-
Childhood Cataract
Wikipedia
Further reading [ edit ] Childhood cataracts at NHS Choices Cataracts in Children, Congenital and Acquired at EyeWiki v t e Diseases of the human eye Adnexa Eyelid Inflammation Stye Chalazion Blepharitis Entropion Ectropion Lagophthalmos Blepharochalasis Ptosis Blepharophimosis Xanthelasma Ankyloblepharon Eyelash Trichiasis Madarosis Lacrimal apparatus Dacryoadenitis Epiphora Dacryocystitis Xerophthalmia Orbit Exophthalmos Enophthalmos Orbital cellulitis Orbital lymphoma Periorbital cellulitis Conjunctiva Conjunctivitis allergic Pterygium Pseudopterygium Pinguecula Subconjunctival hemorrhage Globe Fibrous tunic Sclera Scleritis Episcleritis Cornea Keratitis herpetic acanthamoebic fungal Exposure Photokeratitis Corneal ulcer Thygeson's superficial punctate keratopathy Corneal dystrophy Fuchs' Meesmann Corneal ectasia Keratoconus Pellucid marginal degeneration Keratoglobus Terrien's marginal degeneration Post-LASIK ectasia Keratoconjunctivitis sicca Corneal opacity Corneal neovascularization Kayser–Fleischer ring Haab's striae Arcus senilis Band keratopathy Vascular tunic Iris Ciliary body Uveitis Intermediate uveitis Hyphema Rubeosis iridis Persistent pupillary membrane Iridodialysis Synechia Choroid Choroideremia Choroiditis Chorioretinitis Lens Cataract Congenital cataract Childhood cataract Aphakia Ectopia lentis Retina Retinitis Chorioretinitis Cytomegalovirus retinitis Retinal detachment Retinoschisis Ocular ischemic syndrome / Central retinal vein occlusion Central retinal artery occlusion Branch retinal artery occlusion Retinopathy diabetic hypertensive Purtscher's of prematurity Bietti's crystalline dystrophy Coats' disease Sickle cell Macular degeneration Retinitis pigmentosa Retinal haemorrhage Central serous retinopathy Macular edema Epiretinal membrane (Macular pucker) Vitelliform macular dystrophy Leber's congenital amaurosis Birdshot chorioretinopathy Other Glaucoma / Ocular hypertension / Primary juvenile glaucoma Floater Leber's hereditary optic neuropathy Red eye Globe rupture Keratomycosis Phthisis bulbi Persistent fetal vasculature / Persistent hyperplastic primary vitreous Persistent tunica vasculosa lentis Familial exudative vitreoretinopathy Pathways Optic nerve Optic disc Optic neuritis optic papillitis Papilledema Foster Kennedy syndrome Optic atrophy Optic disc drusen Optic neuropathy Ischemic anterior (AION) posterior (PION) Kjer's Leber's hereditary Toxic and nutritional Strabismus Extraocular muscles Binocular vision Accommodation Paralytic strabismus Ophthalmoparesis Chronic progressive external ophthalmoplegia Kearns–Sayre syndrome palsies Oculomotor (III) Fourth-nerve (IV) Sixth-nerve (VI) Other strabismus Esotropia / Exotropia Hypertropia Heterophoria Esophoria Exophoria Cyclotropia Brown's syndrome Duane syndrome Other binocular Conjugate gaze palsy Convergence insufficiency Internuclear ophthalmoplegia One and a half syndrome Refraction Refractive error Hyperopia Myopia Astigmatism Anisometropia / Aniseikonia Presbyopia Vision disorders Blindness Amblyopia Leber's congenital amaurosis Diplopia Scotoma Color blindness Achromatopsia Dichromacy Monochromacy Nyctalopia Oguchi disease Blindness / Vision loss / Visual impairment Anopsia Hemianopsia binasal bitemporal homonymous Quadrantanopia subjective Asthenopia Hemeralopia Photophobia Scintillating scotoma Pupil Anisocoria Argyll Robertson pupil Marcus Gunn pupil Adie syndrome Miosis Mydriasis Cycloplegia Parinaud's syndrome Other Nystagmus Childhood blindness Infections Trachoma Onchocerciasis v t e Optical illusions ( list ) Illusions Afterimage Ambiguous image Ames room Barberpole Bezold Café wall Checker shadow Chubb Cornsweet Delboeuf Ebbinghaus Ehrenstein Flash lag Fraser spiral Gravity hill Grid Hering Impossible trident Jastrow Lilac chaser Mach bands McCollough Müller-Lyer Necker cube Orbison Penrose stairs Penrose triangle Peripheral drift Poggendorff Ponzo Rubin vase Sander Schroeder stairs Shepard tables Spinning Dancer Ternus Vertical–horizontal White's Wundt Zöllner Popular culture Op art Trompe-l'œil Spectropia (1864 book) Ascending and Descending (1960 drawing) Waterfall (1961 drawing) The dress (2015 photograph) Related Accidental viewpoint Auditory illusions Tactile illusions Temporal illusion This article about the eye is a stub .
