Potassium influx studies demonstrated that, as in other families with pseudohyperkalemia, the net K loss that occurs in the red cells at room temperature or below can be attributed to the disparity in temperature dependence between this 'leak' flux and the opposing NaK pump, which always shows a simple and steep monotonic fall with decreasing temperature.
Familial pseudohyperkalemia (FP) is an inherited, mild, non-hemolytic subtype of hereditary stomatocytosis that is associated with a temperature-dependent anomaly in red cell membrane permeability to potassium that leads to high in vitro potassium levels in samples stored below 37°C. FP is not associated with additional hematological abnormalities, although affected individuals may show some mild abnormalities like macrocytosis. Epidemiology The prevalence is unknown. Less than 15 kindreds have been reported in the literature so far (the majority being from the United Kingdom). The Arg723Gln substitution in ABCB6 leading to FP-Cardiff (a temperature-dependent pattern of cation leak previously recognized in a UK family) may affect 1/500 Europeans. Clinical description FP is an asymptomatic condition that presents through blood tests as unexpectedly high plasma potassium levels, after storage at or below room temperature.
Many later swore they were threatened and beaten during questioning. Government agents cast a wide net, bringing in some American citizens, passers-by who admitted being Russian, some not members of the Russian Workers. ... Hoover later admitted "clear cases of brutality." [17] The raids covered more than 30 cities and towns in 23 states, but those west of the Mississippi and south of the Ohio were "publicity gestures" designed to make the effort appear nationwide in scope. [18] Because the raids targeted entire organizations, agents arrested everyone found in organization meeting halls, not only arresting non-radical organization members but also visitors who did not belong to a target organization, and sometimes American citizens not eligible for arrest and deportation. [19] The Department of Justice at one point claimed to have taken possession of several bombs, but after a few iron balls were displayed to the press they were never mentioned again. All the raids netted a total of just four ordinary pistols. [20] While most press coverage continued to be positive, with criticism only from leftist publications like The Nation and The New Republic , one attorney raised the first noteworthy protest.
In Uganda, it is more practical being the recognition by the (public) government and (public) donor that a (private) philanthropic health facility can receive free test kits for HIV screening, free mosquito nets and water purification to reduce opportunistic infections and free testing and treatment for basic infections of great danger to PLHA. [24] Alternative proposals [ edit ] Several studies, conducted in Uganda and its neighbors, indicate that adult male circumcision may be a cost-effective means of reducing HIV infection. ... The national AIDS Indicator survey in 2011 also indicated that over 48 percent of adult men were willing to be circumcised, generating a critical mass of demand for male circumcision. [26] An economic analysis by Bertran Auvert, a physician from the INSERM U687, Saint-Maurive, France, and colleagues estimated the cost of a roll-out over an initial 5-year period would be $1036 million ($748 – $1319 million) and $965 million ($763 – $1301 million) for private and public health sectors, respectively. The cumulative net cost over the first 10 years was estimated at $1271 million and $173 million for the private and public sectors, respectively. [27] See also [ edit ] HIV/AIDS in Africa Health in Uganda Gideon Byamugisha Noerine Kaleeba Joseph Konde-Lule Philly Lutaaya Roy Mugerwa Fred Nalugoda Sex for Fish Traditional and Modern Health Practitioners Together against AIDS Uganda AIDS Orphan Children Foundation Notes and references [ edit ] ^ Peter Kitonsa Ssebbanja (2007). "4".
Summary Clinical characteristics. Prolidase deficiency is characterized by skin lesions (typically severe, chronic, recalcitrant, and painful skin ulcers of the lower extremities and telangiectasias of the face and hands), recurrent infections (particularly of the skin and respiratory tract), dysmorphic facial features, variable intellectual disability, and hepatomegaly with elevated liver enzymes and splenomegaly. Anemia, thrombocytopenia, hypergammaglobulinemia, and hypocomplementemia are common. An association between systemic lupus erythematosus (SLE) and prolidase deficiency has been described. Diagnosis/testing. The diagnosis of prolidase deficiency is established by detection of either biallelic PEPD pathogenic variants or reduced prolidase enzyme activity in a proband who has characteristic clinical findings and imidodipeptiduria. Management. Treatment of manifestations: No curative treatment is available.
