-
Triple X Syndrome
Wikipedia
It was found in a 35‑year-old, 5 ft. 9 in. (176 cm) tall, 128 lb. (58.2 kg) woman who had premature ovarian failure at age 19; her mother was age 41 and her father was 40 at the time of her conception. [9] Jacobs, et al. called the 47,XXX woman a " superfemale ", a term which was immediately criticized, did not gain acceptance, and was based on the incorrect assumption that the sex-determination system in mammals was the same as in the fruit fly Drosophila . [10] British pathologist and geneticist Bernard Lennox, the principal consultant on medical terms for the Oxford English Dictionary , suggested the term "XXX syndrome". [11] References ^ a b c d e f g h i j k "triple X syndrome" . ... Retrieved 26 September 2016 . ^ a b c d e f g h Tartaglia, NR; Howell, S; Sutherland, A; Wilson, R; Wilson, L (11 May 2010). ... Archived from the original on 2012-02-28. ^ a b c d e f g Otter, M; Schrander-Stumpel, CT; Curfs, LM (March 2010). ... Triple X syndrome Genetics Home Reference v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22

-
Sex Differences In Medicine
Wikipedia
Find sources: "Sex differences in medicine" – news · newspapers · books · scholar · JSTOR ( June 2014 ) Part of a series on Sex differences in humans Physiology Medicine Sexual differentiation Autism Health Mental disorders Narcissism Schizophrenia Stroke care Neuroscience Memory Eyewitness memory Cognition Intelligence Emotional intelligence Psychology Gender psychology Sociology Crime Education Leadership Social capital Suicide v t e Sex differences in medicine include sex-specific diseases or conditions which occur only in people of one sex (for example, prostate cancer in males or uterine cancer in females); sex-related diseases, which are diseases that are more common to one sex (for example, systemic lupus erythematosus occurs predominantly in females); [1] and diseases which occur at similar rates in males and females but manifest differently according to sex (for example, peripheral artery disease ). [2] Sex differences in medicine should not be confused with gender differences. ... ISBN 978-0-89042-555-8 . ^ Di Carlo A, Baldereschi M, Amaducci L, Lepore V, Bracco L, Maggi S, Bonaiuto S, Perissinotto E, Scarlato G, Farchi G, Inzitari D (January 2002). ... PMID 17626963 . ^ Alegria, Analucia A.; Blanco, Carlos; Petry, Nancy M.; Skodol, Andrew E.; Liu, Shang-Min; Grant, Bridget; Hasin, Deborah (July 2013). ... Journal of Clinical Oncology . 31 (36): 4550–9. doi : 10.1200/jco.2013.50.3870 . PMC 3865341 . PMID 24248688 . v t e Sex differences in humans Physiology Medicine Sexual differentiation Autism Health Mental disorders Narcissism Schizophrenia Stroke care Neuroscience Memory Eyewitness memory Cognition Intelligence Emotional intelligence Psychology Gender psychology Sociology Crime Education Leadership Social capital Suicide
-
Trimethylaminuria
Wikipedia
PMID 9536088 . ^ Zschocke J, Kohlmueller D, Quak E, Meissner T, Hoffmann GF, Mayatepek E (1999). ... Aust . 189 (8): 468. doi : 10.5694/j.1326-5377.2008.tb02126.x . PMID 18928446 . ^ Treacy E; Johnson D. Pitt JJ; Danks DM (1995). ... National Library of Medicine and The National Human Genome Research Institute v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria
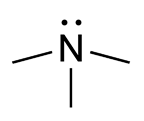
-
Fetal Adenocarcinoma
Wikipedia
Micrograph showing fetal adenocarcinoma. H&E stain . Fetal adenocarcinoma (FA) of the lung is a rare subtype of pulmonary adenocarcinoma that exhibits tissue architecture and cell characteristics that resemble fetal lung tissue upon microscopic examination. ... PMID 20205788 . ^ Longo M, Levra MG, Capelletto E, et al. (April 2008). "Fetal adenocarcinoma of the lung in a 25-year-old woman". ... Pathol . 27 (6): 542–6. doi : 10.1016/S0046-8177(96)90159-8 . PMID 8666362 . ^ a b c d e Cutler CS, Michel RP, Yassa M, Langleben A (February 1998). ... PMID 16387506 . ^ a b Matsuoka T, Sugi K, Matsuda E, et al. (August 2006). "[Clear cell adenocarcinoma with acomponent of well-differentiated fetal adenocareinoma; report of a case]".

-
Tick Paralysis
Wikipedia
External links [ edit ] Classification D ICD - 10 : T63.4 ICD - 9-CM : 989.5 MeSH : D013985 DiseasesDB : 31779 SNOMED CT : 74225001 External resources MedlinePlus : 001359 v t e Poisoning Toxicity Overdose History of poison Inorganic Metals Toxic metals Beryllium Cadmium Lead Mercury Nickel Silver Thallium Tin Dietary minerals Chromium Cobalt Copper Iron Manganese Zinc Metalloids Arsenic Nonmetals Sulfuric acid Selenium Chlorine Fluoride Organic Phosphorus Pesticides Aluminium phosphide Organophosphates Nitrogen Cyanide Nicotine Nitrogen dioxide poisoning CHO alcohol Ethanol Ethylene glycol Methanol Carbon monoxide Oxygen Toluene Pharmaceutical Drug overdoses Nervous Anticholinesterase Aspirin Barbiturates Benzodiazepines Cocaine Lithium Opioids Paracetamol Tricyclic antidepressants Cardiovascular Digoxin Dipyridamole Vitamin poisoning Vitamin A Vitamin D Vitamin E Megavitamin-B 6 syndrome Biological 1 Fish / seafood Ciguatera Haff disease Ichthyoallyeinotoxism Scombroid Shellfish poisoning Amnesic Diarrhetic Neurotoxic Paralytic Other vertebrates amphibian venom Batrachotoxin Bombesin