR23B, BHLHE23, MIR25, CFL1, OSCAR, SPNS2, SEZ6, SLC38A10, MSI2, CFTR, MIR27A, MIR223, ALDH7A1, MIR221, SELENOM, ATP5MC2, CACUL1, HECTD2, SREK1, CTCFL, CBLN4, CDSN, ATP5MC1, DEFB104A, OCIAD2, MAGEC3, MIR210, CEACAM5, CECR, PPARGC1B, ATP5F1B, IL31RA, GNPDA2, SCARB2, NSMCE1, SOCS4, UBE2L1, BNC1, CACNA1C, SLC25A20, SERPINA13P, BLM, SREK1IP1, MIF-AS1, C20orf203, SYPL2, ZNF763, CCL4L1, BID, BGN, ZSCAN1, ZADH2, SMIM20, MILR1, PGP, GOLGA6L2, TMEM189-UBE2V1, TMEM189, IL31, C4BPA, BTK, AMIGO2, HCN1, NHLRC2, ATP9B, SBSN, BMP1, OSTN, C5, CFAP410, BARHL2, NANOS3, C9, STING1, GADL1, ARMH1, VPS51, HCAR2, CAPG, LINC00639, TMEM201, LIN28B, CD5L, MIR127, CD19, KIF6, MS4A1, MS4A3, HYLS1, STOX1, FOLH1B, OR8J1, TRIML2, CD28, KHDRBS2, GAPT, CENPV, KLHDC8B, MIR134, CD86, PIKFYVE, SLC29A4, CCNC, KRIT1, PTF1A, DAOA-AS1, BDKRB2, HCA1, BRD3OS, ASPM, BCS1L, SGMS1, BCR, MIRLET7D, MIR122, RUNX1, ANKK1, BCL6, PHYHD1, BAK1, MIR10A, EBF3, CCKAR, MIR28, FRMD6, MIR613, ADAMTS10, TMEM175, XIAP, MAF1, BIRC3, SLA2, ANTXR1, ASCC2, CRHBP, EVA1A, QRFPR, MAGT1, NCALD, LOC646506, ROPN1L, L3MBTL2, GMNC, SCFV, CSE1L, RNF146, PHF6, HOOK3, BRSK1, MBOAT4, COX15, MIR497, MFSD2A, MIR501, ACCS, ARG2, FAM126A, CPB1, CPN1, ECSCR, MAP1LC3A, MIR484, AQP9, CPS1, SNORD35B, CPT1A, ABLIM2, FASLG, NETO1, DOCK8, RNFT2, C1QBP, TM2D3, ASRGL1, PTGES2, PANK2, MIR590, SCD5, CSNK2A1, VCAN, ZC3H14, CSPG4, CTBS, MIR592, MIR598, CAMKMT, CTNS, SLTM, PTCD2, CTNND1, MIR603, NUBPL, WDR26, SPHKAP, GSTT2B, TMEM163, NCF1, LBH, SPAG11A, SFTPA1, CSF3R, ZNF436, CSN2, NDFIP1, MIR545, SLC44A4, SLC19A3, FAM72B, AIRE, IQCJ, CSNK1G2, DNAJC5, CSNK1G3, LINGO1, ATG4C, ATP2B4, MIR346, SERPINC1, CISH, ASPA, CLC, UCN3, TMEM54, ASS1P1, CLCN3, NACC1, ASIP, STX1B, IFT43, MIR133B, MIR151A, CLK2, TP53INP1, MIR330, MIR335, MIR338, MIR93, SLC26A7, OMA1, CHGB, PLD4, TDRD9, CHD1, MIR299, ATIC, ATHS, H4-16, LRIG3, EXOSC6, CHRM3, MIR30B, MIR30E, AGAP2, MIR31, MIR34C, MIR9-1, GRIN3B, GRIN3A, ASAH1, MIR369, H2BC12, KPRP, LRSAM1, MIR429, H4C15, SHF, GADD45GIP1, COL11A1, ZNF628, MIR431, HNP1, NAV2, MIR409, SLC31A1, RPPH1, SNORD14E, SNORD14D, SNORD14C, SNORD14B, COX6B1, MIR485, CNTN1, DNM1P33, CNTFR, CHRDL1, CLN3, MIR361, MYOCD, PRDM6, DNER, SPECC1, MIR377, CLN5, EXOC3L4, CNP, MIR425, ARRB1, ZNF804A, BDNF-AS, NLRP12, CCR6, ABCC2, DEFB104B, LRRC15, POU3F4, HOOK1, TERC, NAT1, MCM2, EZR, MDH1, VEGFC, MDH2, MDM4, MEF2D, UVRAG, UROD, UQCRC1, UGT1A, SLC35A2, UCP2, UBTF, UBP1, MID1, UBE3A, UBE2V1, CXCL9, ATXN3, UBE2D2, UBE2A, UBC, MAP3K10, MC1R, WARS1, WAS, ZMYM2, MANF, SCG2, FZD5, SMAD7, MAG, MAP3K12, MAP1A, MAP1B, ZNF236, ZNF224, ZNF217, RNF112, WEE1, MZF1, ZIC1, MARS1, MAS1, MAT1A, MAT2A, MAZ, XIST, WT1, WNT2B, WNT5A, UBA52, KMT2A, MLLT3, THBS1, TLR3, TLE3, MSH3, TKT, TIMP4, TIMP3, MSR1, MSRA, THRA, THOP1, THBS4, CYTB, MRE11, NUDT1, TGFBI, TGFB3, ND1, TFF3, TFDP1, MTNR1B, MTRR, TRNL1, MUC1, TERF2, TSPAN7, MRC1, TWIST1, TRAF2, TUBA4A, NR3C2, TTN, TSPY1, TSHR, TSG101, TSC1, MMP7, MMP8, TRPC1, TRH, NR2C2, TNFAIP6, MMP13, TPM1, MOG, MOV10, TP53BP2, TP53BP1, MPG, TNR, TNNI3, MPI, MPST, SLBP, REEP5, DEK, SNX3, TNFRSF6B, RAB11A, LDHA, LEPR, LGALS4, LGALS9, GPAA1, RNMT, GBF1, ADAM19, URI1, TRADD