-
Androgen Insensitivity, Partial
OMIM
., 2015). Clinical Features Reifenstein (1946) reported a family in which 9 of 10 male members over 2 generations exhibited abnormally high follicle-stimulating hormone secretion, hypospadias, sterility, gynecomastia, small testes, absent beard, height of 65 +/- inches, normal sized phallus, normal 17-ketosteroid excretion, late puberty, and normal libido. ... On the basis of this and another reported pedigree, they suggested that the affected members in the kindred reported by Gilbert-Dreyfus et al. (1957), Lubs et al. (1959) and Rosewater et al. (1965) had the same condition as that reported by Reifenstein (1946). Wilson et al. (1974) chose to refer to the condition as type 1 familial incomplete male pseudohermaphroditism (type 2 is autosomal recessive; 264600).

-
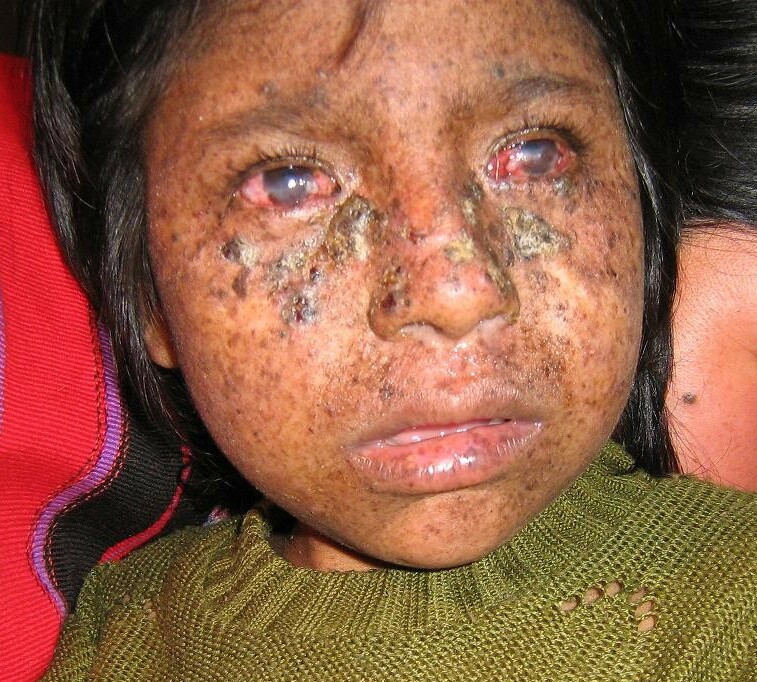
Xeroderma Pigmentosum, Autosomal Dominant, Mild
OMIM
Although the disorder in this kindred was milder than ordinary xeroderma pigmentosum ('George was in a tropical climate for many years, being invalided home on two occasions on account of the disease' and 'Douglas was invalided home from North Africa in 1943'), 'Douglas was considered to be a case of xeroderma pigmentosum by all the members present when he was shown at a meeting of the North British Dermatological Society in 1946.' Imray et al. (1986) described an Australian kindred with persons in 5 generations affected by a mild form of XP.XPA, XPC, POLH, ERCC5, ERCC2, ERCC1, DDB2, ERCC4, ERCC3, TERF2, GTF2H1, TP53, NR1H2, BIVM-ERCC5, GTF2H2, GTF2H3, GTF2H5, GTF2H4, POLQ, CAT, XRCC1, CSH2, XRCC6P5, HPRT1, LIG4, ERCC6, CSH1, HRAS, OGG1, HPGDS, PCNA, PTEN, XRCC3, ENDOV, CDKN2A, GSTM1, SIRT1, MC1R, LCE2B, HSPA9, RAD23A, NRAS, FUS, WDR76, ATR, BRIP1, CHEK1, DDB1, BCL2, DECR1, ERCC8, CCNH, ATM, TYMS, BTN2A2, PTF1A, IRF9, EXO1, HERC2, RAD54L, H3P38, PDIK1L, HFM1, UBE3B, USP7, IFI44, XRCC2, CATSPER1, TDG, SLX4, XYLT2, VPS11, RTEL1, CRLF3, DEFB103A, DEFB103B, MEPE, WDR77, LRRC59, RNU1-1, VEPH1, PRDX5, SMUG1, AAGAB, KAT7, ZC3H12D, NAT1, TALDO1, CGA, FEN1, EGFR, NQO1, TIMM8A, DDX11, GADD45A, CYP1B1, CSNK2A1, CRYZ, COMT, CETN2, SYCP1, CDK7, CDK4, DDR1, BRAF, BRCA1, BAX, ATF3, ASIP, FAS, APEX1, GOT1, GSTM2, GSTT1, HGF, SULT1A1, STAT3, ST13, SMO, RNU2-1, RNU1-4, RECQL, RAD23B, PTCH1, PTAFR, MAPK8, PRKDC, POMC, AKT1, ODC1, NPM1, NCAM1, MYC, MPO, LBR, KIT, H3P33
-
West Nile Fever
Wikipedia