Bufotenin Physalaemin birds / quail Coturnism snake venom Alpha-Bungarotoxin Ancrod Batroxobin Arthropods Arthropod bites and stings bee sting / bee venom Apamin Melittin scorpion venom Charybdotoxin spider venom Latrotoxin / Latrodectism Loxoscelism tick paralysis Plants / fungi Cinchonism Ergotism Lathyrism Locoism Mushrooms Strychnine 1 including venoms , toxins , foodborne illnesses . Category Commons WikiProject v t e Tick-borne diseases and infestations Diseases Bacterial infections Rickettsiales Anaplasmosis Boutonneuse fever Ehrlichiosis ( Human granulocytic , Human monocytotropic , Human E. ewingii infection ) Scrub typhus Spotted fever rickettsiosis Pacific Coast tick fever American tick bite fever rickettsialpox Rocky Mountain spotted fever ) Spirochaete Baggio–Yoshinari syndrome Lyme disease Relapsing fever borreliosis Thiotrichales Tularemia Viral infections Bhanja virus Bourbon virus Colorado tick fever Crimean–Congo hemorrhagic fever Heartland bandavirus Kemerovo tickborne viral fever Kyasanur Forest disease Omsk hemorrhagic fever Powassan encephalitis Severe fever with thrombocytopenia syndrome Tete orthobunyavirus Tick-borne encephalitis Protozoan infections Babesiosis Other diseases Tick paralysis Alpha-gal allergy Southern tick-associated rash illness Infestations Tick infestation Species and bites Amblyomma Amblyomma americanum Amblyomma cajennense Amblyomma triguttatum Dermacentor Dermacentor andersoni Dermacentor variabilis Ixodes Ixodes cornuatus Ixodes holocyclus Ixodes pacificus Ixodes ricinus Ixodes scapularis Ornithodoros Ornithodoros gurneyi Ornithodoros hermsi Ornithodoros moubata Other Rhipicephalus sanguineus Authority control LCCN : sh85135249
-
Blepharospasm
Wikipedia
. ^ Simpson, D. M.; Hallett, M.; Ashman, E. J.; Comella, C. L.; Green, M. W.; Gronseth, G. ... PMID 24682196 . ^ Schellini SA, Matai O, Igami TZ, Padovani CR, Padovani CP (2006). "Blefarospasmo essencial e espasmo hemifacial: características dos pacientes, tratamento com toxina botulínica A e revisão da literatura" [Essential blepharospasm and hemifacial spasm: characteristic of the patient, botulinum toxin A treatment and literature review]. ... External links [ edit ] Classification D ICD - 10 : G24.5 ICD - 9-CM : 333.81 OMIM : 606798 MeSH : D001764 DiseasesDB : 15748 External resources MedlinePlus : 000756 eMedicine : oph/202 Patient UK : Blepharospasm Blepharospasm – Resource Guide from the National Eye Institute (NEI) v t e Diseases of the nervous system , primarily CNS Inflammation Brain Encephalitis Viral encephalitis Herpesviral encephalitis Limbic encephalitis Encephalitis lethargica Cavernous sinus thrombosis Brain abscess Amoebic Brain and spinal cord Encephalomyelitis Acute disseminated Meningitis Meningoencephalitis Brain / encephalopathy Degenerative Extrapyramidal and movement disorders Basal ganglia disease Parkinsonism PD Postencephalitic NMS PKAN Tauopathy PSP Striatonigral degeneration Hemiballismus HD OA Dyskinesia Dystonia Status dystonicus Spasmodic torticollis Meige's Blepharospasm Athetosis Chorea Choreoathetosis Myoclonus Myoclonic epilepsy Akathisia Tremor Essential tremor Intention tremor Restless legs Stiff-person Dementia Tauopathy Alzheimer's Early-onset Primary progressive aphasia Frontotemporal dementia / Frontotemporal lobar degeneration Pick's Dementia with Lewy bodies Posterior cortical atrophy Vascular dementia Mitochondrial disease Leigh syndrome Demyelinating Autoimmune Inflammatory Multiple sclerosis For more detailed coverage, see Template:Demyelinating diseases of CNS Episodic/ paroxysmal Seizures and epilepsy Focal Generalised Status epilepticus For more detailed coverage, see Template:Epilepsy Headache Migraine Cluster Tension For more detailed coverage, see Template:Headache Cerebrovascular TIA Stroke For more detailed coverage, see Template:Cerebrovascular diseases Other Sleep disorders For more detailed coverage, see Template:Sleep CSF Intracranial hypertension Hydrocephalus Normal pressure hydrocephalus Choroid plexus papilloma Idiopathic intracranial hypertension Cerebral edema Intracranial hypotension Other Brain herniation Reye syndrome Hepatic encephalopathy Toxic encephalopathy Hashimoto's encephalopathy Both/either Degenerative SA Friedreich's ataxia Ataxia–telangiectasia MND UMN only: Primary lateral sclerosis Pseudobulbar palsy Hereditary spastic paraplegia LMN only: Distal hereditary motor neuronopathies Spinal muscular atrophies SMA SMAX1 SMAX2 DSMA1 Congenital DSMA Spinal muscular atrophy with lower extremity predominance (SMALED) SMALED1 SMALED2A SMALED2B SMA-PCH SMA-PME Progressive muscular atrophy Progressive bulbar palsy Fazio–Londe Infantile progressive bulbar palsy both: Amyotrophic lateral sclerosis

-
Nonalcoholic Fatty Liver Disease
Mayo Clinic