Prolidase deficiency is a disorder that causes a wide variety of symptoms. The disorder typically becomes apparent during infancy. Affected individuals may have enlargement of the spleen (splenomegaly); in some cases, both the spleen and liver are enlarged (hepatosplenomegaly). Diarrhea, vomiting, and dehydration may also occur. People with prolidase deficiency are susceptible to severe infections of the skin or ears, or potentially life-threatening respiratory tract infections. Some individuals with prolidase deficiency have chronic lung disease. Characteristic facial features in people with prolidase deficiency include prominent eyes that are widely spaced (hypertelorism), a high forehead, a flat bridge of the nose, and a very small lower jaw and chin (micrognathia).
Prolidase deficiency is a rare metabolic condition characterized by skin lesions, recurrent infections, unusual facial features, variable intellectual disability, enlargement of the liver ( hepatomegaly ) with elevated liver enzymes, and enlargement of the spleen ( splenomegaly ). Symptoms typically present during infancy and vary greatly among affected individuals. The condition is caused by mutations in the PEPD gene. It is inherited in an autosomal recessive pattern. Treatment for prolidase deficiency is symptomatic and supportive and often requires the expertise of a multidisciplinary team.
Prolidase deficiency is an inherited disorder of peptide metabolism characterized by severe skin lesions, recurrent infections (involving mainly the skin and respiratory system), dysmorphic facial features, variable cognitive impairment, and splenomegaly. Epidemiology The exact prevalence is unknown but a prevalence estimate of 1/1,235,000 live births has been suggested. A higher carrier frequency of 1/21 has been reported in the Druze community. Approximately 90 patients from different ethnic groups have been reported in the literature to date, but due to underdiagnosis the exact number is probably higher. Clinical description Clinical manifestations and age of onset are quite variable.
A number sign (#) is used with this entry because prolidase deficiency is caused by homozygous or compound heterozygous mutation in the PEPD gene (613230) on chromosome 19q13. Description Prolidase deficiency is a rare autosomal recessive multisystem disorder associated with massive imidodipeptiduria and lack of or reduced prolidase activity in erythrocytes, leukocytes, or cultured fibroblasts. The disorder is clinically heterogeneous and severity varies widely. Features include chronic, slowly healing ulcerations, mainly on the legs and feet. The ulcers are often preceded by other dermatologic manifestations that may occur anywhere and include erythematous papular eruptions, telangiectases with pruritus and photosensitivity, impetigo-like eruptions, pruritic eczematous lesions, and necrotic papules. Mild to severe mental retardation is often a feature, and recurrent respiratory tract infections, sometimes fatal, are common.
It has also been proposed that the distribution of affected blood vessels — predominantly in the superficial subcutaneous plexus (found in the papillary dermis )— results in the net-like pattern of erythema ab igne skin lesions.
