Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Steroid Diabetes
Wikipedia
Mechanism [ edit ] Glucocorticoids oppose insulin action and stimulate gluconeogenesis , especially in the liver , resulting in a net increase in hepatic glucose output. ... Criteria [ edit ] The diagnostic criteria for steroid diabetes are those of diabetes (fasting glucoses persistently above 125 mg/dl (7 mM) or random levels above 200 mg/dl (11 mM)) occurring in the context of high-dose glucocorticoid therapy.
-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... According to the WHO and UNICEF, deaths attributable to malaria in 2015 were reduced by 60% [77] from a 2000 estimate of 985,000, largely due to the widespread use of insecticide-treated nets and artemisinin-based combination therapies. [74] In 2012, there were 207 million cases of malaria.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
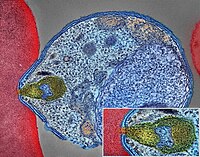
-
Neuroendocrine Tumor
Wikipedia
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Maple Syrup Urine Disease
GeneReviews
Acute metabolic decompensation is corrected by treating the precipitating stress while delivering sufficient calories, insulin, free amino acids, isoleucine, and valine to achieve sustained net protein synthesis in tissues. Some centers use hemodialysis/hemofiltration to remove BCAAs from the extracellular compartment, but this intervention does not alone establish net protein accretion. ... Thus, leucine tolerance reflects a balance between unmeasured protein losses (e.g., sloughed skin, hair, and nails) and the net accretion of body protein, which in turn is linked to growth rate [Strauss et al 2010]. ... The risk for metabolic crisis in any ill person with MSUD depends on residual in vivo BCKD enzyme activity in relation to the net liberation of free leucine from protein catabolism. ... Plasma leucine levels rise predictably as a result of net protein catabolism provoked by a variety of physiologic stresses, including (more...) ... Rather, they are treated with a combination of thiamine (doses ranging from 10 to 1,000 mg/day) and dietary BCAA restriction, making the in vivo contribution of thiamine impossible to discern [Chuang et al 2004].DBT, BCKDHB, BCKDHA, BCAT2, PPM1K, DLD, ARID4B, BDNF, CTSD, SERPINE1, TNS3, CACNA2D2, MKRN3, UMOD, SPN, NME1, PAH, NBN, MEA1, IL1B, GPR4, GLI2, F2, MECP2

-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Pancreatic Neuroendocrine Tumor
Wikipedia
PanNETs are a type of neuroendocrine tumor , representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign , while some are malignant . ... However, morphological imaging alone is not sufficient for a definite diagnosis [14] [16] On biopsy , immunohistochemistry is generally positive for chromogranin and synaptophysin . [17] Genetic testing thereof typically shows altered MEN1 and DAXX / ATRX . [17] Staging [ edit ] The 2010 WHO classification of tumors of the digestive system grades all the neuroendocrine tumors into three categories, based on their degree of cellular differentiation (from well-differentiated "NET G1" through to poorly-differentiated "NET G3"). ... Combinations of several medicines have been used, such as doxorubicin with streptozocin and fluorouracil (5-FU) [12] and capecitabine with temozolomide. [ citation needed ] Although marginally effective in well-differentiated PETs, cisplatin with etoposide has some activity in poorly differentiated neuroendocrine cancers (PDNECs), [12] particularly if the PDNEC has an extremely high Ki-67 score of over 50%. [8] : 30 Several targeted therapy agents have been approved in PanNETs by the FDA based on improved progression-free survival (PFS): everolimus (Afinitor) is labeled for treatment of progressive neuroendocrine tumors of pancreatic origin in patients with unresectable, locally advanced or metastatic disease. [20] [21] The safety and effectiveness of everolimus in carcinoid tumors have not been established. [20] [21] sunitinib (Sutent) is labeled for treatment of progressive, well-differentiated pancreatic neuroendocrine tumors in patients with unresectable locally advanced or metastatic disease. [22] [23] Sutent also has approval from the European Commission for the treatment of 'unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors with disease progression in adults'. [24] A phase III study of sunitinib treatment in well differentiated pNET that had worsened within the past 12 months (either advanced or metastatic disease) showed that sunitinib treatment improved progression-free survival (11.4 months vs. 5.5 months), overall survival , and the objective response rate (9.3% vs. 0.0%) when compared with placebo. [25] Genetics [ edit ] Pancreatic neuroendocrine tumors may arise in the context of multiple endocrine neoplasia type 1 , Von Hippel–Lindau disease , neurofibromatosis type 1 (NF-1) or tuberose sclerosis (TSC) [26] [27] Analysis of somatic DNA mutations in well-differentiated pancreatic neuroendocrine tumors identified four important findings: [28] [6] as expected, the genes mutated in NETs, MEN1 , ATRX , DAXX , TSC2 , PTEN and PIK3CA , [28] are different from the mutated genes previously found in pancreatic adenocarcinoma . [29] [30] one in six well-differentiated pancreatic NETs have mutations in mTOR pathway genes, such as TSC2 , PTEN and PIK3CA . [28] The sequencing discovery might allow selection of which NETs would benefit from mTOR inhibition such as with everolimus , but this awaits validation in a clinical trial . mutations affecting a new cancer pathway involving ATRX and DAXX genes were found in about 40% of pancreatic NETs. [28] The proteins encoded by ATRX and DAXX participate in chromatin remodeling of telomeres ; [31] these mutations are associated with a telomerase -independent maintenance mechanism termed ALT (alternative lengthening of telomeres) that results in abnormally long telomeric ends of chromosomes . [31] ATRX / DAXX and MEN1 mutations were associated with a better prognosis . [28] References [ edit ] ^ Burns WR, Edil BH (March 2012).MEN1, ATRX, DAXX, ELK3, TP53, EPHB1, SLC6A2, CEACAM5, CEACAM7, UQCRFS1, DHDDS, CHPT1, RALBP1, CIB1, SEMA4D, RIPK1, CXCR4, VEGFA, TTR, GNA12, TSC2, TFE3, CDKN1B, PSG2, POMC, MYCN, CEACAM3, GRN, MUC16

- Dowling-Degos Disease GARD
-
Azotemia, Familial
OMIM
Furthermore, urea is reabsorbed actively by the tubule; this process is apparently brought into play particularly in states of low protein intake. Net reabsorption might be due to exaggerated active reabsorption or to deficient secretion.