Prevention [ edit ] Low-cost, ceiling hung mosquito netting for a bed Many of the guidelines for preventing occupational West Nile virus exposure are common to all mosquito-borne diseases . [57] Public health measures include taking steps to reduce mosquito populations. ... DEET formulations as high as 30% are recommended for children over two months of age. [58] The CDC also recommends the use of: IR3535, oil of lemon eucalyptus, para-menthane-diol, or 2-undecanone. [59] Protect infants less than two months of age by using a carrier draped with mosquito netting with an elastic edge for a tight fit. ... Repellents containing permethrin ( e.g. , Permanone) or other insect repellents may be applied to clothing, shoes, tents, mosquito nets, and other gear. (Permethrin is not suitable for use directly on skin.) ... They are now widespread in the United States, and in Florida they have been found in all 67 counties. [60] In an at-risk area, staying in air-conditioned or well- screened room, or sleeping under an insecticide-treated bed net is recommended. Bed nets should be tucked under mattresses, and can be sprayed with a repellent if not already treated with an insecticide. [57] Monitoring and control [ edit ] West Nile virus can be sampled from the environment by the pooling of trapped mosquitoes via ovitraps , carbon dioxide -baited light traps, and gravid traps, testing blood samples drawn from wild birds, dogs, and sentinel monkeys, and testing brains of dead birds found by various animal control agencies and the public. ... Retrieved 28 October 2017 . ^ Gompf, Sandra. "West Nile Virus" . Medicine Net . MedicineNet Inc . Retrieved 15 January 2019 . ^ "Symptoms, Diagnosis, & Treatment" .CCR5, ERVK-32, ROBO3, MAVS, DDX58, PLAAT4, IFIT2, ERVK-6, STAT1, SPP1, OAS1, IL1B, IFNB1, RNASEL, CASP8, HLA-DRB1, PELI1, SELENBP1, ARHGEF2, LRRFIP1, NAMPT, TRAIP, RIPK3, SEC14L2, CSF1R, LAMP3, ERVW-1, FOXP3, ZMYND10, DDX56, CCR7, VCP, CDKN2A, IFIH1, DHX58, ZBP1, HAVCR2, PIK3IP1, NLRP3, TNFRSF13C, TRIM6, RBM45, CCR2, ERVK-20, ERVK-18, VAMP8, TNFRSF1A, IFNA1, TNF, IFNA13, HLA-DQA1, IL1A, HLA-C, IL10, IL17A, IL18, IRF3, IRF5, KIR2DL2, KIR3DL1, KIR3DS1, LSAMP, CD180, SMAD4, MMP9, HLA-A, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PZP, GLS, CASP1, SNCA, GEM, DDX3X, TAP1, TLR3, ATF4

-
Retiform Parapsoriasis
Wikipedia
Retiform parapsoriasis Specialty Dermatology Retiform parapsoriasis is a cutaneous condition, considered to be a type of large-plaque parapsoriasis . [1] It is characterized by widespread, ill-defined plaques on the skin, that have a net-like or zebra-striped pattern. [2] Skin atrophy , a wasting away of the cutaneous tissue , usually occurs within the area of these plaques. [1] See also [ edit ] Parapsoriasis Poikiloderma vasculare atrophicans List of cutaneous conditions References [ edit ] ^ a b Lambert WC, Everett MA (Oct 1981).