But researchers are studying whether some natural compounds could be helpful, such as: Vitamin E. In theory, vitamin E and other vitamins called antioxidants could help protect the liver by reducing or neutralizing the damage caused by inflammation. But more research is needed. Some evidence suggests vitamin E supplements may be helpful for people with liver damage caused by nonalcoholic fatty liver disease. But vitamin E has been linked with increased risk of death and, in men, an increased risk of prostate cancer.TM6SF2, PNPLA3, NFE2L2, LEP, CYP2E1, PPARA, SIRT1, NR1H4, FGF21, ADIPOQ, PPARD, SREBF1, XBP1, LDLR, CAT, FAS, GNMT, CPT1A, PEMT, NR5A2, TGFB1, AHR, GSTP1, GSTM1, PRKCA, IL1A, JAK2, ACE, CD14, KLB, GSTT1, ALDH2, ABCC2, PTEN, PDK4, GSTA1, IL4, LIF, PRKCE, VLDLR, NQO1, PRKCD, FOLR2, EIF2AK1, TNFRSF1B, CYP17A1, CSF2, ALDH4A1, SCARB1, RDX, F2, STC2, LAMA1, IKBKG, CYP1A2, PRKACA, RAG2, B3GAT1, ALDH1B1, ADH1A, IL3, MMP1, ABCB4, TRIB3, ADH4, ADH1B, PRF1, SERPINB2, AHCY, ALDH1A1, GCKR, ATP5F1B, NCAN, PARVB, SAMM50, FDFT1, PTPRD, PLPP7, KRT18, MIR122, TLR4, ZNF512, IGF1, LEPR, MBOAT7, MIR21, SREBF2, CRP, MTTP, IL1B, SP4, ACTR5, IL6, CRACR2A, PRKAA2, SLC5A2, HFE, PRKAA1, COL13A1, GLP1R, GPT, GGT1, GCG, PPARG, PIK3CG, PIK3CD, PIK3CB, GATAD2A, OSBPL5, DPP4, RBP4, NLRP3, EHBP1L1, PIK3CA, SCD, FBL, ERBB4, DDX60L, SHBG, PWWP3A, PRKAB1, EHMT1, LIPA, APOC3, GGTLC3, CREB5, PPARGC1A, ZGLP1, GGTLC4P, CD36, SLC17A5, CNBP, XPR1, GGT2, TNF, FGF19, TP63, GGTLC5P, ALB, MIR34A, AHSG, GH1, GABPA, LBP, HAMP, FASN, REN, LGALS3BP, CCL2, FABP1, NR1H3, IL17A, MLXIPL, HSD17B13, HNF4A, CYP3A4, SIRT3, AGTR1, APOE, PON1, RARRES2, RETN, MTOR, LOC102724197, FETUB, UCP2, GGTLC1, NR0B2, DECR1, MTHFR, IL10, PCSK9, ACACA, TP53, ANGPTL8, SERPINE1, MIR192, SOD1, APOB, VDR, IRS2, HMGB1, LPL, RCBTB1, NR1I2, APRT, CNR1, SERPINA1, SOD2, TRIB1, FADS2, STAT3, IRS1, FOXO1, PPP1R3B, NR3C2, MFAP1, SELENOP, PLIN2, GOLGA6A, AKT1, PNPLA2, MIR33A, TXNIP, ABCD1, CYBB, HIF1A, FGL1, IL18, FLII, APP, SLC27A5, TNFSF10, APOA5, HSD11B1, TMC4, NRG4, IFNG, HP, CCR2, GCK, KLF6, CHPT1, ABCB11, EGFR, EPAS1, CASP1, MAP3K5, CETP, SPP1, FTO, VEGFA, IFNL3, CEBPA, FABP4, INSR, MIR29A, BGLAP, TLR2, COL1A1, MIR140, GDF15, CYP7A1, ABCA1, SMN2, ANPEP, ACLY, BCO2, SMN1, CXCL16, HMOX1, PRRT2, FST, NTS, NR1I3, TIMP2, TIMP1, CPT2, CMKLR1, HPGDS, MIR155, TERT, BMS1, NPC1L1, CTNNB1, GPX1, CYP4F3, SMUG1, BCL2A1, PARP1, MIR451A, BNIP3, NEAT1, MT1B, MMP2, OR10A4, PRL, MAPK8, MIR378A, NAMPT, SLC10A2, SAV1, AFP, BCO1, GIP, FGFR4, ARNTL, CXCL10, PRKCB, CAV1, AGER, BCL2, GOT2, DLAT, ADIPOR2, AGT, ACE2, GPBAR1, MMP13, CORIN, FAM3A, MC4R, YAP1, POSTN, ABCG5, IL25, MAS1, NLRC4, TLR6, MUC1, CCN4, CCN3, ADIPOR1, SCP2, TNFSF11, FAM3B, CCL20, LUC7L3, SELPLG, GDE1, HSD17B7, SLC2A1, SLC2A2, MLYCD, SLPI, TNFRSF11B, IL22, TRPV1, SPARC, SPTBN1, ATRNL1, STAT5A, STAT5B, TBK1, SETD2, TEK, PDLIM3, S100A8, ROCK1, SOCS1, UGT1A1, MYDGF, CCL27, ENPP1, MTMR11, PPIG, CD163, MEG3, NAT10, PTPA, ECD, USP10, SOCS3, SELENBP1, APLN, MAPK1, PRTN3, SIRT2, ELOVL2, RIPK1, PTGS1, PTPN1, LPIN1, MOK, CRNKL1, SPINK1, CEACAM1, CBR1, MIR375, CASP8, CASP3, FOXO3, FOS, CALCR, MIR146B, GC, GCGR, HCC, BCAT1, GLUL, NCF1, GPR119, AZGP1, BHLHE23, LYPLAL1, GRN, NR3C1, PDIA3, AVP, ATHS, ATF3, C1QTNF1, AR, MLKL, FCGR2B, FOXA1, FAT1, MIR20A, MIR222, MIR149, CYBA, CYP1A1, COX8A, MAP3K8, CYP2D6, MIR126, CHIT1, CES1, ENHO, IRGM, ATN1, PLIN5, FFAR4, ELANE, ELAVL2, CD68, SLC13A5, ESR1, ESRRA, CD5L, FNDC5, ACSL4, AQP9, MIR200C, HSP90AA1, STEAP4, ALPP, ALOX15, ALOX5, ALOX12, LCN2, LINC01672, CXCL8, FAS-AS1, IDH2, INS, ACOX1, ACACB, IGFALS, LECT2, DHRS11, ORAI1, HSPA1A, HSPA1B, GRK2, ASRGL1, PSC, ERLIN1, CXCR6, MIR331, TRPC4AP, PRPF31, RNF19A, CAMKK2, SIRT1-AS, EIF2AK4, WWTR1, TOR1AIP1, POLDIP2, MIR330, APPL1, TIPARP, OSBPL3, LINC01684, HULC, MIR30C1, FGF20, AHSA1, MIR206, MIR212, RBMS3, PRDX4, LNCARSR, MIR223, MIR23B, IL37, MIR24-1, MIR27A, NAAA, SORBS1, INTU, MIR27B, LOC110806263, MIR29B1, SIGLEC7, NOX1, MIR29B2, MIR29C, AATF, MIR30B, PRDX5, LATS2, MIR30C2, B4GALT1-AS1, TMEFF2, MIR367, PHLPP2, LOC100505909, MCF2L, TMED2, MIR1224, MPRIP, SLC27A2, LILRB4, SARM1, RIPK3, CD24, MIR190B, DKK1, TNFSF13B, SNHG20, ZHX2, MIR33B, MLXIP, MGLL, MIR590, DNLZ, VSIG4, ADAMTS13, MIR873, DUSP12, MIR205, OCLN, MIR193B, NMU, EBP, IFNL4, CISD2, IL17RA, TREH, YME1L1, MKRN1, DAPK2, MIR361, DDAH1, PARTICL, MMD, SRRM2, BRD4, ERVK-2, ANGPTL2, PLK4, HPSE, PGRMC1, SIRT4, MIR423, MIR20B, CRTC1, DUSP26, NOX4, MAT2B, ERBIN, PAG1, ACOT13, ZNF300, TIMD4, TBL1Y, WNT3A, FAM83A, MIR200B, LGALS14, UBQLN4, PNO1, CARD6, DGAT2, ADAMTS9, ALLC, TXNRD3, C1QTNF3, TWIST2, TRERF1, NLRP2, IFTAP, CHDH, FBXW7, CWF19L1, SLC47A1, CYP2R1, CPT1C, PPARGC1B, NADK2, FGFBP3, SLC38A8, PCBP4, MAK16, SMURF1, NCOA2, FRTS1, ELOVL6, WNK1, APOO, ARV1, GORASP1, XPO4, NSD1, ABCG8, NLRX1, KCTD17, TMBIM1, SMOC2, GSDMD, DHDDS, LRRC7, CIDEC, HKDC1, CPEB4, TRPV4, PGAP6, COASY, SPX, SESN2, TRAPPC9, ITCH, ANGPTL6, ARRDC3, SEMA6A, CHCHD6, ZFP90, CILP2, HJV, MIR142, MBTPS2, IRF2BP2, INSIG2, ABHD5, RTL1, C1QL3, LINC01194, MIR130A, MIR132, SOST, MIR136, MIR141, IL20, AACS, MIR143, ISYNA1, MIR144, MIR146A, DUOX2, SLC40A1, SLC2A8, DMGDH, CERS2, MIR150, MIR15B, MIR17, MIR185, REPIN1, CTNNA3, MYLIP, MOGAT3, CPP, APPL2, EGLN1, NUDT11, PDIK1L, PIWIL2, SIK1, SLC52A1, HORMAD2, SLC2A12, TMPRSS6, TET2, CROT, LINC01554, UNC5B, MARCHF8, UGT1A4, JAZF1, SIRT6, UGT1A6, UGT1A7, UGT1A8, UGT1A10, CCHCR1, ZBTB38, TREM1, BTBD8, DUOX1, BPIFA4P, HCAR2, C1QTNF9, ARID4B, STING1, ATG7, A2M, STK25, GCLC, FGF1, FGR, FOSB, FOSL2, G6PC, GALR1, GDNF, GPC3, GLB1, GLI2, SLC26A3, GLRX, GNA12, GNAI3, GNAO1, GPI, GPR31, FFAR2, GPS2, GSK3B, GPC4, FCGR3B, FCGR3A, FBN1, DUSP9, EDA, S