, LCK, B3GALT4, LIFR, LIG1, SOCS1, NUMB, LIMS1, AOC3, PDE8B, USO1, TNFSF11, STK16, LCT, CES2, KMO, KLRC1, BRSK2, KCNQ1, KIR2DL2, NOL3, ATP6V0E1, SELENBP1, USP13, KLKB1, CDK5R2, RAB29, MBD2, KIF11, TMEM11, ENDOU, EIF2S2, TAX1BP1, NAE1, KRT14, KRT18, LAMC1, LBR, PROM1, LCAT, SOCS2, PRKRA, DEGS1, DDX39B, PABPN1, EOMES, BAP1, LTF, H4C9, COLQ, DYSF, CHAF1B, LYN, NRIP1, COIL, SLC7A5, AD5, H4C1, FGF23, ADAM12, BRD3, PSCA, ARHGEF5, TFPI2, FZD3, GHS, M6PR, MARCKS, SMAD1, LTC4S, H4C4, BHLHE40, IRS4, MAPKAPK5, LMO4, LOXL1, CST7, DDO, DGKZ, GAS7, PIK3R3, PKP4, PPFIA1, LRPAP1, SORBS2, H4C6, CUL4A, GNPAT, LTB, H4C14, H4C13, H4C5, H4C2, H4C8, H4C3, H4C11, H4C12, TERF1, MUC4, KCNMA1, TDO2, PCP4, CDK18, RBM3, RBL2, RBBP6, RB1, RASGRF1, RASA1, RARRES2, RAN, RAF1, RAD52, PCYT1A, RAD23B, RAC2, PDB1, RAB27B, RAB27A, PDC, PDE2A, PURA, PDGFB, ENPP2, PDYN, PTPN13, PC, PAX6, PARN, RPL15, P2RX1, S100A8, P2RX3, P2RX5, P2RY4, RREB1, RPS23, RPS21, RPS6KB2, P2RY6, RPS3A, RPL13, RET, RPA1, PAFAH1B2, RORA, ROM1, SNORD15A, BRD2, RNASE1, RHO, RHD, PAK3, TRIM27, PTPN11, PENK, PTN, PMM2, PLAUR, PLCL1, PLEK, PRKCE, PRKCD, PLP1, PRKAR1B, PRKACB, PRKACA, PLXNB1, PML, PRG2, PLAT, PMP22, PRB1, PPT1, PPP2R5E, POLG, PPP2R1A, PPP1CB, PPL, PPIC, PPIB, POR, MAP2K3, PLAG1, PFDN5, PSMD2, PFKFB3, PTGER2, PTGER1, PGD, PTGDR, PTCH1, PGR, PHB, PSMD9, PSMD7, PSMD3, PSMB2, PITX2, SERPINE2, SERPINI1, KLK10, PIK3C3, PIK3R1, KLK7, PIK3R2, PRS, PROS2P, PIN4, PROC, S100A10, OXT, OXA1L, SQLE, STAR, ST14, ST2, NDUFA6, SSTR3, SSTR2, NDUFA9, NDUFB8, SRM, SRF, NEDD4, SEPTIN2, STC1, SP4, NEU1, SOX5, SOX3, SOS2, SOS1, SOD3, NFE2L1, NFKB2, SNRPG, SNRNP70, NDUFA5, STIM1, NME1, MYH9, TRBV20OR9-2, TCP1, TCN2, MMUT, MUTYH, TCF4, ELOC, TBX2, MX1, MYH6, TARBP2, MAP3K7, STK11, MYO6, TACR2, NACA, VAMP2, VAMP1, SURF1, ABCC8, SUOX, NDP, STXBP1, STX1A, NINJ2, NME2, SAA2, OMP, SFTPC, TRA2B, SRSF6, SRSF5, SRSF3, SRSF1, SFRP1, OCA2, MAP2K4, ODF1, SELE, OPA1, OAS3, CXCL11, CCL21, CCL20, CCL19, CCL8, CCL1, SCP2, SCN1A, ORM2, OSM, TSPAN31, OAT, SGSH, SUMO2, SLC8A3, SMPD2, SLN, SLIT3, SLC22A5, SLC22A2, NQO2, SLC18A1, SLC16A1, SLC12A3, SLC11A1, SLC10A2, SLC8A1, NUP98, SLC6A12, NPHP1, SLC6A1, SLC5A2, NRCAM, NRDC, SLC1A1, NRF1, PMEL, YBX1, NT5E, NAT8, SEMA5A, ESRRA, RAB31, MACF1, HEY2, BRD4, TRAM1, CBX5, ANGPTL2, OPN1MW, GRIP1, KCNH4, MSTN, GFER, GFRA1, SIRT5, COTL1, GFRA3, GHR, ZNF629, UBR4, GHSR, WASHC4, GLI1, NUP160, CLUH, GLI3, SCFD1, KCTD2, CLCF1, SEC14L2, NR5A1, SLC24A2, PRPF6, TFIP11, MAFF, EID1, RAB38, FSHR, TMEFF2, PLA2G15, SLC7A11, FTH1, SNHG1, TSPAN15, GAST, ACKR1, G6PD, GAB1, NTSR2, GABBR1, GAD2, GALNS, DDAH1, PADI4, GART, NBEAL2, GMPR, PLEKHM2, WDHD1, GPR39, CARD8, ARHGEF15, AAK1, SYNPO, GPX4, ECD, PARK7, KLF8, TREX1, WIF1, WDR45, ATF6, CORO1A, TBC1D8, SLC7A9, GRIA4, FAF1, GRIK4, RER1, GRM1, STMN2, RAPGEF4, ADRM1, MSRB2, RAB3GAP1, GNA12, ZNF423, ATG4B, UBXN4, RCOR1, SEPTIN8, STAB1, GNAI1, MAPK8IP3, GRAMD4, GNB3, NFASC, KIF1B, GOLGA2, GPER1, SETX, GOLGA4, PDZD2, SAMD4A, KDM1A, GPM6A, RAB21, GPR6, P2RX2, GPR20, SNW1, FRK, FBXO7, BRI3, SNX12, SOCS7, CD209, FABP6, TBX21, NOP53, FABP7, SLC2A8, UBQLN2, A1CF, PSAT1, FANCG, HOOK2, DELEC1, FASN, SNX8, NPC1L1, BLNK, MS4A2, FCGR1A, FCGR2A, MYLIP, SCG3, DROSHA, TMEM230, NOX4, PCA3, SPCS1, VRK3, ETFA, SLC25A37, TLR8, SLC22A17, ECSIT, CD320, EZH2, DNAJC27, CLEC1B, NT5C3A, MZB1, F2RL2, HP1BP3, F3, F9, IRAK4, SAR1B, UTP11, F13B, SH3GLB1, SIDT2, SHANK1, TRAT1, F11R, RGCC, TMEM176B, NOCT, FBXO2, NPTN, TPK1, VCX, GREM1, FKBP1AP2, FKBP1AP3, AGO1, SEZ6L2, FKBP1AP4, FGF21, CLDN17, NOC2L, SND1, FOXM1, EPC2, ATRNL1, LRP10, FLNB, FMR1, POU2F3, TXN2, FBXL2, FOLR1, FOLR2, FKBP1AP1, B3GAT1, IGHV1-68, TNFRSF21, FEB1, FES, FGF9, RABGEF1, PRPF19, FGF13, FGFR1, FGFR4, PDLIM3, RND1, KLHL20, COQ2, CACYBP, HCAR1, FHL2, VPS4A, IL37, GLS2, NAAA, CYTH4, DKK2, DKK3, BBC3, SDCBP2, GRM3, IL24, SPAG9, CXCL2, PCLAF, IGFBP1, ACAP1, IGFBP5, SART3, KDM4A, IGHG3, SDC3, SH3PXD2A, RGS6, SNCAIP, TCL1B, IL4R, IL7, BCAR1, CCL4L2, CXCR1, BAG2, IL12B, BAG5, TMEM59, TBPL1, GAL3ST1, AKAP5, STX8, PIEZO1, BMS1, PTDSS1, IDH1, SH2D3C, GNE, SH2B3, IRF8, SRA1, TANK, KCNE3, HNRNPDL, CCS, NUP153, MED12, HS3ST1, TOMM20, RBM8A, IDH2, CFI, SV2B, TECPR2, IFI27, TOMM70, KIAA0319, IFIT3, IFNAR1, IGBP1, INSRR, PCYT1B, IL27RA, CCNE2, NOLC1, TIAF1, JUND, ZMYM3, KCNC4, KCNJ13, HGS, SYNGR3, ATG12, CBFA2T2, RABEP1, P2RX6, RAB11B, SLC16A3, SLC16A4, ATP6V0D1, RGN, USP10, USP2, USP14, DNAJA3, CLDN1, CLDN8, ARTN, JUNB, GPR50, PICK1, ITPKB, IRAK1, HOMER2, IREB2, IRF3, IRF6, IRF7, ITGAV, CYP7B1, ITGB3, NRXN1, SLC22A8, ITPR2, AIMP1, JAG2, ITGBL1, LHX2, SLIT2, TAOK2, CD163, JAK2, GPR37L1, PIWIL1, MAPKAPK2, PDLIM7, IARS1, TNC, IL18BP, CCT2, HCK, NRG3, USP39, DCTN6, CD226, HDC, HDLBP, CAMKK2, HIP1, TXNRD2, NPC2, SLC35A1, HBG2, PRDX4, ZNRD2, HYOU1, HLA-DQA1, SEMA4D, HLA-DQB1, NXF1, HLA-DRA, ATP5PD, COG5, GPNMB, CHL1, HAS3, FAM3C, GYPA, GSK3A, COPS5, GSM1, GSTM2, EBNA1BP2, PRSS21, PRDX3, GSTZ1, GTS, GUSB, C1QL1, GYPB, HAS1, GYPC, CCL27, ALDH1L1, GYPE, HAGH, WASF3, HSPH1, GJB6, HARS1, ARPP19, DHS, HLA-DRB4, HLA-G, PQBP1, MPHOSPH6, STAM2, GLYAT, HOXA@, RABEPK, HPCA, CALCOCO2, HRC, OLIG2, DDX39A, TOPORS, EIF1, HSD17B1, RAMP2, WASF2, HSD17B4, HSPA2, HSP90AB1, DNAJB1, NAMPT, BCAP31, CTDSP2, TSPAN3, ACTR2, CERT1, ABCC4, HNRNPK, HMBS, NPM3, CFDP1, PRMT5, HMGB2, YAP1, IFITM3, SPAG11B, PEMT, RACK1, SYCP2, TUBB4B, SEMA3A, WARS2, HNRNPC, FOXA1, CCL26, TLR6, FOXA2, LAMC3, LANCL1, HNF4A, TCIRG1, APBB3, APC2, HNRNPA2B1, MTCH2

-
Wrongful Abortion
Wikipedia
Wrongful birth is a more accurate mirror image of wrongful abortion. [8] The former deals with the non-prevention of the birth of an unwanted child, whereas the latter deals with the prevention of the birth of a wanted child. ... Wrongful Abortion: A Wrong in Search of a Remedy , Yale Journal of Health Policy, Law and Ethics 507 (2005). ^ Baker v. Gordon, 759 S.W.2d 87 (Mo. Ct. App. 1988) (available through LexisNexis Archived 2011-02-04 at the Library of Congress Web Archives and Westlaw ) ^ Appel, J. ... Medicine and Health, Rhode Island . 87 (2): 55–8. PMID 15031969 . ^ Johnson v. United States, 810 F. Supp. 7 (D.D.C. 1993) ^ Breyne v. Potter, 574 S.E.2d 916 (Ga. Ct. App. 2002) ^ Martinez v. Long Island Jewish Hillside Med. ... Medicine and Health, Rhode Island . 87 (2): 55–8. PMID 15031969 . Perry, Ronen & Adar, Yehuda.