External links [ edit ] Classification D ICD - 10 : B50 ICD - 9-CM : 084.8 MeSH : D001742 DiseasesDB : 7751 v t e Malaria Biology Malaria Cerebral Quartan fever Blackwater fever Pregnancy-associated Plasmodium biology life cycle vivax falciparum ovale malariae knowlesi Anopheles mosquito Lifecycle Schizont Merozoite Hypnozoite Gametocyte Control and prevention Public health DDT Mosquito net Malaria prophylaxis Mosquito control Sterile insect technique Genetic resistance Duffy antigen Sickle-cell anaemia Thalassemia G6PDH deficiency Malaria vaccine RTS,S Diagnosis and treatment Diagnosis of malaria Malaria culture Blood film Malaria antigen detection tests Antimalarials Artemisinin Mefloquine Proguanil Society and malaria Diseases of poverty Millennium Development Goals History of malaria Roman fever National Malaria Eradication Program World Malaria Day Epidemiology Malaria and the Caribbean Malaria Atlas Project Organisations Malaria Consortium Against Malaria Foundation Bill & Melinda Gates Foundation Imagine No Malaria Malaria No More Africa Fighting Malaria African Malaria Network Trust South African Malaria Initiative African Leaders Malaria Alliance Amazon Malaria Initiative The Global Fund to Fight AIDS, Tuberculosis and Malaria Medicines for Malaria Venture Category v t e Protozoan infection : SAR and Archaeplastida SAR Alveolate Apicomplexa Conoidasida / Coccidia Coccidia : Cryptosporidium hominis / Cryptosporidium parvum Cryptosporidiosis Cystoisospora belli Isosporiasis Cyclospora cayetanensis Cyclosporiasis Toxoplasma gondii Toxoplasmosis Aconoidasida Plasmodium falciparum / vivax / ovale / malariae / knowlesi Malaria Blackwater fever Babesia Babesiosis Ciliophora Balantidium coli Balantidiasis Heterokont Blastocystis Blastocystosis Pythium insidiosum Pythiosis Archaeplastida Algaemia : Prototheca wickerhamii Protothecosis
Affected individuals exhibited mild anemia and red cell dehydration (xerocytosis), associated with net cation deficiency due to excessive passive K+ efflux.
Dehydrated hereditary stomatocytosis (DHS) is a rare hemolytic anemia characterized by a decreased red cell osmotic fragility due to a defect in cation permeability, resulting in red cell dehydration and mild to moderate compensated hemolysis. Pseudohyperkalemia (loss of potassium ions from red cells on storage at room temperature) is sometimes observed. Epidemiology The prevalence of DHS is unknown but to date, about 20 families with DHS have been described in the literature. Clinical description The clinical presentation of the disease is very heterogeneous. Onset of DHS may occur during the perinatal period with occurrence of edema and ascites (most often not related to an underlying anemia) that usually resolve spontaneously during the first weeks of life but may rarely lead to hydrops fetalis (see this term).
DHS erythrocytes exhibit decreased total cation and potassium content that are not accompanied by a proportional net gain of sodium and water. DHS patients typically exhibit mild to moderate compensated hemolytic anemia, with an increased erythrocyte mean corpuscular hemoglobin concentration and a decreased osmotic fragility, both of which reflect cellular dehydration (summary by Zarychanski et al., 2012).
The mutation involved replacement of a 13-bp sequence in exon 13 by an unrelated 5-bp sequence. The net deletion of 8 bp shifted the reading frame and introduced a premature chain termination downstream.
Leprechaunism is a congenital form of extreme insulin resistance (a group of syndromes that also includes Rabson-Mensenhall syndrome, type A insulin-resistance syndrome, and acquired type B insulin-resistance syndrome; see these terms) characterized by intrauterine and mainly postnatal severe growth retardation. Epidemiology It is a very rare condition with less than 1 case in every million births. Clinical description Leprechaunism is associated with a characteristic dysmorphic facies (resembling that of the 'leprechauns' in Irish folk traditions), atrophic subcutaneous adipose tissue (lipoatrophy) and muscular hypotrophy. Signs of virilization are often observed in young girls. Biologically, episodes of hypo- and hyperglycemia are observed along with marked hyperinsulinemia due to an extreme resistance to insulin. Etiology The syndrome is associated with homozygous or compound heterozygous mutations in the insulin receptor gene ( INSR ; 19p13.3-p13.2).
Leprechaunism is a congenital (present from birth) condition characterized by extreme insulin resistance , pre- and postnatal growth delays, characteristic facial features, skin abnormalities, muscular hypotrophy (reduced muscle mass) and enlarged external genitalia in both males and females. The condition is caused by mutations in the insulin receptor gene ( INSR ) gene. It is inherited in an autosomal recessive manner.