-
Epileptic Encephalopathy, Early Infantile, 45
OMIM
In vitro functional studies in HEK293 cells showed that the mutation altered the kinetic properties of the channel, resulting in the net loss of GABAergic inhibition. In a boy with EIEE45, Lien et al. (2016) identified a de novo heterozygous missense mutation in the GABRB1 gene (T287I; 137190.0002).
-
Head For Heights
Wikipedia
In: DAV Panorama 1/2008, ISSN 1437-5923 Pepi Stückl/Georg Sojer: Bergsteigen: Lehrbuch für alle Spielarten des Bergsteigens , Bruckmann, Munich, 1996, ISBN 3-7654-2859-0 v t e Climbing Types Aid Bouldering Clean Competition Crack Deep-water solo Direttissima Face Free Free solo Grass Ice Indoor Lead Rock Mixed Mountaineering Slab Speed Sport Top rope Trad Tree Lists Alpine clubs Climbers Deaths on eight-thousanders Equipment Everest deaths First ascents Knots Mount Hood incidents Terminology Terminology Abseiling Alpenstock Anchor Approach shoe Ascender Bachar ladder Belay device Belaying Bolt Bouldering mat Cam Carabiner Crampons Dry-tooling Dynamic rope Exposure Fifi hook Grades Grade (bouldering) Harness Head for heights Mountaineering boot Hex Ice axe Ice screw Ice tool Nut Picket Pitch Piton Protection Quickdraw Self-locking device Shoes Sling Snow fluke Snow protection Snowshoe Spotting Sure-footedness Tricam Webbing Media Climbing Rock & Ice Mountain film Companies Black Diamond CAMP Cascade Designs Deuter Early Winters Eastern Mountain Sports Five Ten Frostline Kits GERRY Mountain Sports Grivel Holubar Mountaineering JanSport Kelty La Sportiva Lowe Alpine Mammut Marmot Mountain Works Millet Mountain Safety Research Mountain Equipment Co-op Sierra Designs The North Face Therm-a-Rest Outdoor Research Petzl Rab REI Wild Country Organizations Alpine Club Alpine Club of Canada American Alpine Club Appalachian Mountain Club Austrian Alpine Club Austrian Tourist Club Club Alpin Français Club Alpino Italiano Den Norske Turistforening Federación Española de Deportes de Montaña y Escalada Fédération française de la montagne et de l'escalade German Alpine Club International Federation of Sport Climbing International Mountaineering and Climbing Federation South African National Climbing Federation South Tyrol Alpine Club Swedish Tourist Association Swiss Alpine Club USA Climbing Portal Category Commons WikiProject

-
Biceps Tendon Rupture
Wikipedia
More severe injuries require surgery and post-op physical therapy to regain strength and functionality in the muscle.