-
Cerebral Amyloid Angiopathy, App-Related
OMIM
A number sign (#) is used with this entry because cerebral amyloid angiopathy (CAA) can be caused by mutation in the gene encoding the amyloid precursor protein (APP; 104760). Mutations in the APP gene can also cause autosomal dominant Alzheimer disease-1 (AD1; 104300), which shows overlapping clinical and neuropathologic features. ... Revesz et al. (2003) reviewed the pathology and genetics of APP-related CAA and discussed the different neuropathologic consequences of different APP mutations. ... In 4 affected members of an Italian family with cerebral amyloid angiopathy, Obici et al. (2005) identified a mutation in the APP gene (104760.0019). In 2 brothers from an extensive Iowa kindred with progressive dementia and cerebroarterial amyloidosis, Grabowski et al. (2001) identified a heterozygous mutation in the APP gene (N694D; 104760.0016). ... Human APP mRNA was detected in neurons and neuronal processes, but not in vessel walls. ... Herzig et al. (2006) extended their earlier studies by developing several murine models of APP-related CAA and APP-related parenchymal amyloid deposition.
-
Problematic Smartphone Use
Wikipedia
For 3-4 year-old children: 180 minutes physical activity, 1 hour screen time, 10–13 hours of sleep time per day. [81] Phone settings [ edit ] Many smartphone addiction activists (such as Tristan Harris) recommend turning one's phone screen to grayscale mode, which helps reduce time spent on mobile phones by making them boring to look at. [82] Other phone settings alterations for mobile phone non-use included turning on airplane mode, turning off cellular data and/or Wi-Fi, turning off the phone, removing specific apps, and factory resetting. [83] Phone apps [ edit ] German psychotherapist and online addiction expert Bert te Wildt recommends using apps such as Offtime and Menthal to help prevent mobile phone overuse. [84] In fact, there are many apps available on Android and iOS stores which help track mobile usage. ... In Android a similar feature called "digital wellbeing" has been implemented to keep track of cell phone usage. [85] These apps usually work by doing one of two things: increasing awareness by sending user usage summaries, or notifying the user when he/she has exceeded some user-defined time-limit for each app or app category. ... The researchers implement an Android app that combined these three intervention types and found that users reduced their time with the apps they feel are a poor use of time by 21% while their use of the apps they feel are a good use of time remained unchanged. [86] AppDetox allows users to define rules that limit their usage of specific apps. [87] PreventDark detects and prevents problematic usage of smartphones in the dark. [88] Using vibrations instead of notifications to limit app usage has also been found to be effective. [89] Further, researchers have found group-based interventions that rely on users sharing their limiting behaviors with others to be effective. [90] Bans on mobile phone use [ edit ] See also: Mobile phone use in schools In some places in the world the use of mobile phones was banned in classes during instructional time, for example, in France , Ontario . ... International Journal of Infection Control . 8 (2). doi : 10.3396/ijic.v8i2.014.12 . ^ World Health Organization: International Agency for Research on Cancer (2011). ... "Psychological Predictors of Problem Mobile Phone Use". Cyberpsychology & Behavior . 8 (1): 39–51. CiteSeerX 10.1.1.563.385 . doi : 10.1089/cpb.2005.8.39 .