Protein losing enteropathy Upper and lower human gastrointestinal tract Protein losing enteropathy affects the GI tract [1] Specialty Gastroenterology Symptoms Swelling of the legs [2] Causes Inflammatory bowel disease, Idiopathic ulcerative jejunoileitis [1] Diagnostic method Scintigraphy, Viral serologies [1] Treatment Octreotide, Surgery [1] Protein losing enteropathy refers to any condition of the gastrointestinal tract (e.g. damage to the gut wall) that results in a net loss of protein from the body. [3] Contents 1 Signs and symptoms 2 Causes 3 Mechanism 4 Diagnosis 5 Treatment 6 In animals 7 References 8 Further reading 9 External links Signs and symptoms [ edit ] The signs/symptoms of protein losing enteropathy are consistent with diarrhea, fever, and general abdominal discomfort. [3] Swelling of the legs due to peripheral edema can also occur, however, if the PLE is related to a systemic disease such as congestive heart failure or constrictive pericarditis , then the symptoms could be of the primary disease development. [2] Causes [ edit ] The causes of protein-losing enteropathy can include GI conditions (among other causes), like the following: [1] Inflammatory bowel disease Congenital heart defect (single ventricle following surgical repair resulting in congestive heart failure [4] ) Idiopathic ulcerative jejunoileitis.
Because of inertial forces created by acceleration of the aircraft along with centrifugal force caused by turning, the net gravitoinertial force sensed primarily by the otolith organs is not aligned with gravity, leading to perceptual misjudgment of the vertical.
Fear, hatred towards, demonization of, or prejudice against people generally referred to as Tatars Part of a series on Discrimination General forms Age Class ( Caste ) Physical Disability Education Economic Employment Genetics Hair texture Height Housing Language Looks Race / Ethnicity / Nationality Rank Religion Sanity Sex Sexual orientation Size Skin color Specific forms Social Acephobia Adultism Amatonormativity Anti-albinism Anti-autism Anti-homelessness Anti-intellectualism Anti-intersex Anti-left handedness Anti-Masonry Antisemitism (Judeophobia) Aporophobia Audism Biphobia Clannism Cronyism Drug use Elitism Ephebiphobia Fatism Gerontophobia Heteronormativity Heterosexism HIV/AIDS stigma Homophobia Leprosy stigma Lesbophobia Misandry Misogyny Nepotism Pedophobia Perpetual foreigner Pregnancy Reverse Sectarianism Supremacism Black White Transphobia Non-binary Transmisogyny Vegaphobia Xenophobia Religious Ahmadiyya Atheism Baháʼí Faith Buddhism Catholicism Christianity post–Cold War era Druze Falun Gong Hinduism Persecution Islam Persecution Jehovah's Witnesses Judaism Persecution LDS or Mormon Neopaganism Eastern Orthodox Oriental Orthodox Copts Protestantism Rastafarianism Shi'ism Sufism Sunnism Zoroastrianism Ethnic/national African Albanian American Arab Armenian Australian Austrian Azerbaijani British Canadian Catalan Chechen Chilean Chinese Croat Dutch English Estonian European Filipino Finnish French Georgian German Greek Haitian Hazara Hispanic Hungarian Igbo Indian Indonesian Iranian Irish Israeli Italian Japanese Jewish Khmer Korean Kurdish Malay Manchu Mexican Middle Eastern Mongolian Montenegrin Pakistani Pashtun Polish Portuguese Quebec Romani Romanian Russian Scottish Serb Slavic Somali Soviet Tatar Thai Tibetan Turkish Ukrainian Venezuelan Vietnamese Western Manifestations Blood libel Bullying Compulsory sterilization Counter-jihad Cultural genocide Defamation Democide Disability hate crime Dog-whistle politics Eliminationism Ethnic cleansing Ethnic conflict Ethnic hatred Ethnic joke Ethnocide Forced conversion Freak show Gay bashing Gendercide Genital modification and mutilation Genocide examples Glass ceiling Hate crime Hate group Hate speech online Homeless dumping Indian rolling Lavender scare LGBT hate crimes Lynching Mortgage Murder music Occupational segregation Persecution Pogrom Purge Red Scare Religious persecution Religious terrorism Religious violence Religious war Scapegoating Segregation academy Sex-selective abortion Slavery Slut-shaming Trans bashing Victimisation Violence against women White flight White power music Wife