-
Insulinoma
GARD
Insulinoma is a type of pancreatic neuroendocrine tumor (pancreatic NET), which refers to a group of rare tumors that form in the hormone-making cells of the pancreas.MEN1, RPS15, CDKN2B, CDKN2C, IAPP, GCG, CDKN1B, CDKN1A, SST, FOXM1, GLP1R, PDX1, INS, IL1B, RIT2, PTPRN2, GAD1, EHMT1, IGF2, ZGLP1, CDKN2A, SLC30A8, SLC30A10, GCK, SSTR2, FFAR1, YY1, LEP, DPP4, INSM1, MNX1, HSPD1, GAD2, SLC2A2, CASR, RALBP1, RIPK1, PDHX, BTC, UQCRFS1, TP53, TGM2, SSTR5, CDKN1C, INSR, ABCC8, SLC6A2, SSTR4, SSTR3, WFS1, NIT1, SERPINA1, PTPRN, GIP, GCKR, CORO1A, H3P47, PRL, H3P10, ERBB2, GAST, EGR1, ELK3, CALCA, CASP3, EPHB1, G6PC, DLK1, CCN5, SQSTM1, PTTG1, GCM2, LHX2, KL, MAPK8IP1, INSL5, IRS2, ZNRD2, KHDRBS1, DCTN6, LILRB1, FASTK, CCND1, PDIA5, FAS, ATF6, KDM1A, PDZD2, BCL2, BRCA1, TNKS, PLA2G6, HNF1A, TCF19, TGFA, TGFB1, CASP8, THBD, TKT, TSPAN7, TPD52, TRP-AGG2-5, TRPC1, EIPR1, TXN, TYRP1, UCP2, VDR, CACNA1D, BRAF, STAB1, ERP44, NUP62, KCNH4, CAT, KCNH8, GPR119, STOML3, AKT1, HCAR2, GOLGA6A, TICAM2, HES3, MIR107, MIR144, MIR155, MIR204, MIR21, MIR375, INS-IGF2, ADSS2, TMED7-TICAM2, ECT, LINC02210-CRHR1, H3P23, ADM, SLC22A12, TXNDC5, TRABD, RCBTB1, FGF21, MCAT, MCTS1, TMED7, ADIPOR1, DCTN4, CDKAL1, SLC25A38, BANK1, MEG3, ZC3H12A, APOC2, SOX6, SELENOS, IGSF9, SEMA6A, HAMP, G6PC2, PDIA2, ANGPT2, SYP, STAT5A, STC1, STAT5B, KCNJ1, KCNJ6, KRT8, KRT16, KRT19, DECR1, LEPR, LGALS3, LMO2, EPCAM, SMAD2, SMAD3, SMAD4, MAPT, MC2R, MDK, RAB8A, CUX1, MET, CIITA, MLH1, EGF, EGFR, INPPL1, HK1, MTOR, FGF13, GNA12, GPD2, FBN1, GRN, GSK3B, GSR, GTF2H1, ESR2, ELK1, HLA-DQB1, HMGN2, HNF4A, EPHB2, IFI27, IGFBP1, IGFBP2, IL4, IL10, MRC1, NCAM1, NEDD4, SLC2A1, RAP1A, REG1A, CPE, CMA1, S100A8, SCT, CCL2, CXCL12, SDHD, CHGA, RAB3A, CDKN2D, SLC16A1, SNX1, CDC42, CDK1, CCND3, CCNC, CCK, STAT1, RANBP2, CR2, NF1, PIK3CG, NFE2L1, CTSB, NME1, OPA1, PAX4, PAX6, PCSK1, ENPP1, CTNNB1, PKD1, CRHR1, POLD1, MAPK1, MAPK3, MAPK8, ADCYAP1, PRSS1, PSEN2, PSMD9, PTEN, ACO2
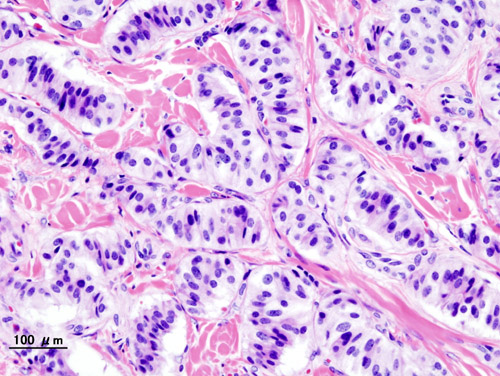
- Sneddon Syndrome GARD
-
Mosquito Bites
Mayo Clinic
Avoid and exclude mosquitoes Limit exposure to mosquitoes by: Repairing any tears in the screens on windows, doors and camping gear Using mosquito netting over strollers and cribs Using mosquito netting when sleeping outdoors Selecting self-care products that don't have scents Use insect repellent Use insect repellent when mosquitoes are active. ... Some sporting goods stores sell clothing pretreated with permethrin. Don't wash bed nets or set them in sunlight, as this breaks down permethrin.
-
Diabetes
Wikipedia
This process is mainly controlled by the hormone glucagon , which acts in the opposite manner to insulin. [72] If the amount of insulin available is insufficient, or if cells respond poorly to the effects of insulin ( insulin resistance ), or if the insulin itself is defective, then glucose is not absorbed properly by the body cells that require it, and is not stored appropriately in the liver and muscles. The net effect is persistently high levels of blood glucose, poor protein synthesis , and other metabolic derangements, such as metabolic acidosis in cases of complete insulin deficiency. [71] When glucose concentration in the blood remains high over time, the kidneys reach a threshold of reabsorption , and the body excretes glucose in the urine ( glycosuria ). [73] This increases the osmotic pressure of the urine and inhibits reabsorption of water by the kidney, resulting in increased urine production ( polyuria ) and increased fluid loss. Lost blood volume is replaced osmotically from water in body cells and other body compartments, causing dehydration and increased thirst ( polydipsia ). [71] In addition, intracellular glucose deficiency stimulates appetite leading to excessive food intake (polyphagia). [74] Diagnosis [ edit ] See also: Glycated hemoglobin and Glucose tolerance test WHO diabetes diagnostic criteria [75] [76] edit Condition 2-hour glucose Fasting glucose HbA 1c Unit mmol/L mg/dL mmol/L mg/dL mmol/mol DCCT % Normal < 7.8 < 140 < 6.1 < 110 < 42 < 6.0 Impaired fasting glycaemia < 7.8 < 140 6.1–7.0 110–125 42–46 6.0–6.4 Impaired glucose tolerance ≥ 7.8 ≥ 140 < 7.0 < 126 42–46 6.0–6.4 Diabetes mellitus ≥ 11.1 ≥ 200 ≥ 7.0 ≥ 126 ≥ 48 ≥ 6.5 Diabetes mellitus is characterized by recurrent or