-
Hereditary Cystatin C Amyloid Angiopathy
Wikipedia
"Stroke in Icelandic patients with hereditary amyloid angiopathy is related to a mutation in the cystatin C gene, an inhibitor of cysteine proteases" . J. Exp. Med . 169 (5): 1771–8. doi : 10.1084/jem.169.5.1771 . PMC 2189307 . ... External links [ edit ] Classification D ICD - 10 : E85.4+ I68.0* OMIM : 105150 External resources Orphanet : 85458 v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2
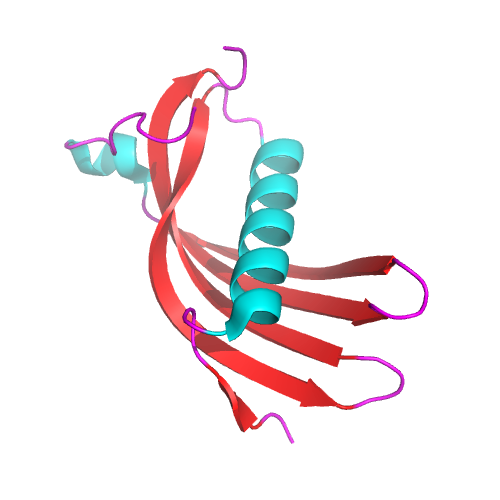
-
Amblyopia
Wikipedia
John Libbey Eurotext. ISBN 978-2-7420-1482-8 . Archived from the original on 8 September 2017 . ... "Conventional occlusion versus pharmacologic penalization for amblyopia" . Cochrane Database Syst Rev . 8 (8): CD006460. doi : 10.1002/14651858.CD006460.pub3 . ... The British Journal of Ophthalmology . 86 (8): 915–9. doi : 10.1136/bjo.86.8.915 . ... Neurorehabilitation and Neural Repair . 27 (8): 760–9. doi : 10.1177/1545968313491006 . ... "Amblyopia therapy: an update". Strabismus . 19 (3): 91–8. doi : 10.3109/09273972.2011.600421 .PPARGC1A, FGFR3, ACVRL1, AP4E1, SMCHD1, PUF60, AP4S1, YME1L1, AP4B1, TUBB3, WARS2, MAFB, CHST3, ARHGEF2, AP4M1, OGT, TWIST1, TRIO, TFAP2A, ADNP, PIGN, PHOX2A, B3GAT3, NPHP3-ACAD11, LAMA1, TSEN54, NPHP4, IRAK1BP1, MTFMT, LOXL3, COL25A1, TRIM8, WDR26, UBA5, XYLT2, PHIP, P4HTM, NDUFB11, ADAMTSL4, GMPPB, TBX1, SMARCB1, PTPN11, COL9A1, DPP6, CRYGC, CRYAA, COX7B, COL11A1, COL9A3, COL9A2, COL4A1, ERF, LYST, CHN1, CBS, CACNA1F, CACNA1E, CACNA1A, C1QBP, ENG, EPRS1, FBN1, LIM2, PITX2, PAX6, OCRL, GPR143, KMT2A, SMAD4, KIF11, HCCS, H1-4, GNAS, GDF2, GALC, FGFR2, VIP, MBD5, IRF1, PLXNA2, RBM19, CRYBB1, BCHE, COMETT

-
Cognitive Deficit
Wikipedia
Cognitive deficit Other names Cognitive impairment Specialty Psychiatry Cognitive deficit is an inclusive term to describe any characteristic that acts as a barrier to the cognition process . [1] The term may describe deficits in overall intelligence (as with intellectual disabilities ), specific and restricted deficits in cognitive abilities (such as in learning disorders like dyslexia ), neuropsychological deficits (such as in attention , working memory or executive function ), or it may describe drug-induced impairment in cognition and memory (such as that seen with alcohol , glucocorticoids , [2] and the benzodiazepines . [3] ) Contents 1 Cause 2 Screening 3 Treatment 4 Other findings 5 See also 6 References 7 Further reading 8 External links Cause [ edit ] It usually refers to a durable characteristic, as opposed to altered level of consciousness , which may be acute and reversible. ... As the maximum cognitive ability that we are able to achieve decreases, it may not actually affect our daily lives, which only require the normal level. [8] Some studies have indicated that childhood hunger might have a protective effect on cognitive decline. ... J Psychiatr Res . 35 (3): 127–45. doi : 10.1016/s0022-3956(01)00018-8 . PMID 11461709 . ^ Kalachnik, JE.; Hanzel, TE.; Sevenich, R.; Harder, SR. ... "Screening for Cognitive Impairment in Older Adults: US Preventive Services Task Force Recommendation Statement" . JAMA . 323 (8): 757–763. doi : 10.1001/jama.2020.0435 .PSEN1, MAPT, APP, APOE, COMT, FMR1, GSK3B, BCHE, PTGS2, DRD2, EPO, CDK5R1, VIP, FGF14, MAOA, EIF2S1, SLC1A1, SLC10A2, MET, DRD3, SMAD4, LAMB2, TSC1, AGT, ZNF41, RAB40AL, SLC51A, NR0B2, IGF2, OTC, SLC51B, CRH, ABCC4, FOSB, OXTR, SLC4A10, AFF4, SLC6A4, CYP2D6, ACHE, DYRK1A, BDNF, PVALB, DISC1, NFE2L2, CHRNA4, BACE1, PIK3CB, PIK3CA, PIK3CD, PIK3CG, SIRT1, GABPA, SNCA, TNF, MTOR, HTT, RELN, DTNBP1, CFP, CDK9, KL, AKT1, NUFIP2, VEGFA, GBA, GRN, GRM5, ACTB, CNR1, CSF2, DLG4, MME, IL6, SYP, LAMC2, DMD, IL4, IL1B, NGF, PREP, PERCC1, MIR34A, PDE4A, PDE2A, EPHB2, FOXP1, MAPK1, TARDBP, SNAP25, ADIPOQ, FUS, HTR2A, NRG1, TCF4, TGFB1, HDAC6, GRM2, GRM3, SUCLA2, ABCA1, CREB1, CDK5, CASP6, CACNA1C, ADAM10, CRP, H3P40, NUBP1, NFKB1, RUNX1T1, NGFR, DCTN4, ASCC1, NOTCH3, MECP2, NTS, UBQLN2, AGTR1, POLDIP2, CD36, CD40, CASP1, AKR1C4, HMGB1, LRRK2, CHRNA7, NLRP3, IGF1, CHRNA5, COASY, SHROOM4, IL10, EHMT1, INSR, CIP2A, CDR1, LEP, RNF19A, PLA2G1B, NUP62, HDAC2, SPP1, SPTBN1, GRAP2, LGI1, TM7SF2, SQSTM1, ARHGEF7, SYNJ1, APLN, UBE3A, BECN1, VCAM1, TP63, VSNL1, YWHAZ, GTF2IRD1, SLC6A8, ANGPT1, PTPN11, PTPA, CALB1, MAPK8, SYNM, PTEN, HRH3, REST, SLC1A2, S100B, NRG3, SCN1A, KHDRBS1, AHSA1, SIGMAR1, TAAR1, TREM2, AIMP2, GAD1, GTF2H1, DAB1, GRIA1, CRK, GH1, MIR132, GFAP, FGFR3, ADCYAP1, CREBBP, CUX1, DBH, CX3CR1, CYBB, ERBB4, DBN1, NR3C1, PLB1, MAPK14, CHL1, MIR361, NPS, SNORD116@, CXCR6, SPAG11A, SFTPA1, PDE10A, TXNRD2, BAIAP2, SPAG11B, TUBA1B, RTN3, MIR574, GJB6, ADAMTS13, PPARGC1A, MIR342, ALDH2, SIRT3, ADNP, MED13L, CRTC1, MIR21, AGTPBP1, ASTN2, MDD1, ARC, PHF8, NMNAT2, VN1R17P, GPR166P, FAM107A, PSIP1, IL1RAPL1, OLIG2, SDS, EHMT2, GADD45G, AMPH, SETD1A, OPN1MW2, PSME3, OCLN, FPGT-TNNI3K, OPN1MW3, CBSL, ANXA1, HDAC3, SYNGAP1, ADA, APOA1, UPK3B, MTCO2P12, PDE5A, NCOA1, H3P45, PSMG1, KHSRP, NCK2, APOB, FZD4, ANP32A, AP3B2, EIF2S2, NOL3, CDKL2, NTN1, KEAP1, NCS1, HDAC9, ABCG1, CCR2, PTGES, HOMER2, AD10, GS1-600G8.3, C20orf181, SEMA5A, NRXN1, ANK3, ANPEP, TGM5, SRSF11, NOG, ECT, LPAR2, USP2, MIR188, OR2AG1, MIR181C, ALB, CALHM1, SPG11, PCSK9, TNFAIP8L2, PLEKHF1, LYNX1, DCLRE1C, LGR6, ACE2, NGB, PCDH10, SEMA6A, MRGPRX1, ADRA2B, PCDH19, KIDINS220, COPD, ADRB1, PCBP4, ACKR3, NPAS4, GPRC6A, DIPK2A, SRCIN1, ADCYAP1R1, GPR151, VPS13B, HSPB6, ADRA1A, DEGS2, OXER1, MRGPRX4, MRGPRX3, OMA1, KLHDC8B, TRAPPC9, CDCA5, FOXP2, CHRDL1, SLC39A13, CHRFAM7A, PTPN5, C9orf72, ADRA2A, KLF16, PAG1, NSUN5, CCDC141, AUTS2, IRF2BP2, RTL1, IGHD4-11, TMEM97, SRPX2, AGER, AHR, SIGLEC7, HES5, NECTIN3, DNAAF3, MIR137, TPSG1, SLC24A2, BRD1, FRRS1L, PDIK1L, AIF1, LPAR3, MAPK8IP2, HDGFL3, TNNI3K, CHD7, FAM3B, SMPD3, CCDC91, RCBTB1, CC2D1A, PGPEP1, AHI1, TMEM106B, EPM2A, NANS, SETD4, GEN1, GOLGA6A, CINP, HOOK1, ZDHHC3, MCIDAS, NRN1, CD320, CRBN, TUBB2B, MIR181A2, DCX, LRP8, IFIT1, IL18, IL13, IL12A, CHAT, IL2RB, CHI3L1, IKBKB, IGFALS, IFNG, IFNA13, IFNA1, IAPP, IRS1, HTR4, HTR2C, HTR1A, HSPA4, PRMT2, AGFG1, HPRT1, HOXA1, HMOX1, HIF1A, CLCN3, IDO1, ITGAM, CLU, MDM2, RNR2, COX2, COX1, MS, MPO, CD200, MNAT1, MMP9, MMP2, CBS, MAP3K5, MAOB, ITPR1, CDH1, CAPRIN1, LIMK1, LGALS9, LGALS3, LCN2, CEACAM4, KCNQ2, KCNJ6, JAK2, ITPR3, HDAC1, HCRT, FAS, CYP1A2, FGFR1, CXADR, FES, FDPS, FCN2, FAT1, PTK2B, FABP5, FABP3, ETS2, ESR1, EPHA4, FOXO3, ENO2, EIF4E, EIF2S3, EGR1, EDNRA, EDN1, TOR1A, DAO, DUSP2, DUSP1, DRD4, FOXO1, AFF2, HCLS1, GLUL, HCFC1, HARS1, GZMH, GTF2I, KLF6, GRPR, GRP, CORT, GRIN2B, GRIA2, GPR42, GLI3, FOLR1, GJB2, GIP, MSTN, GDF2, OPN1MW, GCHFR, GCG, FYN, FUT1, CST3, CTRL, MTRR, CEACAM6, NEUROD1, BRCA1, BMI1, STAT3, SSTR4, SST, SREBF1, SOX4, SOD1, SOAT1, BMP1, BMP2, SMARCA4, BRAF, ATP5F1A, SLC2A3, BRS3, SFRP1, SETMAR, SET, SELP, CX3CL1, CCL11, CCL2, SCN8A, SCN2A, TAT, TCF3, NF1, UBC, XPNPEP1, CLIP2, WAS, TRPV1, VLDLR, AQP4, AR, VDR, VCP, UFD1, SUMO1, TXN, TCN2, ARSD, TPT1, TPM2, TNFRSF1B, ASL, TLR4, TLR2, TJP1, THY1, DRD1, TDO2, ATXN1, TSPAN31, S100A1, PAFAH1B1, CALB2, CAMK4, CAPN1, CAST, PEPD, PDE9A, PDE4D, CASP2, CASP3, PAK3, SERPINE1, CAT, RXRA, ORM1, OPRL1, OPHN1, NTRK1, NRGN, NOVA2, NOTCH1, NOS2, NOS1, NMB, CBR1, PITX2, PLAT, PLD2, PNOC, RPS27A, RPS6KB1, OPN1LW, RBM3, RARB, MOK, RAC1, BUB1B, PTPRG, ACAT1, PTGS1, PTGER3, PTGER2, PSMC1, PSEN2, C5AR1, PRNP, MAPK10, MAPK3, CAD, PRKCA, PRCP, PPARA, ATP1A3
-
Trichotillomania
Wikipedia
Other individuals may have focused, or conscious, rituals associated with hair pulling, including seeking specific types of hairs to pull, pulling until the hair feels "just right", or pulling in response to a specific sensation. [8] Knowledge of the subtype is helpful in determining treatment strategies. [8] Treatment [ edit ] Treatment is based on a person's age. ... Several mobile apps exist to help log behavior and focus on treatment strategies. [38] There are also wearable devices that track the position of a user's hands. ... Expert Review of Neurotherapeutics . 11 (8): 1165–1174. doi : 10.1586/ern.11.93 . ... The Journal of Clinical Psychiatry . 57 Suppl 8: 42–7, discussion 48–9. PMID 8698680 . ^ Keuthen NJ, Makris N, Schlerf JE, et al. (2007). ... "A double-blind comparison of clomipramine and desipramine in the treatment of trichotillomania (hair pulling)". N. Engl. J. Med . 321 (8): 497–501. doi : 10.1056/NEJM198908243210803 .