selling Witch-hunt Policies Age of candidacy Blood purity Blood quantum Crime of apartheid Disabilities Catholic Jewish Ethnocracy Ethnopluralism Gender pay gap Gender roles Gerontocracy Gerrymandering Ghetto benches Internment Jewish quota Jim Crow laws Law for Protection of the Nation McCarthyism MSM blood donation restrictions Nonpersons Numerus clausus (as religious or racial quota) Nuremberg Laws One-drop rule Racial quota Racial steering Redlining Same-sex marriage (laws and issues prohibiting) Segregation age racial religious sexual Sodomy law State atheism State religion Ugly law Voter suppression Countermeasures Affirmative action Anti-discrimination law Cultural assimilation Cultural pluralism Diversity training Empowerment Feminism Fighting Discrimination Hate speech laws by country Human rights Intersex rights LGBT rights Masculism Multiculturalism Nonviolence Racial integration Reappropriation Self-determination Social integration Toleration Related topics Allophilia Anti-cultural, anti-national, and anti-ethnic terms Bias Christian privilege Civil liberties Cultural assimilation Dehumanization Diversity Ethnic penalty Eugenics Internalized oppression Intersectionality Male privilege Masculism Medical model of disability autism Multiculturalism Net bias Neurodiversity Oikophobia Oppression Police brutality Political correctness Polyculturalism Power distance Prejudice Prisoner abuse Racial bias in criminal news Racism by country Religious intolerance Second-generation gender bias Snobbery Social exclusion Social model of disability Social stigma Stereotype threat The talk White privilege v t e Anti-Tatar sentiment or Tatarophobia ( Russian : Татарофобия , romanized : Tatarofobiya ) refers to the fear, hatred towards, demonization of, or prejudice against people generally referred to as Tatars , including but not limited to Volga , Siberian , and Crimean Tatars , although negative attitudes against the latter are by far the most severe, largely in part due to the long history of Soviet media only depicting them in a negative way and promoting negative stereotypes to help politically justify their deportation and marginalization.
However, recent research shows that increased access to healthcare weakens the urban association with these diseases, [6] and the net effect is still unclear. Interestingly, many mental health disorders have also been associated with urban areas, especially in low socioeconomic areas. [7] Increased levels of stress, air & light & noise pollution, and reduced "green" space are all urban-associated environmental effects that are adversely linked to mental health. [2] Though urban areas are often correlated with dirtiness and disease, they are likely to have more access to higher quality health care which can lead to more positive health outcomes.
One example of a macro-botellón was on 17 March 2006, "Half of Spain [met] on the net to organize a macro-botellón". [13] The macro-botellón was organized in cities around Spain, such as Madrid, Barcelona, Sevilla, Oviedo, Murcia, Vitoria, Málaga, Córdoba, Granada, and Jaén. [14] One of the purposes of the macro-botellón on 17 March 2006, near the Faro de Moncloa in Madrid, Spain, was to protest against the municipal restrictions on drinking alcohol in the streets.
Thus, the redox state of Quinone A is no longer active and there is, again, no change in the concentration of carbon dioxide in the intracellular airspaces of the leaf. All these factors work to have a net decrease of stomatal conductance. ... Photoinhibition damages PSII at the same rate whether the leaf stalk is in water or lincomycin, but, in the “leaf stalk in water” sample, repair is so rapid that no net decrease in (F V /F M ) occurs Photoinhibition can be measured from isolated thylakoid membranes or their subfractions, or from intact cyanobacterial cells by measuring the light-saturated rate of oxygen evolution in the presence of an artificial electron acceptor ( quinones and dichlorophenol-indophenol have been used).
Jejunal perfusion studies showed the jejunum to be in a net secretory state with intact hexose transport, but with an anomalous relation between jejunal sodium and hydrogen transport.