persistent high blood sugar, and is diagnosed by demonstrating any one of the following: [65] Fasting plasma glucose level ≥ 7.0 mmol/L (126 mg/dL) Plasma glucose ≥ 11.1 mmol/L (200 mg/dL) two hours after a 75 gram oral glucose load as in a glucose tolerance test (OGTT) Symptoms of high blood sugar and casual plasma glucose ≥ 11.1 mmol/L (200 mg/dL) Glycated hemoglobin (HbA 1C ) ≥ 48 mmol/mol (≥ 6.5 DCCT %). [77] A positive result, in the absence of unequivocal high blood sugar, should be confirmed by a repeat of any of the above methods on a different day. It is preferable to measure a fasting glucose level because of the ease of measurement and the considerable time commitment of formal glucose tolerance testing, which takes two hours to complete and offers no prognostic advantage over the fasting test. [78] According to the current definition, two fasting glucose measurements above 7.0 mmol/L (126 mg/dL) is considered diagnostic for diabetes mellitus. Per the WHO, people with fasting glucose levels from 6.1 to 6.9 mmol/L (110 to 125 mg/dL) are considered to have impaired fasting glucose . [79] People with plasma glucose at or above 7.8 mmol/L (140 mg/dL), but not over 11.1 mmol/L (200 mg/dL), two hours after a 75 gram oral glucose load are considered to have impaired glucose tolerance . Of these two prediabetic states, the latter in particular is a major risk factor for progression to full-blown diabetes mellitus, as well as cardiovascular disease. [80] The American Diabetes Association (ADA) since 2003 uses a slightly different range for impaired fasting glucose of 5.6 to 6.9 mmol/L (100 to 125 mg/dL). [81] Glycated hemoglobin is better than fasting glucose for determining risks of cardiovascular disease and death from any cause. [82] Prevention [ edit ] See also: Prevention of type 2 diabetes There is no known preventive measure for type 1 diabetes. [2] Type 2 diabetes—which accounts for 85–90% of all cases worldwide—can often be prevented or delayed [83] by maintaining a normal body weight , engaging in physical activity, and eating a healthy diet. [2] Higher levels of physical activity (more than 90 minutes per day) reduce the risk of diabetes by 28%. [84] Dietary changes known to be effective in helping to prevent diabetes include maintaining a diet rich in whole grains and fiber , and choosing good fats, such as the polyunsaturated fats found in nuts, vegetable oils, and fish. [85] Limiting sugary beverages and eating less red meat and other sources of saturated fat can also help prevent diabetes. [85] Tobacco smoking is also associated with an increased risk of diabetes and its complications, so smoking cessation can be an important preventive measure as well. [86] The relationship between type 2 diabetes and the main modifiable risk factors (excess weight, unhealthy diet, physical inactivity and tobacco use) is similar in all regions of the world.INS, PPARG, CP, RAC1, APPL1, EIF2S3, FN1, ADIPOQ, SIRT1, SOD1, NR1I2, LEPR, IRS1, POMC, ATP6, PON1, CAT, PTGS2, AOC3, PAX6, IL2RA, CYBB, NR1I3, MAP3K5, MT2A, NCF1, MAFA, ALPK1, CPT1A, KIF1A, THBS2, NR0B2, ADRB1, GAD1, KIF5B, ZFP57, CLOCK, GLP1R, SERPINE1, PPARA, PTEN, ARNTL, ABCG2, ICA1, PROM1, LRP1, SLC22A12, KCNJ11, HNF4A, TCF7L2, UMOD, GLIS3, ABCC8, AQP4, AQP1, PTPN22, CDKN2A, STAT3, WFS1, HFE, IGF1R, CASR, APOE, F7, HNF1B, ASIP, GCKR, LMNA, IL6, PDX1, CFTR, LPA, INSR, IFIH1, PAX4, SLC19A2, HNF1A, FXN, HLA-DQB1, FTO, GCK, SRD5A2, HLA-DRB1, TP53, APOA5, HAMP, FOXP3, ATM, CDKN2B-AS1, COX2, CISD2, HLA-B, BRCA1, LIPC, BMP2, CAV1, GATA6, SPINK1, CUBN, PNPLA2, ARG1, CNBP, WRN, DNAJC3, GATA3, PRKAR1A, DNM1L, MLXIPL, GJA1, TRNL1, BLK, LIPE, TERT, PLIN1, IER3IP1, KRAS, ZFHX3, ITPR3, ELN, BSCL2, AIRE, SLC25A4, TLR1, SLC22A2, PDE4D, PRSS1, SLC30A3, DCAF17, NDUFS7, BRCA2, AGPAT2, C2, ND1, CPS1, HBB, ND5, SMAD4, PTF1A, MKKS, PRKAG2, HS6ST1, FOXC2, CYTB, FOS, SLC29A3, COX1, GNAS, FADS2, PIK3R1, MTNR1B, ND3, DKC1, CTLA4, TRNW, THSD7B, HPSE, PNPLA3, TRNV, HLA-DQA1, POLG2, ENPP1, NUBPL, HP, TXNIP, MAPK14, ACE2, TRNF, TRIM31, CYP2C19, RNASEH2B, HPN, SERPINF1, OR2B2, HLA-A, PALB2, PPARGC1A, CCDC28B, ND6, PARN, CCN2, SLC39A8, CTNNB1, TRNK, CDH23, TERC, HMOX1, EHMT1, HMGB1, TRNS1, ACE, CYP21A2, TREX1, USB1, ARMC5, DBP, CTC1, CAPN10, TRNQ, ELMO2, CST3, DAB2, NDUFAF5, DECR1, TRNS2, CARMIL1, SUGP1, RRM2B, NDUFA1, NDUFS6, MTOR, NEUROG3, PDE11A, TPRKB, NDUFS8, NDUFAF3, NDUFV2, FOXO1, ADIPOR1, NDUFAF1, NDP, NEUROD1, NFATC1, UCP1, FGF2, NFE2L2, UCP2, TIMMDC1, POLK, TTPA, TMEFF2, NGF, NHS, NOX4, KDSR, HIF1A, GLUL, TINF2, GIP, GJB3, NDUFB3, GH1, NDUFB9, ZBTB20, NDUFB10, NDUFS1, GLO1, GCGR, GCG, NDUFS2, NDUFS3, GATM, NDUFAF4, NDUFV1, NDUFA6, SAMHD1, CD274, GRHL1, GAD2, GABPA, NDUFS4, G6PD, FFAR1, RTEL1, PLA2G15, UCP3, MAML3, FOXRED1, ZCCHC8, CRP, NHP2, SIK3, KAZN, CELSR2, TRPV1, PALLD, TMEM126B, EDN1, OPA1, ACSS2, ANGPTL8, TNFRSF11B, CTRC, TWNK, RETN, DSPP, TGFB1, ATN1, SHROOM3, HGF, DPP4, SLC30A10, BEST1, NOP10, VEGFA, GPT, GPX1, FABP4, SEMA5B, NOS3, CCHCR1, NDUFB11, VDR, NPY, LEMD3, BCAS3, CDKAL1, TLR4, SARS2, ESR1, ROS1, TNF, WRAP53, RCBTB1, GSK3B, CHDH, ZFYVE26, EPO, GSTM1, OR12D3, PIK3CA, GPR101, APOA1, BBS1, GDF15, EIF2AK3, MIR146A, AVP, SLC5A2, ATP2B1, SLC5A1, SLC2A4, SLC2A2, SLC2A1, PTH, MIR21, ARSA, ST3GAL4, KL, SI, PTPN1, KCNQ1, APRT, APOC3, BBS2, BCHE, PRSS2, NSMCE2, PCSK9, PRKACA, WDR72, COPD, CAD, OR10A4, CAVIN1, MMP9, MAPK1, C9, MIR126, RAPGEF5, GOLGA6A, MMP2, MAPK8, NR3C2, ITPK1, LINC02694, BGLAP, BDNF, APOB, HMGA2, PRKAA2, SHBG, REN, ADRB3, RENBP, ZGLP1, HPN-AS1, PARP1, FADS1, CCL2, ALDH1A2, LPL, LRP6, ADAR, LIPC-AS1, ADA, LOC102723407, MC4R, PDE8B, ABO, ABCA1, IRS2, MBL2, AGER, LHX1, AGT, AKR1B1, MFAP1, PIP5K1B, USP8, AIP, BAZ1B, MOK, SELENBP1, MGAM, LCN2, CXCL12, AGTR1, ALDH2, ZPR1, LDLR, ALB, RBP4, LEP, AKT1, AHSG, LGALS3, PRKAB1, SLC34A1, IL1B, IL4, RNASEH2A, RBM45, SPP1, PLEKHH2, IAPP, LMNB2, IGFBP3, IL1A, COX3, PMM2, CPA1, ACCS, IL1RN, ND2, RNASEH2C, ARL6, POLD1, GJB4, FGF21, PIK3CB, PIK3CG, IDE, NLRP3, TRNC, UBE2Q2, PLCG1, IFNG, IGF1, PLG, NDUFA11, CETP, PIK3CD, NDUFAF2, MTHFR, XRCC4, UBXN11, ICAM1, POLG, FOXP2, SOD2, UBR1, CXCL8, IL17A, SLC30A8, PEX6, MPO, CASP3, PRKAA1, PEX1, PDILT, CERT1, HJV, IL10, IL18, CD34, CD36, HMGCR, ADAMTS13, CYP2E1, GHR, CYP3A4, EGFR, P2RY12, VWF, FGF23, TAC1, ALOX15, IFNA1, PDHX, SST, GPBAR1, P2RX7, IFNA13, HSPD1, DIANPH, TCF7, IGF2BP2, HSD11B1, SOST, SOCS3, RNF19A, ANGPT1, CALCA, KLF11, BCL2, SREBF1, UTS2, ADM, HPGDS, GSR, AHSA1, CRK, AIMP2, INSM2, GRAP2, SIRT3, ATP6AP2, MALAT1, HHEX, POLDIP2, TRBV20OR9-2, ACHE, AFP, DMPK, APP, FABP2, S100A8, APOL1, GAST, GFAP, S100B, REG1A, TNFSF10, MSTN, APLN, ADH1B, TRPM2, SELE, VCAM1, PLA2G7, G6PC2, CCK, FFAR4, PPARD, MME, TLR2, GSTT1, CD59, DDIT3, MIR155, TNFRSF1B, F3, IGFBP2, PYY, AR, PTPN6, PLAT, LAD1, SGK1, HSPA14, PTPRN, ADIPOR2, IGF2, EGR1, RFX6, KLK3, CXCR4, FGF19, FOXA2, MAPT, IDO1, CXCL10, MAP6, HBA1, KDR, OGA, NTN1, AKR1A1, HSPA5, HDAC3, KNG1, XBP1, PADI4, VEGFC, GNB3, IL2, LTA, SOCS1, SOAT1, CEL, PDK4, C20orf181, SCD, DMD, FBL, F2, NOS2, SERPINA5, MIR375, MIR223, PTK2B, NFKB1, ACR, SIRT6, PRKCB, NQO1, SPARC, ANPEP, RUNX2, PLXNA2, ANGPT2, G6PC, CCR2, IL22, AOC2, CYP19A1, APOA4, MIR34A, CTSD, PAEP, MIR29A, JAK2, CUX1, CYBA, DHX40, PINK1, B2M, PCK2, ATF4, MIR27A, PSIP1, PTX3, PTPRN2, MIR132, PCK1, BTF3P11, SPX, CCR5, ENTPD1, NAMPT, CD68, PPARGC1B, JAZF1, IGFBP1, PRKCA, SREBF2, IFNL3, BTBD8, SORCS1, INPPL1, TRMT10A, HSPA4, CNR1, MAPK7, TSPO, GSTK1, CNDP1, ISG20, SORBS1, CPOX, PECAM1, HSP90AA1, HSPB1, BMP7, MET, SLC17A5, CCL5, ANGPTL2, SORT1, BECN1, CLEC16A, ACACB, MTCO2P12, AGRP, NUCB2, ADRB2, OLR1, ERBB2, TNFSF11, TFPI, NR3C1, SMUG1, PERCC1, ACTB, ANGPTL3, S100A9, PDE5A, DPT, TFRC, HAVCR1, THBS1, TIMP1, APOM, S100A1, NRG1, SFRP5, P4HB, AMBP, SIGLEC7, MMRN1, HCAR2, RN7SL263P, ACP1, VPS51, NPHS2, KHSRP, MAPK3, ABCB6, SMAD3, MMP14, SMAD2, NAT2, TNFRSF9, SNCA, RIPK1, PEA15, CALR, MMP3, DGCR2, CASP8, ANXA1, LINC01672, LRP2, PPIG, LGALS1, SLC6A2, AHR, AKT2, HSPB3, MIR204, KMT2D, DCAF1, PTPN2, SELP, ARTN, MGP, POR, KLK1, AGTR2, SLC9A3, OGT, JAG1, BAX, GRK2, BRS3, SUMO4, DEGS1, LOX, MMP1, MICA, CARTPT, ERVK-18, S100A12, ABCG1, NOS1AP, FZD4, CEACAM5, PON2, HK2, SELENOS, DKK1, SIRT2, ATF6, EDNRA, HCLS1, HCRT, TH, TRIB3, DNMT1, HLA-C, TM7SF2, TREH, MNX1, WDHD1, REG3A, CD38, FN3K, HMGA1, HHIP, NOX5, CYP11B2, TKT, GSTP1, CYP2C9, ANGPTL4, GGT1, GFER, NOX1, SETD2, GAPDH, GAP43, NR5A2, FLNA, GOT2, FOXM1, UCHL1, NPPC, TXN, TTR, TRPC6, ADA2, ACSL1, TRAF6, NOTCH1, TNFRSF1A, ERN1, NPPB, CYP2J2, PRKN, RORC, GPR119, IL33, CLU, STK11, CHGA, FBXO32, IARS1, CECR, GIPR, TRIM63, RPAIN, CREBBP, SLCO6A1, CSF2, CCN1, HSPB2, STAT5B, CSF3, AGFG1, TRIM13, TCF3, IL2RB, PPP1R3B, PLA2G2A, PKD1, VIM, CCL4, PPIA, NPPA, MYD88, PLA2G1B, CCN3, MANF, RYR2, TNNI3, TYK2, SPG7, PLTP, PPP1R3C, ZFP36, PIN1, ST8SIA4, SMS, MT1A, PPID, PSMD9, MMP8, PDR, SLC6A3, PTHLH, ZEB1, PC, MIP, SLC6A4, TEK, PCSK1, PDCD1, PROX1, TFAM, PRL, TFE3, SLC22A1, MEN1, TIMP4, SELENOP, SYP, PGF, PGM1, RAF1, SELL, PLA2G6, RARRES2, CXCL5, SERPINA1, CCL21, SLC2A3, OGG1, TIMP3, TIMP2, DDAH2, PSAT1, ELANE, CASP1, ARID4B, UBASH3A, TLR9, CCKAR, TET2, ETV3, SLC52A1, SLC35G1, SLC47A1, IMPACT, EPHB2, CBLL2, EPHB1, MEG3, ENO2, ELK3, IL23A, CAPN1, ISYNA1, CFB, MIR15B, CABIN1, MIR15A, DDAH1, MIR145, CD2AP, MIR143, BGN, CANX, GNAO1, DESI1, SGSM3, FLT1, GAL, CA2, GP6, CD86, NRG4, ATP2A2, CISD1, CYP2D6, CYP2B6, CXADR, CX3CR1, CTSS, CTSB, HDAC11, ZC3H12A, CSN2, PRRT2, MUC16, SLC2A10, CMKLR1, CNTF, CPE, COL9A3, COPA, MUL1, CYP7A1, CHRM3, TRPM6, EDNRB, CD40, CD40LG, SLC2A9, DYRK1A, CD44, ACKR3, CIP2A, C1QTNF5, IL21, CDK4, TRPV4, DLD, CDKN1C, CHI3L1, DCN, CELA3B, COMT, CFH, KIR2DS2, MIR27B, FST, FOXO6, ITGAM, IDDM7, MIR29B1, GGTLC4P, IFNAR1, MIR29B2, GGT2, KIR2DL2, TUBB4B, KCNMA1, GGTLC3, AMPD1, IL6R, GGTLC5P, CXADRP1, MIR146B, PPIF, IL15, SH2B3, ELMO1, KEAP1, ITGB2, BMS1, SOX13, ADORA1, ADRA2B, INSRR, PGR-AS1, PER3, CDC123, HRC, MIR200B, TNDM1, MIR210, ARR3, KAT2B, ACACA, LNPEP, MIR217, HLA-DPB1, HSPA1A, HSPA1B, HLA-DQB2, ADCY5, C2CD4A, GGTLC1, GPR151, H3P10, MIR486-1, RPS6, BPIFA2, PWAR1, ABCC11, WNT3A, DNER, MIR483, RPS6KB1, C1QTNF3, CCL20, DBA2, TMX2-CTNND1, SALL1, SRL, UCN3, STAT1, SSTR5, LOC102724197, STAT5A, TMEM18, SSTR4, CCL11, SLC26A9, TRIM21, MRGPRX3, ST2, NOXO1, MRGPRX4, SOX2, MBOAT4, SLC18A2, SMPD1, MIR24-1, MIR22, GPIHBP1, PWAR4, STX4, ENHO, TBPL2, C2CD4B, C1QTNF12, LINC01194, SLC11A1, FRMD3, MIR203A, MIR122, MIR130A, MIR140, SLC6A8, MIR200C, MIR149, MIR19A, MIR199A2, MIR199A1, SLC5A3, MRGPRX1, MIR25, MIR503, MIR93, LYPD4, SP1, MIR499A, MIR184, PLB1, CREBRF, GPR166P, VN1R17P, MIAT, MIR17HG, SFTPD, SLC16A11, SHC1, MIR30D, TMPRSS6, OXER1, MIR302A, CRTC2, ZBTB7C, SNAP25, SNAI1, TPCN2, GPRC6A, IL27, TP63, CIDEC, STXBP1, SCGN, LILRB1, TCFL5, WNT1, NFAT5, CXCR6, KHDRBS1, CCT2, KCNQ1OT1, PRDX4, HYOU1, NSA2, RACK1, NOD1, SEMA3A, SLC22A7, BTN2A1, KLF2, BACE1, PAMR1, UBE2I, POC1A, TXN2, PRPF6, NUP62, UTRN, PDAP1, VDAC1, DAPK2, LPAR3, VGF, PES1, LPIN1, YY1, YWHAZ, STXBP3, F2RL3, TAM, S1PR2, GPR55, MSC, SLC33A1, LPAR2, WASF1, CD163, PLA2G10, SQSTM1, ARHGEF7, DENR, CES2, TNFRSF6B, KLF4, SLC7A5, RIDA, TNDM, TSHZ1, ZMYM2, AIR, CELA3A, ALMS1, NR1H4, HDAC4, GLP2R, BCAR1, NPEPPS, NR4A3, TBPL1, AIM2, COX5A, SH2B1, FETUB, FOXD3, IGAN1, EBF2, COP1, ABCG8, NOD2, IDDM17, SRR, BACH2, CORO7, LGR6, TFF3, UBL5, JPH3, TG, CAMK1D, SLC52A2, TCF21, INTU, TXNDC5, UBASH3B, SULT1A1, PPP1R15B, CCDC8, ANGPTL6, MFRP, SETD7, VTCN1, COL18A1, TACR1, TAPBP, TAT, TBP, LIN28A, TGM2, FAM20C, TRPV5, TWIST1, GHRL, UFM1, TSC2, TTN, DTL, TLR7, CYB5R4, ZNF395, DCTN4, SDF4, ASCC1, IL20, TBK1, IL17B, NR2C2, TPO, TPD52, ERRFI1, TREM2, KRT20, DLL4, TRPM7, ANO1, TMSB4X, TSPAN8, TSPAN7, TLR3, SOX6, STAP2, KDM3A, PAG1, SYN2, H3P40, HLA-DQA2, CELA1, NFKBIA, NGFR, NIDDM2, EIF5A, C3, NNMT, NOTCH3, EPOR, NPHS1, SLC11A2, NRF1, HDAC2, KLF9, BTC, NFKB2, PTTG1IP, NEFL, ECE1, CALCR, MYH9, CAPN2, MYC, MVD, DUSP1, CASP9, CASQ1, PRMT1, CAV3, MSX2, MST1, MSMB, BSG, OGN, HSF1, GYPB, PCNT, PCSK2, PCYT1A, F2R, GYPE, F2RL1, ATF3, ERBB3, GYPA, GUSB, GTF2H1, ARNT, F8, PGC, ATP2B2, EZH2, ATP5PF, PBX1, ATR, GZMB, PAWR, P2RY1, BCL2A1, EREG, HAS2, P2RX3, ERCC2, BMP4, BMP6, ERBB4, BMPR2, CD3E, CD14, SLC25A3, LAMP2, CTNND1, KRT16, COL1A1, CTSL, CYP1A1, LAMC2, CYP8B1, LIF, LBP, LCK, IL13, LETM1, CYP27B1, CXCR1, COL11A2, KRT5, COMP, KLF6, KISS1, IMPDH2, COX8A, CPB1, CSTA, KCNJ1, IRAK1, JUND, JUN, CSE1L, CRYZ, CRH, CRMP1, CD55, LIG4, CD19, DIO2, MDM2, CDK5, MECP2, DHPS, MEF2A, MEFV, MIF, LRP5, CD69, CD80, MS4A1, DPYD, HSPA9, DPYS, CDKN2B, DEFB1, IGFALS, MCL1, MC3R, DDT, IGFBP7, CEBPB, MB, MAS1, MAOA, MAFD2, CETN1, CTSC, CHAT, IL3, LYZ, PHB, ITPR1, FBN1, PMP22, FGFR4, GPER1, FASN, PON3, GPR35, FDPS, ADH1C, AMD1P2, ALAD, PVR, ADORA2A, AMH, RNASE3, RAD1, FAP, GCLC, PVT1, PROS1, GATA4, FKBP5, ADCYAP1, ALCAM, GLRX, FLG, FOXO3, ADD1, FLII, FMO3, GC, FKBP4, PSPN, ALOX12, ALOX5, GCH1, PROC, ALOX5AP, PSMA6, AMD1, PLAG1, ANXA2, GJA3, PTGS1, APOA2, ADRA2A, ADRA1A, GRN, F10, APLNR, PSPH, ADORA2B, FAS, ANXA6, GK, F11, CBLIF, FES, GPR42, FABP1, XIAP, GAS5, MFSD1, TFB1M, PROK2, GORASP1, PDIA2, WDR13, FGFR1, DAPK1, NIF3L1, ELOVL5, DIO3, THADA, GNPNAT1, SLC25A19, DLAT, AGXT2, DGKQ, FGF13, SCPEP1, DIH1, FBRS, DAPK3, DBH, TSPYL2, DES, DMRTA1, LHPP, TNMD, PCIF1, WNK1, DEFA3, DEFA1, DIAPH1, UBE2O, FGL1, STRA6, INSIG2, FHL1, ABCG5, DDOST, FHL2, PRDM16, DCX, RMDN1, METTL9, IKZF5, RTN4R, MOV10L1, UBE2Z, CSK, CERS2, CSH2, PDGFD, NRBP1, FLAD1, CDK5RAP3, SCUBE1, CTAA1, CYP4F12, SLC37A4, SH3KBP1, MOGAT2, ASRGL1, ERVW-1, CTBS, BICC1, COASY, CSH1, PADI1, CSF2RA, NME7, SEC61A1, MED25, CRY1, GPR132, MAP1LC3B, TSC22D4, CS, SLC38A1, KCNH6, NENF, CSF1, DSE, ZNF436, CSF1R, NT5C, PIK3R4, FUT2, CYP3A5, CYP4A11, F11R, CYP24A1, FOLR1, FOLH1, PAGR1, CYP27A1, FGF1, FSD1, MBOAT7, FLT4, DAB1, TMED7, TVP23B, ASPSCR1, AK3, NAA16, CTF1, CYP2D7, CTNS, NR5A1, FSHMD1A, VASH2, FOSB, SHCBP1, CTRB1, CTSG, MBL3P, CTSK, FOLR2, NLRX1, CYC1, ASAP1, CYP1A2, DLX3, CLEC1B, GER, SF3B6, ENG, MPC1, ZFR, ENO1, ENPEP, EP300, EMCN, HEMGN, FAT1, GIMAP5, FBXW7, RHOT1, ERCC1, RMDN3, MSTO1, RASD1, ELAVL2, SARDH, PLXNA3, EIF4E, ENAH, EIF4EBP1, SYBU, EIF4EBP2, FERMT1, ITLN1, GDE1, RNPC3, PIAS4, MIOX, TRIM33, NBAS, ATP6V1H, SERPINB1, MTPAP, FBLN1, MAP3K20, ERG, FABP3, FBLIM1, FEV, ARL15, H2BS1, EPB41L4B, TREM1, DDIT4, F5, ROBO4, SIAE, APBB1IP, SMOX, YIPF1, F9, CNNM2, FAM3B, SLCO1C1, CASZ1, ESD, MOCOS, MARCHF1, TUG1, ESRRA, ETS1, IL17D, DYM, NCAPG2, VPS13C, FABP5, HDL3, EXT2, IL20RA, FBN2, ELOVL3, EIF2S1, AKR1B10, SEMA6A, AHRR, AS3MT, DACT1, GJD2, DSC3, ADAMTSL3, PELI1, GOPC, RCAN1, KMT5AP1, DSG3, GPC4, PCBP4, DCDC2, CFAP97, FGB, NT5C3A, NEUROD4, DMRT1, DNAH8, ARHGAP22, OPRPN, GLRX5, PCTP, ZNF410, MZB1, DNASE1, DNM2, DNMT3A, MARK4, DOCK3, RNF213, COQ9, FCGRT, EGF, HBEGF, NSFL1C, EBF1, LANCL2, SIRT7, ZC4H2, EDA, S1PR1, GPRC5C, GSDMB, EEF2, ACOT13, USE1, EFNB2, EFEMP1, RAB14, MYDGF, PCDHGA4, ZNF253, SUCNR1, ADAMTS9, CHPT1, ENY2, NMUR2, SPHK2, DUSP4, DUSP9, E2F1, HSD17B7, FBP1, CLDND1, ASAH2, TOR1A, CYP26B1, RP9, LTB4R, SESN2, MIR200A, MIR214, MIR211, ARRB2, STS, MIR205, SERPINC1, ATIC, RERE, ATP12A, AREG, FXYD2, ATP2A1, MIR190A, MIR185, MIR182, MIR181C, ATP4A, MIR152, MIR216A, MIR221, RNASEK, MIR30A, MIR99A, MIR98, MIR96, ANG, MIR320A, MIR31, MIR30E, ANXA5, MIR29C, MIR222, APC, APCS, MIR296, APOH, AQP7, MIR23B, MIR23A, AQP9, MIR150, AVPR1A, MIR148A, GPR142, TMEM189, DST, LINC-PINT, KLF5, BUB1, TICAM2, SLCO4C1, ZACN, C1QBP, AVPR1B, SOX2-OT, CYCSP51, NANOS3, C4A, VWA2, TSPAN33, GADL1, ARMH1, TMEM189-UBE2V1, SERPINA13P, LINC01550, BNIP3L, AVPR2, BAAT, MIR142, BAD, MIR134, CCND1, HCN2, BCR, MIR107, MIRLET7G, GTF2H5, BID, SMIM10L2A, HES3, C1QL3, LIN28B, CCL4L1, AMY2A, CDNF, TMEM119, DNM3OS, ADAM10, TRPM2-AS, EMSLR, LINC01150, OPN1MW3, ADCY3, ARAP1-AS1, ADCY8, ERVK-20, ACVR2B, KLRC4-KLRK1, RBM14-RBM4, VIM-AS1, OCLN, ARAP1-AS2, PLIN2, MIR466, AAA1, MIR6835, ACP3, MIR330, LOC110806263, SERPINA3, H3P42, H3P28, H3P8, ABL2, LOC113664106, LINC02605, LOC111674464, ERVK-32, LOC102724334, ACADSB, ASIC1, CST12P, CERNA3, THRA1/BTR, ACP5, TP53COR1, CBSL, TMED7-TICAM2, C4B_2, H3C9P, MIR433, ALDH1A1, HCC, DELYQ11, ABCD2, NPS, MIR497, MIR409, HNP1, MIR449A, IBD20, MIR429, MIR424, MIR377, ALPI, ALPP, TRIM72, PLF, MIR338, ECSCR, SMIM10L2B, ALAS1, SFTPA1, KIF28P, SOD2-OT1, HOTAIR, TLE5, MIR675, PSS, AFM, AGA, OPN1MW2, POTEF, AIC, INS-IGF2, MIR618, MIR615, MIR579, H3P37, AIF1, BRINP3, C4B, CRISPLD2, AZIN2, CISH, MYSM1, CYGB, CKM, ERCC8, TXNRD3, CLCN7, TMEM54, SLC46A1, CHIT1, IL17F, CLK2, TP53INP1, CLPS, CCR1, ACKR2, GPSM2, SLC38A5, CHRNA4, C1QTNF7, OSCAR, THEM4, CD52, FOPNL, DEGS2, CYP2R1, MSS51, RLN3, CEBPA, DCD, TADA1, MARCHF3, ARAP1, LRG1, SNAP47, CENPA, ABHD15, RMI2, SLC5A11, GPR146, CMM, NAF1, MTG1, SLC41A2, KISS1R, ZGPAT, HOPX, SYVN1, CPT2, NLRC5, ASCC2, CR2, CREB1, CNP, TKTL2, MAGT1, ATF2, MIXL1, FSD1L, CREM, SETDB2, KCNK16, SPZ1, CLDN4, CPD, CPB2, CTBP1-DT, ARRDC4, CNR2, TIMD4, RFT1, COL6A3, PYGO2, BMF, KNSTRN, UCN2, GALP, KRT90P, CORT, PLXDC2, ORAI1, HAVCR2, ATP5MD, MSI2, CDKN3, C5AR1, FOLH1B, FNDC5, CAPN3, CAPS, CAST, CARS1, HOXA11-AS, PHACTR1, RANP1, LRRC55, CNIH2, NRK, PRSS55, CBLB, CBS, PAOX, ARID2, PTCRA, NLRP6, ANGPTL5, CALM3, TSACC, KSR2, MMAB, BPIFA4P, CA1, CACNA1A, RSPO1, ZNF763, CACNA1E, CHAMP1, HECTD4, TCERG1L, DDR1, NEAT1, SLC25A20, HS6ST3, CALB1, NEGR1, CALM1, CALM2, CCKBR, CD1D, SGMS2, UPRT, SERPINA12, PIWIL4, CACUL1, SIRPA, HT, ADGRE5, CDC42, CDH1, FUNDC1, CD5L, KLF14, TAAR1, WDR36, CDH5, CDH11, CDH13, CDK2, CDKN1B, CD47, SCARB1, WIPF2, CD33, CD8A, MARCHF10, SPRED1, LINC00599, AMOT, SLC2A12, PPM1K, KLB, UPP2, RMDN2, CD27, ERFE, CD28, SIK1, PDIK1L, ZNF569, CILP2, EEF2K, GAPDHS, NPC1L1, MTNR1A, WNT6, WNT5A, WNT2, MTTP, NSD2, MUC1, LAT2, MUSK, MMUT, VPREB1, MUTYH, VIP, EZR, VHL, MYF6, VEGFB, MYL2, PPP1R12A, NAB2, WNT7A, MTM1, MPI, XBP1P1, SCG2, FZD5, MPST, PAX8, TUBA1A, MPZ, PXDN, MRE11, ABCC1, MS, ZNF236, MSH3, SF1, MT1E, YES1, MT1JP, XRCC1, XPC, XK, VASP, KDM6A, NAGLU, USF1, NNAT, CRISP2, NM, TPM4, NME1, NOS1, NOTCH2, TNFAIP3, NRTN, YBX1, TLR5, NTS, NTSR1, NUMA1, OAS3, TLE3, TLE1, OAT, TJP1, NINJ1, TRPC3, NIDDM1, NCL, UPK2, UPP1, NUBP1, UGCG, NBN, NCAM1, NCK1, UCN, UBE2V1, TSC1, NDN, NFATC3, TNFSF4, NFATC4, NFIL3, TTC4, TST, TSHR, SLMAP, MPG, SLC16A4, LTBR, CCN5, NRP1, DPM1, TRIM24, SUCLA2, TNFRSF10B, TNFRSF11A, SIGLEC5, FADD, MXD1, SMAD1, TNFSF14, HRK, SMAD7, DGAT1, MAP2, VAMP4, DYNLL1, MBNL1, CCN4, LRPAP1, DDX39B, ASAP2, KRT18, SLC7A7, GPRC5A, KTN1, LAMP1, RPL14, CH25H, RPSA, TRPA1, LCN1, LCP1, EIF2S2, LDHB, FUBP1, SPHK1, LGALS2, LGALS3BP, LGALS4, LOXL2, NCOA1, ABCB11, MBP, MC2R, UBL4A, MGAT2, KITLG, AKAP1, HBHR, CXCL9, AAAS, MITF, MLH1, ECB2, MAP3K9, MAP3K10, STAM, MLN, FZD3, FOXO4, NTT, MMP10, MNAT1, TKTL1, AXIN1, BRAP, CST7, STC2, SMCP, MADD, ADAM11, DNAJB9, MAPKAPK5, LGR5, MDH2, ELP1, MFGE8, IRS4, MDM4, CUL1, MEHMO, EEA1, TAGLN2, MEOX2, LOH19CR1, ODC1, ODF1, ORM1, PSEN1, SLC5A5, PSMD7, PSMD10, SLC3A1, PTBP1, PTCH1, PTGDS, PTGIS, SLC1A3, SKP2, SIX1, PMEL, ST6GAL1, PTH1R, PTPRD, PTPRF, SGCA, SFTPC, SFTPB, PSEN2, PRSS8, TIE1, SLC7A2, POU4F1, SNRNP70, PPP1R3A, PTPA, PPY, PREP, PRKCD, SMO, SMARCA4, MAP2K3, MAP2K6, SLC20A2, SLC20A1, SLC19A1, SLC14A1, SLC12A3, SLC10A2, MASP1, SLC9A1, SRSF6, SRSF5, NECTIN1, SEMA3F, SATB1, RHEB, ACSM3, RHO, RIT2, S100A10, RNASE2, BRD2, RNY1, RYR3, RNY3, RXRG, RREB1, RPS19, ROBO1, RPS6KA1, ROCK1, RPL36A, RPL29, RHD, CLEC11A, SCN2A, CCL22, SELPLG, RAC2, RAG2, RAN, RAPSN, SDC4, RBP3, CXCL11, RCN2, SCN7A, OPN1LW, CCL16, REG1B, RELA, REST, RFC2, RGS1, SCN10A, POU2F2, POU2F1, SOD3, ADAM17, PAX2, TDO2, TDGF1P3, TRD, PCBD1, TCF19, PCDH8, PCMT1, PCNA, TCF4, TBXA2R, PDB1, TBX1, PDC, PDE7A, TAP2, TAP1, TALDO1, TAGLN, TERF1, PAPPA, PAM, TGFBR2, KLF10, THRB, THRA, THBS4, CLDN11, OXA1L, THBD, OXTR, TGFBR1, PRDX1, TGFBI, TGFB3, TGFB2, P2RX4, TGFA, P2RX5, PA2G4, FURIN, PDGFRA, PER1, SORD, SYT5, PLCB3, ITPRID2, PLEK, SRY, SERPINF2, PLK1, AKR1D1, PLN, SPRR2A, SPR, PLXNA1, SPINT1, PNOC, PODXL, SOX9, SOX4, POU1F1, SOS1, SORL1, PLAUR, PLAGL1, ST13, SERPINB6, SYT1, PF4, A2M, VAMP2, SUV39H1, PF4V1, ABCB1, STX1A, STIM1, ST14, ELOVL4, STC1, PIM1, PKLR, STAT4, PKM, STAT2, PKNOX1, MTA1, SLC16A3, ICOS, NFASC, UNC13A, H1-2, PDCD11, TPX2, HABP2, HARS1, MSRB2, HBG2, KLRK1, SACM1L, CARD8, INPP5F, NLRP1, ANKRD26, AAK1, VASH1, PUF60, MRAS, TUSC2, MYO16, KDM6B, NNT, PASK, GSN, GSTA4, BRD4, TRAM1, NPTXR, ABCB10, GTF2H4, TARDBP, SIRT4, ZNF629, CUX2, MCF2L, GYS1, PHLPP1, PLCB1, FBXO28, NBEAL2, TBC1D1, ATG4B, HCRTR1, ACOT7, PHB2, STK38, ESM1, SLC29A2, RAPGEF4, LIAS, KIF2C, SLC27A2, IMMT, METAP2, HNRNPK, EBNA1BP2, ERP29, PDIA5, BTG3, PRSS21, RALBP1, SUB1, PAPOLA, HOXA3, CPSF4, ONECUT1, NUP42, RPP14, AKAP13, RBPJL, PARK7, SLCO2B1, KLF12, UBE2K, DUSP12, PTGDR2, HLA-DMA, IRAK3, HNF4G, MAP4K5, SLC2A6, HLA-DMB, HMGCS2, SLC7A9, NR4A1, FAF1, PTPRT, CXCL1, RBFOX2, ARHGEF1, RPS6KA6, PDCD4, GPR162, HCAR1, GDF2, GLS2, NAAA, GDF10, CACYBP, TAF5L, FOXP1, SND1, LAT, GDNF, GEM, ATP2C1, GFPT1, GHRH, GHSR, GREM1, OPN1MW, GBE1, GRM5, MAT2B, TRPM5, GALNS, GALNT3, GAS1, DROSHA, CNIH4, STXBP6, GAS6, DBNL, FLVCR1, OSTM1, LAMTOR2, DEXI, REM1, TRBV7-8, DLL1, IGHD1-7, SLCO1B3, TOR2A, RPL10, GJA4, ANKRD2, GJA5, TPSG1, ARIH1, TAFA5, GCA, ZNF318, PANX1, TMEM245, RAB38, GPR39, FFAR2, GPX3, SH3BP1, GPX4, GRIA2, ELP5, GRIN1, GRIN2A, GRIN2B, GRM2, BACE2, MCHR1, PART1, GORASP2, FBXO8, FBXO25, PHGDH, GLA, LRIT1, GLI2, ACOT11, TKFC, SNED1, UTS2R, CHMP2B, TIPARP, KANK2, TPGS2, GLUD2, GNAT1, GP1BA, CXCR3, MMP24, MALT1, HOXC4, ING2, INSL3, RASSF2, INSM1, HDAC9, SART3, SEMA3E, IRF1, LPIN2, SOCS5, IKBKE, IRF7, ISL1, PRDX6, ITGAX, ITGB7, ITIH1, H6PD, CCL4L2, CIR1, PIEZO1, IP6K1, MRPS30, GIT2, PARP2, CHAF1A, INSL5, IL15RA, BCL2L11, HDAC6, CASP8AP2, FOXK2, CCS, ILF3, ILK, TNFSF15, IMPDH1, MVP, MFN2, RNF10, NUAK1, TBC1D4, ING1, EI24, BAG3, IVD, JAK1, LONP1, KCNJ9, SLIT2, KCNN4, B4GALT5, KIR2DL3, KIR3DL1, KLKB1, PIWIL1, STK17B, MFHAS1, NOG, PTTG1, HACD1, KRT8, ARHGEF2, PCSK7, HGS, PDCD5, KCNJ6, KCNJ5, KCNJ3, FHL5, GAL3ST1, ADAMTS1, ADAMTS4, MAPK8IP1, NAPSA, ROCK2, CHST3, PCYT1B, HOMER1, CIAO1, MAP4K4, JUNB, NCR1, KCNC2, CYP7B1, KCNE1, KCNH2, CD101, MAMLD1, GJC1, ABCC5, IDDM3, HSPG2, NDST1, HTC2, GNLY, HTR2A, CCT4, HTR2C, SLC35A1, TNC, IBD2, UBD, IBSP, ID1, DEAF1, CIB2, FBLN5, SEMA4D, CARM1, CAP1, SLCO1B1, SH2B2, IVNS1ABP, TRAF3IP2, USP19, HPX, HRH1, PGRMC1, WASF3, SEPTIN9, NEK6, DHS, GIPC1, HSP90AB1, MASP2, HES1, HSD3B1, HSD11B2, TNFSF13B, CELF1, HSPA2, POSTN, TIMM44, TOMM40, IL12B, VAV3, IL1R1, RABEPK, MRPS31, GDF11, DDX39A, NUTF2, IL4R, WASF2, IL6ST, MBNL2, IL7, IL7R, LRPPRC, RASGRP1, HIPK3, IL9, PTPRU, IL18BP, NR1H3, ABCC4, CDK2AP2, STUB1, IFNAR2, C1D, LYPLA1, RBM14, IDDM4, RAPGEF3, CPQ, PEMT, IDDM11, CORO2B, SIGMAR1, IGF2R, CITED2, MICU1, IGFBP5, TFG, LAMC3, TNIP1, LANCL1, A1BG

-
Clanging
Wikipedia
. ^ Spitzer, Manfred (1999). The mind within the net: Models of learning, thinking, and acting .
-
Ocular Myasthenia
Wikipedia
MG may be limited to the muscles of the eye (ocular MG), leading to abrupt onset of weakness/fatigability of the eyelids or eye movement. MG may also involve other muscle groups ( generalized MG ). ... Diagnosis [ edit ] The variable course of MG may make the diagnosis difficult. ... Treatment [ edit ] The prognosis tends to be good for patients with MG. It is often best not to treat mild cases of MG. ... The symptoms of ocular MG can also be addressed by non-medicinal means.
-
Chronic Hallucinatory Psychosis
Wikipedia
Others, again, might be swept into the widespread net of dementia praecox . This state of affairs cannot be regarded as satisfactory, for they are not truly cases of melancholia, paranoia, dementia praecox or any other described affection.
-
Androgen Deficiency
Wikipedia
The Food and Drug Administration (FDA) stated in 2015 that neither the benefits nor the safety of testosterone have been established for low testosterone levels due to aging . [6] The FDA has required that testosterone pharmaceutical labels include warning information about the possibility of an increased risk of heart attacks and stroke. [6] v t e Androgen replacement therapy formulations and dosages used in men Route Medication Major brand names Form Dosage Oral Testosterone a – Tablet 400–800 mg/day (in divided doses) Testosterone undecanoate Andriol, Jatenzo Capsule 40–80 mg/2–4x day (with meals) Methyltestosterone b Android, Metandren, Testred Tablet 10–50 mg/day Fluoxymesterone b Halotestin, Ora-Testryl, Ultandren Tablet 5–20 mg/day Metandienone b Dianabol Tablet 5–15 mg/day Mesterolone b Proviron Tablet 25–150 mg/day Buccal Testosterone Striant Tablet 30 mg 2x/day Methyltestosterone b Metandren, Oreton Methyl Tablet 5–25 mg/day Sublingual Testosterone b Testoral Tablet 5–10 mg 1–4x/day Methyltestosterone b Metandren, Oreton Methyl Tablet 10–30 mg/day Intranasal Testosterone Natesto Nasal spray 11 mg 3x/day Transdermal Testosterone AndroGel, Testim, TestoGel Gel 25–125 mg/day Androderm, AndroPatch, TestoPatch Non-scrotal patch 2.5–15 mg/day Testoderm Scrotal patch 4–6 mg/day Axiron Axillary solution 30–120 mg/day Androstanolone ( DHT ) Andractim Gel 100–250 mg/day Rectal Testosterone Rektandron, Testosteron b Suppository 40 mg 2–3x/day Injection ( IM or SC ) Testosterone Andronaq, Sterotate, Virosterone Aqueous suspension 10–50 mg 2–3x/week Testosterone propionate b Testoviron Oil solution 10–50 mg 2–3x/week Testosterone enanthate Delatestryl Oil solution 50–250 mg 1x/1–4 weeks Xyosted Auto-injector 50–100 mg 1x/week Testosterone cypionate Depo-Testosterone Oil solution 50–250 mg 1x/1–4 weeks Testosterone isobutyrate Agovirin Depot Aqueous suspension 50–100 mg 1x/1–2 weeks Testosterone phenylacetate b Perandren, Androject Oil solution 50–200 mg 1x/3–5 weeks Mixed testosterone esters Sustanon 100, Sustanon 250 Oil solution 50–250 mg 1x/2–4 weeks Testosterone undecanoate Aveed, Nebido Oil solution 750–1,000 mg 1x/10–14 weeks Testosterone buciclate a – Aqueous suspension 600–1,000 mg 1x/12–20 weeks Implant Testosterone Testopel Pellet 150–1,200 mg/3–6 months Notes: Men produce about 3 to 11 mg testosterone per day (mean 7 mg/day in young men).