-
Ideas And Delusions Of Reference
Wikipedia
Contents 1 Psychoanalytic views 2 Anti-psychiatry 3 Delusions of reference 4 Examples 5 Literary analogues 6 See also 7 References Psychoanalytic views [ edit ] Main article: Psychoanalytic theory In Sigmund Freud 's view, "Delusions of being watched present this power in a regressive form, thus revealing its genesis...voices, as well as the undefined multitude, are brought into the foreground again by the [ paranoid ] disease, and so the evolution of conscience is reproduced regressively." [7] As early as 1928, Freud's contemporary, Carl Jung , introduced the concept of synchronicity , a theory of "meaningful coincidences". [8] In 1946, Otto Fenichel concluded that "the projection of the superego is most clearly seen in ideas of reference and of being influenced....Delusions of this kind merely bring to the patient from the outside what his self-observing and self-critical conscience actually tells him." [9] Jacques Lacan similarly saw ideas of reference as linked to "the unbalancing of the relation to the capital Other and the radical anomaly that it involves, qualified, improperly, but not without some approximation to the truth, in old clinical medicine, as partial delusion" [10] —the "big other, that is, the other of language, the Names-of-the-Father , signifiers or words", [11] in short, the realm of the superego. ... ISBN 978-0-670-03292-1 . ^ Fenichel, Otto (1946). The Psychoanalytic Theory of Neurosis (London) pp. 430–1 ^ Jacques Lacan, Ecrits: A Selection (London 1996) p. 214 ^ Hill, Philip (1997).
-
Muselmann
Wikipedia
Boder assisted in identifying the term musselman when in 1946 he conducted interviews with camp survivors in Europe. ... ISBN 978-0-19-533955-0 . ^ Alan Rosen (18 October 2010). The Wonder of Their Voices: The 1946 Holocaust Interviews of David Boder .

-
Pott Disease
Wikipedia
Imogen, in the novella "The Princess with the Golden Hair", part of Memoirs of Hecate County by Edmund Wilson (1946), has Pott disease. Jane Addams , social activist and Nobel Peace Prize winner, had Pott disease. ... Willem Ten Boom, brother of Corrie Ten Boom , died of tuberculosis of the spine in December 1946 [5] English writer Denton Welch (1915–1948) died of spinal tuberculosis after being involved in a motor accident (1935) that irreparably damaged his spine.TNF, CCL2, IL10, VDR, IL4, MAP3K7, THEMIS, SNHG15, SIL1, NCAPG2, SMUG1, PHB2, VCAM1, NR2C2, TLR2, BMP4, SPP1, CD14, P2RX7, MT1JP, MMP13, MMP9, MMP1, MBL2, IL6, IFNG, HSPE1, HLA-DQA1, MIR155

-
Murrain
Wikipedia
Retrieved 28 July 2008 . ^ Mullett, Charles F. (1946). "The Cattle Distemper in Mid-Eighteenth-Century England" .
-
Nipples, Supernumerary
OMIM
Rather extensive literature supporting dominant inheritance was reviewed by Gates (1946). Klinkerfuss (1924) found polymastia in 5 females in 4 generations.GPC3, ACTB, PORCN, PGAP2, RNF216, TMCO1, PIGV, HDAC8, KLHL7, ARHGAP31, COLEC11, COLEC10, CSPP1, PIGO, PIGY, PGAP3, CKAP2L, ASXL1, PNPLA6, ZEB2, TFAP2A, KIAA0586, DHODH, MEGF8, GPC4, KRAS, NONO, MASP1, TCF4, PIGW, TFAP2B, KAT6A, TRRAP, IKBKG, TP63, PIGL, HDAC4, BRCA2

-
Maple Syrup Urine Disease
GeneReviews
Acute metabolic decompensation is corrected by treating the precipitating stress while delivering sufficient calories, insulin, free amino acids, isoleucine, and valine to achieve sustained net protein synthesis in tissues. Some centers use hemodialysis/hemofiltration to remove BCAAs from the extracellular compartment, but this intervention does not alone establish net protein accretion. ... Thus, leucine tolerance reflects a balance between unmeasured protein losses (e.g., sloughed skin, hair, and nails) and the net accretion of body protein, which in turn is linked to growth rate [Strauss et al 2010]. ... The risk for metabolic crisis in any ill person with MSUD depends on residual in vivo BCKD enzyme activity in relation to the net liberation of free leucine from protein catabolism. ... Following the neonatal period, acute metabolic intoxication (leucinosis) and neurologic deterioration can develop rapidly at any age as a result of net protein degradation precipitated by infection, surgery, injury, or psychological stress (see Figure 1). ... Plasma leucine levels rise predictably as a result of net protein catabolism provoked by a variety of physiologic stresses, including (more...)DBT, BCKDHB, BCKDHA, BCAT2, PPM1K, DLD, ARID4B, BDNF, CTSD, SERPINE1, TNS3, CACNA2D2, MKRN3, UMOD, SPN, NME1, PAH, NBN, MEA1, IL1B, GPR4, GLI2, F2, MECP2

-
Afterdepolarization
Wikipedia
They are due to elevated cytosolic calcium concentrations, classically seen with digoxin toxicity. [3] [4] The overload of the sarcoplasmic reticulum may cause spontaneous Ca 2+ release after repolarization, causing the released Ca 2+ to exit the cell through the 3Na + /Ca 2+ -exchanger. This results in a net depolarizing current. The classical feature is Bidirectional ventricular tachycardia .
