Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Neuroendocrine Tumor
Wikipedia
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Deafness, Autosomal Recessive 88
OMIM
A number sign (#) is used with this entry because of evidence that autosomal recessive deafness-88 (DFNB88) is caused by homozygous mutation in the ELMOD3 gene (615427) on chromosome 2p11.
-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Mental Retardation, X-Linked 88
OMIM
Clinical Features Vervoort et al. (2002) reported 9 male patients with a mutation in the AGTR2 gene who had moderate to severe mental retardation. Five of the patients had seizures and, with the exception of 1 patient, they were not hypertensive. Two patients also showed autistic behavior. Cytogenetics Vervoort et al. (2002) analyzed a de novo balanced translocation 46,X,t(X;7)(q24;q22) in a female patient with moderate mental retardation. This patient had completely skewed X inactivation. They mapped the X-chromosome breakpoint and demonstrated by RT-PCR that the AGTR2 gene was silenced in this patient. History Vervoort et al. (2002) screened affected males from 33 families with possible X-linked MR but no definitive linkage data, and a large cohort of 552 unrelated male patients with MR of unknown cause but negative for the FMR1 expansion.IQSEC2, DLG3, GDI1, PAK3, ACSL4, ARX, MECP2, RPS6KA3, HCFC1, IL1RAPL1, TSPAN7, FTSJ1, ZNF41, DMD, CNKSR2, MID2, AGTR2, UPF3B, CXorf56, FRMPD4, ALG13, SLC9A7, RAB39B, PTCHD1, ZNF81, MED12, ZNF711, ARHGEF6, SYP, CLCN4, USP27X, USP9X, ABCG2, FRAXE, AFF2, STS, OPHN1, ALAS2, POU3F4, SERPINA4, RPS6KA6, THOC2, ANOS1, FMR1, ELK1
-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Appendix Cancer
Wikipedia
Appendix cancer Other names Appendiceal cancer Specialty Oncology , general surgery Symptoms Bloating , discomfort in lower right abdomen , shortness of breath , loss of appetite [1] Usual onset ~50-55 years old [2] Types Colonic-Type Adenocarcinoma, Non-carcinoid Appendix Tumors, Signet-Ring Cell Adenocarcinoma [3] Risk factors Smoking , family history , Multiple endocrine neoplasia type 1 [4] Diagnostic method Biopsy , CT Scan , MRI [5] Differential diagnosis Acid reflux , Irritable bowel syndrome , Lactose intolerance , Stomach cancer [6] Treatment Appendectomy , chemotherapy , radiation therapy [7] Prognosis Five-year survival rate 25-88% (U.S.) [8] Frequency ~1,000 cases per year (U.S.) [9] Deaths Unknown Appendix cancer are very rare cancers of the vermiform appendix . ... A carcinoid is a neuroendocrine tumor (NET) of the intestines. Incidence rates among carcinoids occur at about .15 per 100,000 per year.
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... According to the WHO and UNICEF, deaths attributable to malaria in 2015 were reduced by 60% [77] from a 2000 estimate of 985,000, largely due to the widespread use of insecticide-treated nets and artemisinin-based combination therapies. [74] In 2012, there were 207 million cases of malaria.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
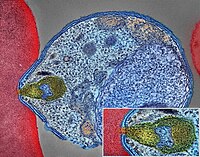
-
Abdominal Obesity
Wikipedia
Ghroubi et al. (2007) concluded that a high abdominal circumference is associated with great functional repercussion. [37] Causes [ edit ] Diet [ edit ] See also: Diet and obesity The currently prevalent belief is that the immediate cause of obesity is net energy imbalance—the organism consumes more usable calories than it expends, wastes, or discards through elimination . ... The absolute waist circumference 102 centimetres (40 in) in men and 88 centimetres (35 in) in women and the waist-hip ratio (>0.9 for men and >0.85 for women) [55] are both used as measures of central obesity. ... Journal of Clinical Endocrinology and Metabolism . 88 (11): 5452–5455. doi : 10.1210/jc.2002-021808 . ... "Resistin and type 2 diabetes: regulation of resistin expression by insulin and rosiglitazone and the effects of recombinant resistin on lipid and glucose metabolism in human differentiated adipocytes" . J. Clin. Endocrinol. Metab . 88 (6098–6106): 6098–106. doi : 10.1210/jc.2003-030898 . ... Endocrine Updates . 4. 30 . Springer. pp. 355–365. doi : 10.1007/978-1-4419-7034-3_17 .EIF2S3, HSD11B1, USP8, PWRN1, SNORD115-1, PWAR1, RNPC3, MAGEL2, NPAP1, MKRN3-AS1, DYRK1B, AIP, HERC2, CUL4B, MKRN3, SNRPN, NDN, MTTP, LIPE, IPW, SNORD116-1

-
Xh Antigen
OMIM
The Xh antigen was first described by Bundschuh (1966), who suggested X-linkage because the antigen is more frequent in women (97%) than in men (88%). The antigen is demonstrated with antiserum produced by injecting rabbits with pooled serum from healthy women and absorption of the immune serum with selected male sera.