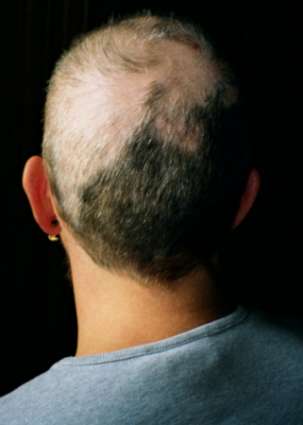
-
Endogenous Depression
Wikipedia
Since symptoms are due to a biological phenomenon, prevalence rates tend to be higher in older adults. [7] Due to this fact, biological-focused treatment plans are often used in therapy to ensure the best prognosis. [2] Contents 1 Signs and symptoms 2 Risk factors 3 Treatment 4 Prevalence 5 History 6 See also 7 References 8 External links Signs and symptoms [ edit ] The forefront indication that a depressive episode is manifesting is the sudden loss of energy or motivation in daily routines. [8] [9] When this occurs, it is not uncommon for individuals to seek medical attention with excessive worrying or anxiety that a more severe, physiological disease may be the underlying issue. [8] However, without an actual disease present, this neurotic thinking often results in severe anxiety, sleep disturbance, and mood swings which may hinder social relationships. Individuals with endogenous depression may experience inconsistencies in symptom severity [10] which is often the reason for delayed treatment. [8] If left untreated, symptoms may progress to a major depressive episode.TPH2, DISC1, CRH, POMC, ADORA2A, PTGS2, MIF, MTHFR, NOS1, NPY, NPY1R, PDCD2, PON1, RELN, SELP, S100A10, ALB, SLC6A4, SNAP25, SOD1, TG, PDCD6, CADM1, GHRL, DIXDC1, KCNK2, IL6, IFNA2, FTH1, APP, BDNF, CNR2, CREBBP, CSF3, DRD5, ACSL4, HTR7, FGFR1, FGF2, FTL, GLO1, GNAS, GNB1, NR3C1, GSK3A, GSK3B, HAP1, MORC1, DBH, COMT, HLA-B, HLA-C, ECT
-
Color Blindness
Wikipedia
Specialty Ophthalmology Symptoms Decreased ability to see colors [3] Duration Long term [3] Causes Genetic ( inherited usually X-linked ) [3] Diagnostic method Ishihara color test [3] Treatment Adjustments to teaching methods, mobile apps [1] [3] Frequency Red–green: 8% males, 0.5% females (Northern European descent) [3] Color blindness ( color vision deficiency ) is the decreased ability to see color or differences in color . [3] It can impair such tasks as selecting ripe fruit, choosing clothing, and reading traffic lights. [3] Color blindness may make some educational activities more difficult. [3] However, problems are generally minor, and most color-blind people adapt. [3] People with total color blindness (achromatopsia) may also be uncomfortable in bright environments [3] and have decreased visual acuity . ... Non-color-blind females can carry genes for color blindness and pass them on to their children. [3] Males only have one X chromosome and therefore always express the genetic disorder if they have the recessive gene. [3] Color blindness can also result from physical or chemical damage to the eye , the optic nerve , or parts of the brain . [3] Diagnosis is typically with the Ishihara color test ; other methods include genetic testing . [3] [4] There is no cure for color blindness. [3] Diagnosis may allow a person's teacher to change the teaching method to accommodate the handicap. [1] Special lenses may help people with red–green color blindness in bright light. [3] Mobile apps can help people identify colors. [3] Red–green color blindness is the most common form, followed by blue–yellow color blindness and total color blindness. [3] Red–green color blindness affects up to 8% of males and 0.5% of females of Northern European descent. [3] [5] The ability to see color also decreases in old age. [3] In certain countries, color blindness may make people ineligible for certain jobs, [1] such as those of aircraft pilots , train drivers , crane operators , and people in the armed forces . [1] [6] The effect of color blindness on artistic ability is controversial. [1] [7] The ability to draw appears to be unchanged, and a number of famous artists are believed to have been color blind. [1] [8] Contents 1 Signs and symptoms 2 Causes 2.1 Genetics 2.2 Other causes 3 Mechanism 4 Diagnosis 4.1 Classification 4.1.1 Red–green color blindness 4.1.2 Blue–yellow color blindness 4.1.3 Total color blindness 5 Management 5.1 Lenses 5.2 Apps 6 Epidemiology 7 History 8 Society and culture 8.1 Design implications 8.2 Occupations 8.3 Driving 8.4 Piloting aircraft 8.5 Art 8.6 Rights of the color blind 8.6.1 Brazil 8.6.2 United States 9 Research 10 See also 11 References 12 Further reading 13 External links Signs and symptoms [ edit ] Simulation of the normal (above) and dichromatic (below) perception of red and green apples Horizontal traffic light in Halifax, Nova Scotia, Canada In almost all cases, color blind people retain