-
Krukenberg's Spindle
Wikipedia
The sign was described in 1899 by Friedrich Ernst Krukenberg (1871-1946), who was a German pathologist specialising in ophthalmology . [1] Contents 1 Diagnosis 1.1 Differential diagnosis 1.1.1 Iritis 1.1.2 Vortex keratopathy 1.1.3 Corneal guttata 2 See also 3 References 4 External links Diagnosis [ edit ] Differential diagnosis [ edit ] Iritis [ edit ] Painful red eye with photophobia associated with inflammation Vortex keratopathy [ edit ] Corneal deposits also known as cornea verticillata , caused by netarsudil eye drops or chronic amiodarone use for cardiac arrhythmias . [2] Corneal guttata [ edit ] Non-transparent collagen deposits appearing following loss of corneal endothelial cells [3] See also [ edit ] Pigment dispersion syndrome References [ edit ] ^ Krukenberg F. (1899) Beiderseitige angeborene Melanose der Hornhaut.

-
Bugchasing
Wikipedia
, 10 April 2006 v t e Lesbian , gay , bisexual , and transgender ( LGBT ) slang List Ace Bareback Banjee Bear Beard Beat Bi-curious Boi Top, bottom and versatile Bottom surgery Breeder Bugchasing Bulldagger Butch Castro clone Chicken Chickenhawk Chub Chubby chaser Cottaging Cruising Daddy Down-low Drag Dyke En femme En homme Fag (Faggot) Fag hag Fag stag Faux queen F2M Femme Flagging (hanky code) Friend of Dorothy Fruit Fruit fly Gay-for-pay Gaydar Gaymer Genderfuck Gold star lesbian Glory hole Heteroflexibility Lesbian until graduation Lipstick lesbian M2F Non-op Packing Party and play Passing Poppers Post-op Pre-op Queen RLE Shemale Soft butch Scissoring SRS Stone butch Stealth Swish T Tea-room TERF Top surgery Trache shave Trade Tranny Transfan Transition Tribbing Troll Twink U-Haul lesbian Womyn-born womyn Related Polari LGBT linguistics Terminology of homosexuality Category v t e Sexual slang General Anilingus Banjee Bareback Baseball metaphors for sex Blue balls Bottom Camel toe Chickenhead Circle jerk Cock tease Cornhole Cougar Cunt Deep-throating Dick Dirty Sanchez Dogging Donkey punch Douche Felching Fuck Girlfriend experience Glory hole Hogging Hot Karl Italian profanity Latin profanity Mama-san Mammary intercourse Mat Mile high club Motherfucker Nookie Party and play Pearl necklace Pegging Pirate Pussy Quickie Red wings Rusty trombone Serosorting Shemale Slut Snowballing Soggy biscuit Switch Teabagging Tits Top Top, bottom and versatile Turkey slap Twat Voulez-vous coucher avec moi?
-
Nephrolithiasis, Calcium Oxalate
OMIM
In vitro flux studies indicated that mice lacking Slc26a6 have a defect in intestinal oxalate secretion resulting in enhanced net absorption of oxalate. Jiang et al. (2006) concluded that the anion exchanger, SLC26A6, has a major constitutive role in limiting net intestinal absorption of oxalate, thereby preventing hyperoxaluria and calcium oxalate urolithiasis.
-
Steroid Diabetes
Wikipedia
Mechanism [ edit ] Glucocorticoids oppose insulin action and stimulate gluconeogenesis , especially in the liver , resulting in a net increase in hepatic glucose output.
- Naegeli-Franceschetti-Jadassohn Syndrome/dermatopathia Pigmentosa Reticularis MedlinePlus