-
Lichenoid Trikeratosis
Wikipedia
"Lichenoid tri-keratosis (Kaposi-Bureau-Barrière-Grupper)". Dermatologica . 148 (3): 179–88. doi : 10.1159/000251619 . PMID 4836959 .
-
Rectal Pain
Wikipedia
Emerg. Med. Clin. North Am . 14 (4): 757–88. doi : 10.1016/S0733-8627(05)70278-9 . ... Pelvic Floor Disorders: Imaging and Multidisciplinary Approach to Management . Springer. pp. 601–603. ISBN 978-88-470-1542-5 . External links [ edit ] Classification D ICD - 9-CM : 569.42 v t e Pain By region/system Head and neck Headache Neck Odynophagia (swallowing) Toothache Respiratory system Sore throat Pleurodynia Musculoskeletal Arthralgia (joint) Bone pain Myalgia (muscle) Acute Delayed-onset Neurologic Neuralgia Pain asymbolia Pain disorder Paroxysmal extreme pain disorder Allodynia Chronic pain Hyperalgesia Hypoalgesia Hyperpathia Phantom pain Referred pain Congenital insensitivity to pain congenital insensitivity to pain with anhidrosis congenital insensitivity to pain with partial anhidrosis Other Pelvic pain Proctalgia Back Low back pain Measurement and testing Pain scale Cold pressor test Dolorimeter Grimace scale (animals) Hot plate test Tail flick test Visual analogue scale Pathophysiology Nociception Anterolateral system Posteromarginal nucleus Substance P Management Analgesia Anesthesia Cordotomy Pain eradication Related concepts Pain threshold Pain tolerance Suffering SOCRATES Philosophy of pain Cancer pain Drug-seeking behavior
- Squamous Cell Papilloma Wikipedia
-
Leishmaniasis
Wikipedia
Leishmaniasis Other names Leishmaniosis Cutaneous leishmaniasis in the hand of a Central American adult Pronunciation Leishmaniasis / ˌ l iː ʃ m ə ˈ n aɪ ə s ɪ s / leishmaniosis / l iː ʃ ˌ m eɪ n i ˈ oʊ s ɪ s , - ˌ m æ n i -/ [1] Specialty Infectious disease Symptoms Skin ulcers , fever , low red blood cells , enlarged liver [2] [3] Causes Leishmania parasites spread by sandflies [2] Prevention Bug nets , insecticide [2] Frequency 4–12 million [4] [5] Deaths 24,200 (2015) [6] Leishmaniasis is a disease caused by parasites of the Leishmania type. [2] It is spread by the bite of certain types of sandflies , and occurs most frequently in the tropics and sub-tropics of Africa, Asia, the Americas, and southern Europe. [2] The disease can present in three main ways: cutaneous , mucocutaneous, or visceral . [2] The cutaneous form presents with skin ulcers, while the mucocutaneous form presents with ulcers of the skin, mouth, and nose. The visceral form starts with skin ulcers and later presents with fever, low count of red blood cells, and enlarged spleen and liver. [2] [3] Infections in humans are caused by more than 20 species of Leishmania . [2] Risk factors include poverty , malnutrition , deforestation , and urbanization . [2] All three types can be diagnosed by seeing the parasites under microscopy. [2] Additionally, visceral disease can be diagnosed by blood tests. [3] Leishmaniasis can be partly prevented by sleeping under nets treated with insecticide . [2] Other measures include spraying insecticides to kill sandflies and treating people with the disease early to prevent further spread. [2] The treatment needed is determined by where the disease is acquired, the species of Leishmania , and the type of infection. [2] Some possible medications used for visceral disease include liposomal amphotericin B , [7] a combination of pentavalent antimonials and paromomycin , [7] and miltefosine . [8] For cutaneous disease, paromomycin, fluconazole , or pentamidine may be effective. [9] About 4 to 12 million people are currently infected [4] [5] in some 98 countries. [3] About 2 million new cases [3] and between 20 and 50 thousand deaths occur each year. [2] [10] About 200 million people in Asia, Africa, South and Central America, and southern Europe live in areas where the disease is common. [3] [11] The World Health Organization has obtained discounts on some medications to treat the disease. [3] It is classified as a neglected tropical disease . [12] The disease may occur in a number of other animals, including dogs and rodents . [2] Contents 1 Signs and symptoms 2 Cause 2.1 Vector 2.2 Organisms 2.3 Risk factors 3 Diagnosis 4 Prevention 5 Treatment 6 Epidemiology 7 History 8 Society and culture 9 Research 10 See also 11 References 12 External links Signs and symptoms [ edit ] Cutaneous leishmaniasis ulcer The symptoms of leishmaniasis are skin sores which erupt weeks to months after the person is bitten by infected sand flies. ... Prevention [ edit ] Using insecticide can reduce phlebotomine sandfly numbers which in turn reduces the risk of cutaneous leishmaniasis infection. [18] Leishmaniasis can be partly prevented by using nets treated with insecticide while sleeping. [2] To provide good protection against sandflies, fine mesh sizes of 0.6 mm or less are required, but a mosquito net with 1.2mm mesh will provide a limited reduction in the number of sandfly bites. [19] Finer mesh sizes have the downside of higher cost and reduced air circulation which can cause overheating. Many Phlebotomine sandfly attacks occur at sunset rather than at night, so it may also be useful to put nets over doors and windows or to use insect repellents . ... The American Journal of Tropical Medicine and Hygiene . 88 (1): 157–61. doi : 10.4269/ajtmh.2012.11-0717 .TNF, IFNG, IL10, IL6, ARG1, IL18, CRP, TNFRSF18, MCL1, HSPA4, IL1B, SLC11A1, CXCL10, NLRP3, IL17A, TLR2, CCR5, TLR4, IL32, PRDX2, LEP, TGFB1, CD274, FCN2, CD163, MTOR, HM13, IL4, BCL2, BAX, LMLN, IGF1, HIF1A, ANXA1, VDR, UNG, TAM, NR0B2, EZR, ADA, TLR3, STAT1, MAPK3, MAPK4, EIF2AK2, PSG5, PSMD7, PTHLH, PTPN1, PTPN2, PTPN6, RPA1, RPS6, CCL2, CCL8, CXCL11, SLC1A5, SLC1A7, SNAP25, SOAT1, SPP1, TP63, EIF2S2, CDK5R1, GOPC, FOXP3, HSPA14, CD244, TOLLIP, FBLIM1, MSTO1, FBXW7, ACSS2, PDXP, SLC52A2, ALDH1A2, TMPRSS13, DCLK3, IL33, CDCA5, PWAR1, ARMH1, HNP1, CCR2, UPK3B, DLL1, SGSM3, NOX1, PABPC1, NR1I2, SPHK1, EIF2B4, EIF2B2, PRKAB1, HSPB3, SLC7A6, ARHGEF2, AIM2, H6PD, RABEPK, LANCL1, TNFSF13B, EBNA1BP2, CD160, GABARAPL2, GABARAPL1, PRDX5, POLR1A, MAPK1, NOS2, PRKAA2, PRKAA1, CST3, CTLA4, CTSB, CTSL, CYP51A1, DDT, DHFR, DPAGT1, DPP4, DSPP, DUSP4, EEF1B2, EEF2, EGFR, EIF2B1, F2R, FCGR2A, FECH, FLI1, CPB1, CCR7, LRBA, ATR, AKT1, ALDH1A1, APEX1, APRT, AQP1, ATM, ATP2A3, ATP2B4, PRDM1, CD69, BRCA1, CAPN1, CD1A, CD28, CD86, CD40, CD40LG, CD44, FPR2, G6PD, GAPDH, CYTB, MNAT1, CD200, MPG, MPL, MPST, MRC1, MSMB, MST1, AHR, MFAP1, PAEP, PHB, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PLP1, PNOC, MAP3K10, MBL2, GCHFR, IFNB1, GCK, GTF3C1, HLA-C, HMOX1, HSPD1, IFN1@, IFNA1, IFNA13, IL1A, LTA, IL9, IL12A, IL12RB1, IL13, ITGA4, ITGAL, JAK2, RPSA, H3P28

-
Lichen Verrucosus Et Reticularis
Wikipedia
"Lichenoid tri-keratosis (Kaposi-Bureau-Barrière-Grupper)". Dermatologica . 148 (3): 179–88. doi : 10.1159/000251619 . PMID 4836959 .
-
Morel's Ear
Wikipedia
Haeberle, "'Stigmata of Degeneration': Prisoner Markings in Nazi Concentration Camps," Journal of Homosexuality , vol. 6, 1980/81, 135-139, available online Archived 2011-04-21 at the Wayback Machine , accessed January 3, 2012 ^ Patrick Alexander, Marcel Proust's Search for Lost Time: A Reader's Guide (), 88, availbel online , accessed January 3, 2012 This medical sign article is a stub .
-
Endogenous Infection
Wikipedia
Springer Science & Business Media. p. 64. ISBN 978-88-470-2242-3 . ^ Laura Ester Ziady; Nico Small (December 2005).
-
Accelerated Tumor Formation, Susceptibility To
OMIM
They demonstrated that SNP309 was associated with accelerated tumor formation in both hereditary and sporadic cancers in humans. Bond et al. (2004) studied 88 individuals who were members of Li-Fraumeni syndrome (LFS1; 151623) families and had germline mutations in 1 allele of p53. The frequency of SNP309 in these individuals was similar to that found in the 50 normal volunteers. Of the 88 individuals in the Li-Fraumeni cohort, 66 were diagnosed with at least 1 cancer at a median age of 22 years old.