1PR1, EEF1A1, EGF, EGR1, EIF4E, ENO3, EPHB2, EPHB6, EPO, ERN1, ESRRB, EZH2, F2RL1, FABP2, FABP5, ACSL1, PTK2B, GSTM2, H2AX, HADHB, IGF2, IGFBP2, IGFBP4, IGFBP7, IKBKB, IL2, CXCR2, INPP5D, INSIG1, IRF3, ITGAM, ITGB1, ITIH4, ITPR2, JAK1, JUN, JUNB, JUND, CD82, KRT8, IGFBP1, IFNA13, HADH, IFNA2, HCFC1, HLA-A, HLA-C, HLA-DQA1, HLA-DQB1, HLA-DQB2, HLA-DRB1, NR4A1, FOXA2, HOXD13, HRG, PRMT1, HSPA4, HSPA5, HSPA8, HSPG2, HTR2A, TNC, IFNA1, DSPP, DPYSL3, LCP1, BMP4, ARG2, ARNT, ARRB1, ASNS, ATF4, BAAT, BAX, BCHE, BLVRA, BMP8B, DMD, TSPO, C3, CALCA, RUNX2, CCNC, CD44, CDC42, CDH1, CDK4, AREG, AQP7, KLK3, APOA2, ABO, ACAT1, ACTB, ADM, ADRB2, AEBP1, JAG1, AKT2, ALDOB, AKR1B1, AMD1, AMD1P2, ANGPT1, ANGPT2, AOX1, APOF, AIRE, XIAP, APOA1, CDK8, CDKN2A, CEL, CTNNA1, CTSB, CTSD, CTSG, CTSS, CYP2A6, CYP2B6, CYP2B7P, CYP2D7, CYP2J2, CYP4A11, CYP8B1, CYP19A1, CYP27B1, CYP51A1, DBP, DDOST, TIMM8A, DHCR7, DIO3, CTRL, CCN2, CHRM3, CTF1, CHUK, CIDEA, CLCN2, CNR2, COL3A1, SLC31A1, CP, CPN1, CPOX, CPS1, CREB1, ATF2, CREBBP, CRK, CRMP1, MAPK14, CSF3, CSF3R, CTAA1, LCAT, LGALS3, CAP1, VEGFC, UCP1, UCP3, SLC35A2, UGT1A, UGT2B4, USP4, UROD, UTRN, VCAM1, VIP, STAT1, BEST1, YY1, ZBTB16, LAP, PLA2G7, TFEB, AIMP2, STAM, FOSL1, TRAF3, TPO, TP53BP1, TNFRSF1A, SULT1E1, STK11, SYT1, ADAM17, MAP3K7, TAZ, TCF7L2, TFAM, TFF3, TG, TGFB1I1, THRSP, TIMP3, TLL1, TLR1, NR2E1, TM7SF2, TMPO, TNFAIP3, FGF23, SLC7A5, BAS, TMPRSS11D, GGPS1, ATG5, LITAF, CLOCK, ABCG1, KEAP1, PLPPR4, MFN2, PARP2, DNM1L, PPIF, LRPPRC, SLC25A13, PRMT3, PLIN3, FSTL3, ZNF267, AKR1A1, MERTK, EIF2AK3, GRAP2, PLA2G6, COX5A, PIK3R3, CYP4F2, DENR, RTCA, DGAT1, ABCC3, MBTPS1, TNFSF14, DLK1, TRIM24, CES2, HDAC3, SQSTM1, MBD2, CCRL2, MYOM2, HACD1, NOG, GLP2R, STAT6, STAR, LIPE, P2RX5, NBN, NEU1, NHS, NNMT, NOS2, NOS3, NOTCH1, NRF1, NUCB2, P4HA1, AKR1D1, PAPPA, PC, PCK1, PDGFRB, SERPINA4, SERPINB6, PIGR, PITX3, PKM, HNRNPM, MYD88, MYC, ND6, FADS1, LNPEP, LOXL2, LRP6, LTF, LUM, SMAD7, MAT1A, DNAJB9, MEFV, MET, MIF, CXCL9, MMP9, MPI, MPO, MPST, MRC1, MST1, PKNOX1, PLCG1, PLG, CCL19, SELL, SELP, SETMAR, SFRP4, SFRP5, SRSF3, ST3GAL4, SLC2A4, SLC3A2, SLC6A2, SLC10A1, SLC13A1, SMPD1, SIGLEC1, SNAI1, SORD, SP1, SPRR2A, SQLE, CXCL5, CCL17, PLTP, CCL5, PMCH, PPP1R3C, PRKDC, MAPK3, MAP2K7, PRLR, PTBP1, PTGFRN, PTPN6, PTPRC, PTX3, RARA, OPN1LW, RELA, RLN2, BRD2, RPS6KB1, SORT1, S100A4, H3P10

-
Hyperplasia
Wikipedia
Aplasia (organ or part of organ missing) Desmoplasia (connective tissue growth) Dysplasia (change in cell or tissue phenotype ) Hyperplasia (proliferation of cells) Hypoplasia (congenital below-average number of cells, especially when inadequate) Metaplasia (conversion in cell type) Neoplasia (abnormal proliferation) Prosoplasia (development of new cell function) Abiotrophy (loss in vitality of organ or tissue) Atrophy (reduced functionality of an organ, with decrease in the number or volume of cells) Hypertrophy (increase in the volume of cells or tissues) Hypotrophy (decrease in the volume of cells or tissues) Dystrophy (any degenerative disorder resulting from improper or faulty nutrition) v t e Contents 1 Causes 2 Mechanism 3 Diagnosis 3.1 Types 4 Treatment 5 See also 6 References 7 Further reading 8 External links Causes [ edit ] Hyperplasia may be due to any number of causes, including proliferation of basal layer of epidermis to compensate skin loss, chronic inflammatory response , hormonal dysfunctions , or compensation for damage or disease elsewhere. [9] Hyperplasia may be harmless and occur on a particular tissue. ... ISBN 978-1416026129 . ^ a b Dunphy, Lynne M.; Winland-Brown, Jill E. (2011-04-06). Primary Care: The Art and Science of Advanced Practice Nursing . ... External links [ edit ] Classification D MeSH : D006965 SNOMED CT : 76197007 External resources MedlinePlus : 003441 Scholia has a topic profile for Hyperplasia . v t e Overview of tumors , cancer and oncology Conditions Benign tumors Hyperplasia Cyst Pseudocyst Hamartoma Malignant progression Dysplasia Carcinoma in situ Cancer Metastasis Primary tumor Sentinel lymph node Topography Head and neck ( oral , nasopharyngeal ) Digestive system Respiratory system Bone Skin Blood Urogenital Nervous system Endocrine system Histology Carcinoma Sarcoma Blastoma Papilloma Adenoma Other Precancerous condition Paraneoplastic syndrome Staging / grading TNM Ann Arbor Prostate cancer staging Gleason grading system Dukes classification Carcinogenesis Cancer cell Carcinogen Tumor suppressor genes / oncogenes Clonally transmissible cancer Oncovirus Carcinogenic bacteria Misc. Research Index of oncology articles History Cancer pain Cancer and nausea v t e Pathology Principles of pathology Disease Infection Neoplasia Cause Pathogenesis Hemodynamics Ischemia Inflammation Cell damage Wound healing Cellular adaptation Atrophy Hypertrophy Hyperplasia Dysplasia Metaplasia Squamous Glandular Cell death Necrosis Coagulative necrosis Liquefactive necrosis Gangrenous necrosis Caseous necrosis Fat necrosis Fibrinoid necrosis Myocytolysis Programmed cell death Apoptosis Pyknosis Karyorrhexis Karyolysis Accumulations pigment Hemosiderin Lipochrome / Lipofuscin Melanin Steatosis Anatomical pathology Surgical pathology Cytopathology Autopsy Molecular pathology