A number sign (#) is used with this entry because of evidence that congenital secretory sodium diarrhea (DIAR8) is caused by homozygous or compound heterozygous mutation in the NHE3 gene (SLC9A3; 182307) on chromosome 5p15. For discussion of genetic heterogeneity of diarrhea, see DIAR1 (214700). Clinical Features Holmberg and Perheentupa (1985) described a 9-year-old Finnish girl with congenital diarrhea that seemed to be caused by a specific defect in intestinal sodium absorption. The pregnancy was complicated by maternal polyhydramnios, and in the delivery room the patient had a distended abdomen and passed a voluminous watery stool, indicative of intrauterine onset of the diarrhea. Stool analysis showed an abnormally high chloride level, but an even higher sodium concentration.
A rare, genetic, non-syndromic intestinal transport defect characterized by congenital onset of severe watery diarrhea containing high concentrations of sodium, hyponatremia and metabolic acidosis. Epidemiology Less than 50 cases have been described to date. Clinical description Presentation is typically prenatal with polyhydramnios, prominent abdominal distension due to dilated fluid-filled loops of the intestine. A watery diarrhea is present after birth, independent of oral feeding (breast or formula) or nil by mouth. The diarrhea can be described as 'non-stopping', and can be mistaken for urine. There are increased bowel sounds at examination, and passing of meconium is never reported.
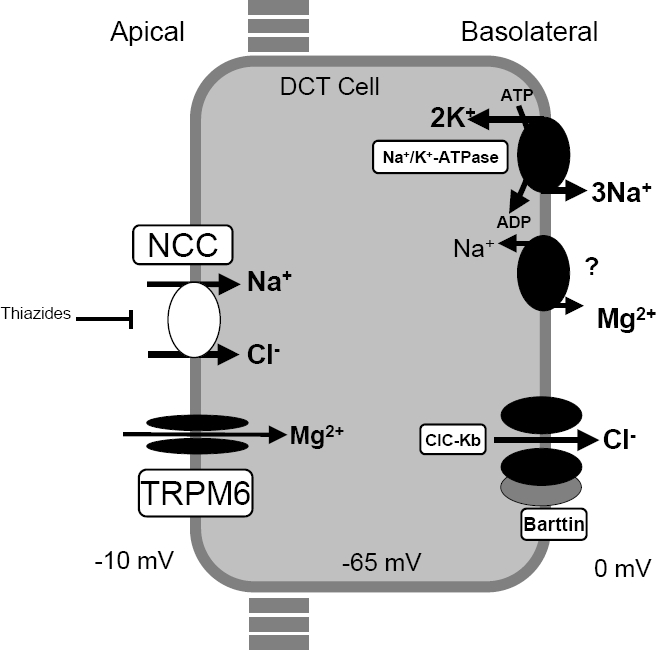
Mapping An attractive candidate gene for one form of Bartter syndrome is the thiazide-sensitive Na-Cl cotransporter of the distal convoluted tubule (SLC12A3; 600968), which is believed to be the principal mediator of sodium and chloride reabsorption in this segment of the nephron, accounting for a significant fraction of net renal sodium reabsorption. This cotransporter is the target of thiazide diuretics, one of the major classes of agents used in the treatment of hypertension.
A rare syndrome characterized by hypokalemic metabolic alkalosis in combination with significant hypomagnesemia and low urinary calcium excretion. Epidemiology Gitelman syndrome (GS) prevalence is estimated at 1 to 10 per 40,000 and potentially higher in Asia. GS is arguably the most frequent inherited tubulopathy. Clinical description GS presents mainly in adolescents and adults but also encountered in children, as early as in the neonatal period. The diagnosis may be incidental, due to blood tests obtained for unrelated reasons. Clinical symptoms may include salt craving, thirst and nocturia, transient periods of muscle weakness and tetany, sometimes accompanied by abdominal pain.