blue–yellow discrimination, and most color blind individuals are anomalous trichromats rather than complete dichromats. ... Men do not have a second X chromosome to override the chromosome that carries the mutation. If 8% of variants of a given gene are defective, the probability of a single copy being defective is 8%, but the probability that two copies are both defective is 0.08 2 , i.e. 0.64%. ... People in whom deuteranopia or deuteranomaly manifest are sometimes referred to as deutans, those with protanopia or protanomaly as protans. [43] Since deuteranomaly is by far the most common form of red–green blindness among men of northwestern European descent (with an incidence of 8%) it follows that the proportion of carriers (and of potential deuteranomalous tetrachromats) among the females of that genetic stock is 14.7% (i.e. 92% × 8% × 2), based on the Hardy–Weinberg principle . [41] Illustration of the distribution of cone cells in the fovea of an individual with normal color vision (left), and a color blind (protanopic) retina. ... As of 2012, sunglasses that notch out color wavelengths are available commercially. [57] Apps [ edit ] Many mobile and computer applications have been developed to help color blind individual to view better differentiate between colors.CNGA3, PDE6H, GNAT2, CNGB3, ATF6, G6PD, F9, PTCH1, RET, TAS2R38, CCDC6, SART3, AIFM1, SND1, TPX2, PAG1, PCBP4, NAA50, AMN, DYRK3, OPN1SW, PMEL, OPN1LW, OPA1, OAS3, NFKB2, MYP1, INS, IK, EIF4E, CUX1, CREBBP, ASCC2

-
Equine Protozoal Myeloencephalitis
Wikipedia
There are currently three FDA approved treatments available in the US: ReBalance ( sulfadiazine and pyrimethamine ), [7] [8] Marquis ( ponazuril ), [9] and Protazil ( diclazuril ). ... The Journal of Parasitology . 77 (2): 212–8. doi : 10.2307/3283084 . JSTOR 3283084 . ... Differentiation of Sarcocystis neurona from eight related coccidia by random amplified polymorphic NA assay. J Molec Cellular Probes 8:353-356, 1994. Hamir AN, Moser G, Galligan DT, et al. ... S P Ellison, T Kennedy, KK Brown. 4, 2003, J App Res Vet Med, Vol. 1, pp. 318–327 Experimental infection of horses with S. neurona merozoites as a model for Equine Protozoal Myeloencephalitis. ... P., Greiner, E., Brown, K K., Kennedy, T. 2, 2004, J App Res Vet Med, Vol.2, pp. 79–89. Characterization of a Sarcocystis neurona isolate from a Missouri horse with equine protozoal myeloencephalitis.

-
Human Genetic Enhancement
Wikipedia
Also changes in the mystatin gene [8] may alter appearance. Behavior [ edit ] Further information: Behavioural genetics Further information: Genetics of aggression Behavior may also be modified by genetic intervention. [9] Some people may be aggressive, selfish, ... and may not be able to function well in society. [ clarification needed ] There is currently research ongoing on genes that are or may be (in part) responsible for selfishness (i.e. ruthlessness gene , aggression (i.e. warrior gene ), altruism (i.e. ... "Current anti-doping policy: a critical appraisal" . BMC Medical Ethics . 8 (1): 2. doi : 10.1186/1472-6939-8-2 . ... "Long-term neprilysin gene transfer is associated with reduced levels of intracellular Abeta and behavioral improvement in APP transgenic mice" . BMC Neuroscience . 9 : 109. doi : 10.1186/1471-2202-9-109 . ... "Adeno-associated Viral (AAV) Serotype 5 Vector Mediated Gene Delivery of Endothelin-converting Enzyme Reduces Aβ Deposits in APP + PS1 Transgenic Mice" . Molecular Therapy . 16 (9): 1580–1586. doi : 10.1038/mt.2008.148 .

-
Cerebral Amyloid Angiopathy, Cst3-Related
OMIM
By 1986, the Icelandic experience included 128 affected members in 8 families originating from the same geographic area of Iceland (Jensson et al., 1986). ... A critical piece of evidence indicating a difference between the 2 diseases is the finding by van Duinen et al. (1987) that in the Dutch form of the disease the vascular amyloid deposits have immunohistochemical characteristics of Alzheimer disease-related beta-protein (APP; 104760). Molecular Genetics In Icelandic patients with hereditary cerebral hemorrhage with amyloidosis, Abrahamson et al. (1987) identified a heterozygous mutation in the CST3 gene (L68Q; 604312.0001). Palsdottir et al. (1988) identified this mutation in affected members of 8 families, establishing incontrovertibly that the mutation is the cause of this disorder.CST3, ITM2B, APP, CTSD, APOE, ACTB, PTPRC, TSHZ1, SYP, SPP1, SDC2, HSPG2, SMAD2, ITGAX, GFAP, FN1, FAP, CD68, HSPB8