Forensic pathology Oral and maxillofacial pathology Gross examination Histopathology Immunohistochemistry Electron microscopy Immunofluorescence Fluorescence in situ hybridization Clinical pathology Clinical chemistry Hematopathology Transfusion medicine Medical microbiology Diagnostic immunology Immunopathology Enzyme assay Mass spectrometry Chromatography Flow cytometry Blood bank Microbiological culture SerologyTGFB1, CCND1, MMP9, HMOX1, IL13, LEP, OGG1, PTGS2, HIF1A, TCF7L2, ZFP36, MAPK6, GLP1R, PRG4, NFE2L2, MYCN, MAT1A, KDM1A, LDLR, KCNK1, BRD4, IL9, HSPB1, RLN3, PIK3CA, AKT1, MIR340, COL2A1, EGFR, MMP2, PAM, TGFBR2, CCNB1, MMP1, CDKN1A, ASCL1, IKBKB, CSK, CDKN2A, TP53, KRAS, PTEN, CAV1, VDR, PLAU, LHCGR, IRS1, GJA1, STAR, CASR, IRS2, PDE11A, RET, XIAP, BCL2, IL25, TGFA, KRT75, TERT, NOD2, SOD1, SHBG, SCG5, SCN9A, SAA1, S100A9, BLM, TNFRSF13B, TNF, TNFAIP3, AREG, TNFRSF10D, TNFSF10, TP63, TNFSF13B, IRS4, AR, SLC7A5, CXCR4, VGF, VEGFC, VEGFA, RXRA, KLK3, TPMT, TP73, CCN5, RXRG, EPOR, RLN2, HGF, IL15, CHGA, IL4, CCN1, IGFBP3, IGF1, HTR4, HBEGF, GPX1, RLN1, GNAS, EPO, FPR2, FN1, FOXO3, FOXO1, FGF2, EZH2, INSL3, ITGB1, CDKN2B, KRT5, PTK2, BMPR2, PTGIR, CALB2, PRKAR1A, ESR2, PLAUR, PAPPA, SERPINE1, P2RX7, OXTR, CD34, MUC5AC, APEX1, MEN1, CDH1, MARCKS, POMC

-
Cholinergic Urticaria
Wikipedia
Archives of Dermatology . 123 (4): 462–467. doi : 10.1001/archderm.1987.01660280064024 . PMID 3827277 . ^ Poon, E.; Seed, P. T.; Greaves, M. W.; Kobza-Black, A. (1999). ... "Successful treatment of cholinergic urticaria with anti-immunoglobulin E therapy". Allergy . 63 (2): 247–249. doi : 10.1111/j.1398-9995.2007.01591.x . ... PMID 2973859 . ^ Ammann, P.; Surber, E.; Bertel, O. (1999). "Beta blocker therapy in cholinergic urticaria". ... External links [ edit ] Classification D ICD - 10 : L50.5 ICD - 9-CM : 708.5 DiseasesDB : 29573 External resources eMedicine : derm/442 v t e Urticaria and erythema Urticaria ( acute / chronic ) Allergic urticaria Urticarial allergic eruption Physical urticaria Cold urticaria Familial Primary cold contact urticaria Secondary cold contact urticaria Reflex cold urticaria Heat urticaria Localized heat contact urticaria Solar urticaria Dermatographic urticaria Vibratory angioedema Pressure urticaria Cholinergic urticaria Aquagenic urticaria Other urticaria Acquired C1 esterase inhibitor deficiency Adrenergic urticaria Exercise urticaria Galvanic urticaria Schnitzler syndrome Urticaria-like follicular mucinosis Angioedema Episodic angioedema with eosinophilia Hereditary angioedema Erythema Erythema multiforme / drug eruption Erythema multiforme minor Erythema multiforme major Stevens–Johnson syndrome , Toxic epidermal necrolysis panniculitis ( Erythema nodosum ) Acute generalized exanthematous pustulosis Figurate erythema Erythema annulare centrifugum Erythema marginatum Erythema migrans Erythema gyratum repens Other erythema Necrolytic migratory erythema Erythema toxicum Erythroderma Palmar erythema Generalized erythema

-
Hypogonadotropic Hypogonadism
Wikipedia
The mechanism for this reversal is unknown but there is believed to be some neuronal plasticity within GnRH releasing cells. [4] See also [ edit ] Isolated hypogonadotropic hypogonadism Hypergonadotropic hypogonadism Kallmann syndrome Hypothalamic–pituitary–gonadal axis GnRH and gonadotropins ( FSH and LH ) androgens and estrogens References [ edit ] ^ a b c d e f Basaria S (2014). "Male hypogonadism". ... The New England Journal of Medicine . 366 (7): 629–635. doi : 10.1056/NEJMoa1111184 . hdl : 2263/18519 . PMID 22335740 . ^ a b c d e f g h i Boehm U, Bouloux P, Dattani M, de Roux N, Dodé C, Dunkel L, Dwyer A, Giacobini P, Hardelin J, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J (2015). ... PMC 3583156 . PMID 23503957 . ^ a b c d e f g h Silveira L, Latronico A (2013). ... External links [ edit ] Classification D ICD - 10 : N91.1 External resources MedlinePlus : 000390 v t e Gonadal disorder Ovarian Polycystic ovary syndrome Premature ovarian failure Estrogen insensitivity syndrome Hyperthecosis Testicular Enzymatic 5α-reductase deficiency 17β-hydroxysteroid dehydrogenase deficiency aromatase excess syndrome Androgen receptor Androgen insensitivity syndrome Familial male-limited precocious puberty Partial androgen insensitivity syndrome Other Sertoli cell-only syndrome General Hypogonadism Delayed puberty Hypergonadism Precocious puberty Hypoandrogenism Hypoestrogenism Hyperandrogenism Hyperestrogenism Postorgasmic illness syndrome Cytochrome P450 oxidoreductase deficiency Cytochrome b5 deficiency Androgen-dependent condition Aromatase deficiency Complete androgen insensitivity syndrome Mild androgen insensitivity syndrome Hypergonadotropic hypogonadism Hypogonadotropic hypogonadism Fertile eunuch syndrome Estrogen-dependent condition Premature thelarche Gonadotropin insensitivity Hypergonadotropic