Specialty Endocrinology Anatomy of a Nephron; functional unit of the kidney [1] Gitelman syndrome ( GS ) is an autosomal recessive kidney tubule disorder characterized by low blood levels of potassium and magnesium , decreased excretion of calcium in the urine , and elevated blood pH . [2] The disorder is caused by genetic mutations resulting in improper function of the thiazide -sensitive sodium-chloride symporter (also known as NCC, NCCT, or TSC) located in the distal convoluted tubule of the kidney . [2] The distal convoluted tubule of the kidney serves a minimal role in salt absorption and a greater role in managing the excretion of electrolytes like magnesium and calcium to produce more concentrated urine. [3] Genetic mutations along the sodium chloride symporter, lead to inadequate transport of multiple electrolytes along this channel such as sodium , chloride , calcium , magnesium , and potassium . The net effect is an electrolyte imbalance consistent with thiazide diuretic therapy Gitelman syndrome was formerly considered a subset of Bartter syndrome until the distinct genetic and molecular bases of these disorders were identified.
Gitelman syndrome is a kidney function disorder that causes an imbalance of charged atoms (ions) in the body, including ions of potassium , magnesium , and calcium . It is usually diagnosed during late childhood or adulthood. More common symptoms include fatigue, salt craving, thirst, frequent urination, muscle cramping, muscle weakness, dizziness, tingling or numbness, low blood pressure, and heart palpitations. Gitelman syndrome can be caused by changes (mutations) in the SLC12A3 or CLCNKB genes and is inherited in an autosomal recessive manner. Treatment may include supplementation of potassium and magnesium, and a high sodium and high potassium diet.
Gitelman syndrome is a kidney disorder that causes an imbalance of charged atoms (ions) in the body, including ions of potassium, magnesium, and calcium. The signs and symptoms of Gitelman syndrome usually appear in late childhood or adolescence. Common features of this condition include painful muscle spasms (tetany), muscle weakness or cramping, dizziness, and salt craving. Also common is a tingling or prickly sensation in the skin (paresthesias), most often affecting the face. Some individuals with Gitelman syndrome experience excessive tiredness (fatigue), low blood pressure, and a painful joint condition called chondrocalcinosis.
INHERITANCE - Autosomal dominant HEAD & NECK Ears - Hearing loss, initially conductive, later mixed conductive-sensorineural - Absence of middle ear ossicles (in some patients) Teeth - Resorption of cervical region of the teeth - Progressive tooth mobility - Spontaneous tooth fracture - Early loss of dentition SKELETAL - Severe bone pain - Pathologic fractures - Osteolytic lesions mainly affecting the appendicular skeleton - Multinuclear osteoclasts - Medullary expansion - Abundant osteoblasts - Increased bone turnover and remodeling Limbs - Bowing of the long bones - Abnormal modeling, onset before focal disease (humerus, radius, ulna, tibia) - Coarse 'fish net' bone trabeculae, onset before focal disease - Rounded areas of trabecular loss (juxta-articular, metaphyseal, diaphyseal, early stage) - Enlargement of affected area (intermediate stage) - Cortical thinning (late stage) - Bone expansion (late stage) - Complete loss of trabecular pattern (late stage) LABORATORY ABNORMALITIES - Elevated serum alkaline phosphatase - Elevated urinary hydroxyproline MISCELLANEOUS - Onset of bone disease in second decade (range 18-44 years) - Onset of hearing loss in childhood - Progressive disorder MOLECULAR BASIS - Caused by mutation in the tumor necrosis factor receptor superfamily, member 11A gene (TNFRSF11A, 603499.0001 ) ▲ Close
A rare primary bone dysplasia characterized by abnormal bone metabolism with bone pain, deformity, pathological fractures, early conductive hearing loss, and dental abnormalities. Focal bone lesions are typically found in the appendicular skeleton and consist of progressively expanding lytic areas, while generalized disordered bone modeling and altered trabecular pattern are the result of the multifocal, progressive nature of the disease. Age of onset is variable, mode of inheritance is autosomal dominant.