hypergonadismTACR3, GNRHR, GNRH1, TAC3, NR0B1, LHB, FSHB, KISS1R, LEP, LEPR, NR5A1, KISS1, PRL, CYP19A1, SLC29A3, POLD1, CYP17A1, CGB3, CSHL1, FGFR1, GHRH, SOX2, ANOS1, POLR3A, PROKR2, FGF8, PROP1, CHD7, PROK2, POLR3B, PNPLA6, NSMF, NDN, SEMA3A, RNF216, HS6ST1, FGF17, SRY, GJB2, AXL, CCDC141, TYMP, WDR11, UBA6-AS1, TFR2, SRA1, PWAR1, SEMA3E, GTF2IRD1, NTN1, HJV, SNORD116-1, SNORD115-1, CTDP1, PWRN1, AIP, BAZ1B, HERC2, MKRN3-AS1, HESX1, PTCH2, TP63, LZTR1, MKRN3, WT1, CLIP2, FEZF1, DNAL4, ZMPSTE24, A2ML1, RBM28, IL17RD, MAGEL2, HDAC8, CDH23, DCAF17, SUFU, SOX10, SPRY4, LAS1L, DHH, RRM2B, TBL2, GPR101, RAB3GAP2, FLRT3, NPAP1, KAT6B, PLXND1, RAB3GAP1, MRAS, CBX2, LHX4, SOS1, RNU4ATAC, SOX9, OTX2, POU1F1, ELN, POLG, PMM2, PDGFB, PCSK1, GLI2, GTF2I, SIX6, PTPN11, HFE, NRAS, HSD17B3, NF2, IPW, MEN1, MAP3K1, KRAS, LMNA, PTCH1, RAD51, RAF1, SOX3, SOS2, SNRPN, ANK1, SMARCB1, BMP2, BRAF, RRAS, BRD2, RIT1, CTNNB1, DCC, RFC2, REV3L, DMRT1, DUSP6, RASA2, LIMK1, AR, GH1, ESRRB, GK, MPZ, SERPINA4, VDR, SHBG, PMP22, GGN, NRP2, BECN1, DAZ1, PDE5A, CTLA4, CBLL2, NEUROG3, S1PR1, CPE, IGSF10, S100A4, TYRO3, AMH, NRP1, POMC, CD274, SCO2, FGF2, FGF3, MUL1, FGF9, FGF10, MSTN, TUBB3, PRKN, NT5E, NGF, IGF1, STAR, MC4R, ADH5
-
Muir–torre Syndrome
Wikipedia
Muir–Torre syndrome Other names MTS Micrograph of a sebaceous adenoma , as may be seen in Muir–Torre syndrome. H&E stain . Specialty Oncology , dermatology , medical genetics Muir–Torre syndrome is a rare hereditary, autosomal dominant cancer syndrome [1] : 663 that is thought to be a subtype of HNPCC . ... CS1 maint: multiple names: authors list ( link ) ^ Kleinerman R, Marino J, Loucas E (May 2012). "Muir–Torre Syndrome / Turcot Syndrome overlap? ... CS1 maint: multiple names: authors list ( link ) External links [ edit ] Classification D OMIM : 158320 MeSH : D055653 DiseasesDB : 31385 External resources eMedicine : derm/275 Orphanet : 587 v t e Cancers of skin and associated structures Glands Sweat gland Eccrine Papillary eccrine adenoma Eccrine carcinoma Eccrine nevus Syringofibroadenoma Spiradenoma Apocrine Cylindroma Dermal cylindroma Syringocystadenoma papilliferum Papillary hidradenoma Hidrocystoma Apocrine gland carcinoma Apocrine nevus Eccrine / apocrine Syringoma Hidradenoma or Acrospiroma / Hidradenocarcinoma Ceruminous adenoma Sebaceous gland Nevus sebaceous Muir–Torre syndrome Sebaceous carcinoma Sebaceous adenoma Sebaceoma Sebaceous nevus syndrome Sebaceous hyperplasia Mantleoma Hair Pilomatricoma / Malignant pilomatricoma Trichoepithelioma Multiple familial trichoepithelioma Solitary trichoepithelioma Desmoplastic trichoepithelioma Generalized trichoepithelioma Trichodiscoma Trichoblastoma Fibrofolliculoma Trichilemmoma Trichilemmal carcinoma Proliferating trichilemmal cyst Giant solitary trichoepithelioma Trichoadenoma Trichofolliculoma Dilated pore Isthmicoma Fibrofolliculoma Perifollicular fibroma Birt–Hogg–Dubé syndrome Hamartoma Basaloid follicular hamartoma Folliculosebaceous cystic hamartoma Folliculosebaceous-apocrine hamartoma Nails Neoplasms of the nailbed v t e Metabolic disease : DNA replication and DNA repair-deficiency disorder DNA replication Separation/initiation: RNASEH2A Aicardi–Goutières syndrome 4 Termination/ telomerase : DKC1 Dyskeratosis congenita DNA repair Nucleotide excision repair Cockayne syndrome / DeSanctis–Cacchione syndrome Thymine dimer Xeroderma pigmentosum IBIDS syndrome MSI / DNA mismatch repair Hereditary nonpolyposis colorectal cancer Muir–Torre syndrome Mismatch repair cancer syndrome MRN complex Ataxia telangiectasia Nijmegen breakage syndrome Other RecQ helicase Bloom syndrome Werner syndrome Rothmund–Thomson syndrome / Rapadilino syndrome Fanconi anemia Li-Fraumeni syndrome Severe combined immunodeficiency

-
Neuroacanthocytosis
Wikipedia
Known genetic mutations provide a basis for studying some of the conditions. [5] References [ edit ] ^ Reiss, Ulrike M., Pedro A. De Alacron, and Frank E. Shafer. " Acanthocytosis: eMedicine Pediatrics: General Medicine. " EMedicine - Medical Reference (2010). 7 August 2008. 8 February 2010. ^ a b c d e Walker, RH; Jung, HH; Danek, A (2011). ... Chicago. 19 (4): 403–409. doi : 10.1001/archneur.1968.00480040069007 . PMID 5677189 . ^ E. M. R. Critchley, et al. "Acanthocytosis, normolipoproteinemia and multiple tics" Postgraduate Medical Journal, Leicester, 1970, 46: 698-701. ^ Ole Daniel Enersen. " Levine Critchley syndrome ." ... Chromosome+disorders at the US National Library of Medicine Medical Subject Headings (MeSH) v t e Chromosome abnormalities Autosomal Trisomies /Tetrasomies Down syndrome 21 Edwards syndrome 18 Patau syndrome 13 Trisomy 9 Tetrasomy 9p Warkany syndrome 2 8 Cat eye syndrome / Trisomy 22 22 Trisomy 16 Monosomies / deletions ( 1q21.1 copy number variations / 1q21.1 deletion syndrome / 1q21.1 duplication syndrome / TAR syndrome / 1p36 deletion syndrome ) 1 Wolf–Hirschhorn syndrome 4 Cri du chat syndrome / Chromosome 5q deletion syndrome 5 Williams syndrome 7 Jacobsen syndrome 11 Miller–Dieker syndrome / Smith–Magenis syndrome 17 DiGeorge syndrome 22 22q11.2 distal deletion syndrome 22 22q13 deletion syndrome 22 genomic imprinting Angelman syndrome / Prader–Willi syndrome ( 15 ) Distal 18q- / Proximal 18q- X / Y linked Monosomy Turner syndrome (45,X) Trisomy / tetrasomy , other karyotypes / mosaics Klinefelter syndrome (47,XXY) XXYY syndrome (48,XXYY) XXXY syndrome (48,XXXY) 49,XXXYY 49,XXXXY Triple X syndrome (47,XXX) Tetrasomy X (48,XXXX) 49,XXXXX Jacobs syndrome (47,XYY) 48,XYYY 49,XYYYY 45,X/46,XY 46,XX/46,XY Translocations Leukemia / lymphoma Lymphoid Burkitt's lymphoma t(8 MYC ;14 IGH ) Follicular lymphoma t(14 IGH ;18 BCL2 ) Mantle cell lymphoma / Multiple myeloma t(11 CCND1 :14 IGH ) Anaplastic large-cell lymphoma t(2 ALK ;5 NPM1 ) Acute lymphoblastic leukemia Myeloid Philadelphia chromosome t(9 ABL ; 22 BCR ) Acute myeloblastic leukemia with maturation t(8 RUNX1T1 ;21 RUNX1 ) Acute promyelocytic leukemia t(15 PML ,17 RARA ) Acute megakaryoblastic leukemia t(1 RBM15 ;22 MKL1 ) Other Ewing's sarcoma t(11 FLI1 ; 22 EWS ) Synovial sarcoma t(x SYT ;18 SSX ) Dermatofibrosarcoma protuberans t(17 COL1A1 ;22 PDGFB ) Myxoid liposarcoma t(12 DDIT3 ; 16 FUS ) Desmoplastic small-round-cell tumor t(11 WT1 ; 22 EWS ) Alveolar rhabdomyosarcoma t(2 PAX3 ; 13 FOXO1 ) t (1 PAX7 ; 13 FOXO1 ) Other Fragile X syndrome Uniparental disomy XX male syndrome / 46,XX testicular disorders of sex development Marker chromosome Ring chromosome 6 ; 9 ; 14 ; 15 ; 18 ; 20 ; 21 , 22VPS13A, XK, ORAI1, PANK2, JPH3, ACTB, STIM1, ATXN7, ERAL1, VAMP8, TBP, TAC1, SLC6A2, SELP, PIK3CG, CACNA1A, PIK3CD, PIK3CB, PIK3CA, ATXN3, KCNA6, KCNA5, KCNA1, GCH1, ESR1, EPHB1, ELK3, CRP, TRAP

-
Junctional Epidermolysis Bullosa (Medicine)
Wikipedia
The graft had integrated into the lower layers of skin within a month, and the modified epidermal stem cells sustained this transgenic epidermis, curing the boy. [11] The introduction of genetic changes could increase the chances of skin cancer in other patients, but if the treatment is deemed safe in the long term, scientists believe the approach could be used to treat other skin disorders. [12] The use of gentamicin has been shown to provide some attenuation of this disease. [13] See also [ edit ] Epidermolysis bullosa Junctional epidermolysis bullosa (veterinary medicine) Skin lesion References [ edit ] ^ a b c d e Freedberg IM, Fitzpatrick TB (2003). ... Peter; Marshall, John F.; McGrath, John A.; Mellerio, Jemima E. (2020-09-24). "Epidermolysis bullosa" . ... Dermatology 1-8 External links [ edit ] Classification D ICD - 10 : Q81.8 ( ILDS Q81.850) ICD - 9-CM : 757.39 OMIM : 226700 226650 226730 MeSH : D016109 DiseasesDB : 29579 External resources GeneReviews : Junctional Epidermolysis Bullosa GeneReview/NIH/UW entry on Junctional Epidermolysis Bullosa v t e Congenital malformations and deformations of integument / skin disease Genodermatosis Congenital ichthyosis / erythrokeratodermia AD Ichthyosis vulgaris AR Congenital ichthyosiform erythroderma : Epidermolytic hyperkeratosis Lamellar ichthyosis Harlequin-type ichthyosis Netherton syndrome Zunich–Kaye syndrome Sjögren–Larsson syndrome XR X-linked ichthyosis Ungrouped Ichthyosis bullosa of Siemens Ichthyosis follicularis Ichthyosis prematurity syndrome Ichthyosis–sclerosing cholangitis syndrome Nonbullous congenital ichthyosiform erythroderma Ichthyosis linearis circumflexa Ichthyosis hystrix EB and related EBS EBS-K EBS-WC EBS-DM EBS-OG EBS-MD EBS-MP JEB JEB-H Mitis Generalized atrophic JEB-PA DEB DDEB RDEB related: Costello syndrome Kindler syndrome Laryngoonychocutaneous syndrome Skin fragility syndrome Ectodermal dysplasia Naegeli syndrome / Dermatopathia pigmentosa reticularis Hay–Wells syndrome Hypohidrotic ectodermal dysplasia Focal dermal hypoplasia Ellis–van Creveld syndrome Rapp–Hodgkin syndrome / Hay–Wells syndrome Elastic / Connective Ehlers–Danlos syndromes Cutis laxa ( Gerodermia osteodysplastica ) Popliteal pterygium syndrome Pseudoxanthoma elasticum Van der Woude syndrome Hyperkeratosis / keratinopathy PPK diffuse : Diffuse epidermolytic palmoplantar keratoderma Diffuse nonepidermolytic palmoplantar keratoderma Palmoplantar keratoderma of Sybert Meleda disease syndromic connexin Bart–Pumphrey syndrome Clouston's hidrotic ectodermal dysplasia Vohwinkel syndrome Corneodermatoosseous syndrome plakoglobin Naxos syndrome Scleroatrophic syndrome of Huriez Olmsted syndrome Cathepsin C Papillon–Lefèvre syndrome Haim–Munk syndrome Camisa disease focal : Focal palmoplantar keratoderma with oral mucosal hyperkeratosis Focal palmoplantar and gingival keratosis Howel–Evans syndrome Pachyonychia congenita Pachyonychia congenita type I Pachyonychia congenita type II Striate palmoplantar keratoderma Tyrosinemia type II punctate : Acrokeratoelastoidosis of Costa Focal acral hyperkeratosis Keratosis punctata palmaris et plantaris Keratosis punctata of the palmar creases Schöpf–Schulz–Passarge syndrome Porokeratosis plantaris discreta Spiny keratoderma ungrouped: Palmoplantar keratoderma and spastic paraplegia desmoplakin Carvajal syndrome connexin Erythrokeratodermia variabilis HID / KID Other Meleda disease Keratosis pilaris ATP2A2 Darier's disease Dyskeratosis congenita Lelis syndrome Dyskeratosis congenita Keratolytic winter erythema Keratosis follicularis spinulosa decalvans Keratosis linearis with ichthyosis congenita and sclerosing keratoderma syndrome Keratosis pilaris atrophicans faciei Keratosis pilaris Other cadherin EEM syndrome immune system Hereditary lymphedema Mastocytosis / Urticaria pigmentosa Hailey–Hailey see also Template:Congenital malformations and deformations of skin appendages , Template:Phakomatoses , Template:Pigmentation disorders , Template:DNA replication and repair-deficiency disorder Developmental anomalies Midline Dermoid cyst Encephalocele Nasal glioma PHACE association Sinus pericranii Nevus Capillary hemangioma Port-wine stain Nevus flammeus nuchae Other/ungrouped Aplasia cutis congenita Amniotic band syndrome Branchial cyst Cavernous venous malformation Accessory nail of the fifth toe Bronchogenic cyst Congenital cartilaginous rest of the neck Congenital hypertrophy of the lateral fold of the hallux Congenital lip pit Congenital malformations of the dermatoglyphs Congenital preauricular fistula Congenital smooth muscle hamartoma Cystic lymphatic malformation Median raphe cyst Melanotic neuroectodermal tumor of infancy Mongolian spot Nasolacrimal duct cyst Omphalomesenteric duct cyst Poland anomaly Rapidly involuting congenital hemangioma Rosenthal–Kloepfer syndrome Skin dimple Superficial lymphatic malformation Thyroglossal duct cyst Verrucous vascular malformation Birthmark v t e Diseases of collagen , laminin and other scleroproteins Collagen disease COL1 : Osteogenesis imperfecta Ehlers–Danlos syndrome, types 1, 2, 7 COL2 : Hypochondrogenesis Achondrogenesis type 2 Stickler syndrome Marshall syndrome Spondyloepiphyseal dysplasia congenita Spondyloepimetaphyseal dysplasia, Strudwick type Kniest dysplasia (see also C2/11 ) COL3 : Ehlers–Danlos syndrome, types 3 & 4 Sack–Barabas syndrome COL4 : Alport syndrome COL5 : Ehlers–Danlos syndrome, types 1 & 2 COL6 : Bethlem myopathy Ullrich congenital muscular dystrophy COL7 : Epidermolysis bullosa dystrophica Recessive dystrophic epidermolysis bullosa Bart syndrome Transient bullous dermolysis of the newborn COL8: Fuchs' dystrophy 1 COL9: Multiple epiphyseal dysplasia 2, 3, 6 COL10: Schmid metaphyseal chondrodysplasia COL11: Weissenbacher–Zweymüller syndrome Otospondylomegaepiphyseal dysplasia (see also C2/11 ) COL17: Bullous pemphigoid COL18: Knobloch syndrome Laminin Junctional epidermolysis bullosa Laryngoonychocutaneous syndrome Other Congenital stromal corneal dystrophy Raine syndrome Urbach–Wiethe disease TECTA DFNA8/12, DFNB21 see also fibrous proteins
-
Nonbacterial Thrombotic Endocarditis
Wikipedia
. ^ " nonbacterial thrombotic endocarditis " at Dorland's Medical Dictionary External links [ edit ] Classification D MeSH : D059905 DiseasesDB : 29250 External resources eMedicine : article/155230 v t e Cardiovascular disease (heart) Ischaemic Coronary disease Coronary artery disease (CAD) Coronary artery aneurysm Spontaneous coronary artery dissection (SCAD) Coronary thrombosis Coronary vasospasm Myocardial bridge Active ischemia Angina pectoris Prinzmetal's angina Stable angina Acute coronary syndrome Myocardial infarction Unstable angina Sequelae hours Hibernating myocardium Myocardial stunning days Myocardial rupture weeks Aneurysm of heart / Ventricular aneurysm Dressler syndrome Layers Pericardium Pericarditis Acute Chronic / Constrictive Pericardial effusion Cardiac tamponade Hemopericardium Myocardium Myocarditis Chagas disease Cardiomyopathy Dilated Alcoholic Hypertrophic Tachycardia-induced Restrictive Loeffler endocarditis Cardiac amyloidosis Endocardial fibroelastosis Arrhythmogenic right ventricular dysplasia Endocardium / valves Endocarditis infective endocarditis Subacute bacterial endocarditis non-infective endocarditis Libman–Sacks endocarditis Nonbacterial thrombotic endocarditis Valves mitral regurgitation prolapse stenosis aortic stenosis insufficiency tricuspid stenosis insufficiency pulmonary stenosis insufficiency Conduction / arrhythmia Bradycardia Sinus bradycardia Sick sinus syndrome Heart block : Sinoatrial AV 1° 2° 3° Intraventricular Bundle branch block Right Left Left anterior fascicle Left posterior fascicle Bifascicular Trifascicular Adams–Stokes syndrome Tachycardia ( paroxysmal and sinus ) Supraventricular Atrial Multifocal Junctional AV nodal reentrant Junctional ectopic Ventricular Accelerated idioventricular rhythm Catecholaminergic polymorphic Torsades de pointes Premature contraction Atrial Junctional Ventricular Pre-excitation syndrome Lown–Ganong–Levine Wolff–Parkinson–White Flutter / fibrillation Atrial flutter Ventricular flutter Atrial fibrillation Familial Ventricular fibrillation Pacemaker Ectopic pacemaker / Ectopic beat Multifocal atrial tachycardia Pacemaker syndrome Parasystole Wandering atrial pacemaker Long QT syndrome Andersen–Tawil Jervell and Lange-Nielsen Romano–Ward Cardiac arrest Sudden cardiac death Asystole Pulseless electrical activity Sinoatrial arrest Other / ungrouped hexaxial reference system Right axis deviation Left axis deviation QT Short QT syndrome T T wave alternans ST Osborn wave ST elevation ST depression Strain pattern Cardiomegaly Ventricular hypertrophy Left Right / Cor pulmonale Atrial enlargement Left Right Athletic heart syndrome Other Cardiac fibrosis Heart failure Diastolic heart failure Cardiac asthma Rheumatic fever v t e Paraneoplastic syndromes Endocrine Hypercalcaemia SIADH Zollinger–Ellison syndrome Cushing's syndrome Hematological Multicentric reticulohistiocytosis Nonbacterial thrombotic endocarditis Neurological Paraneoplastic cerebellar degeneration Encephalomyelitis Limbic encephalitis Opsoclonus Polymyositis Transverse myelitis Lambert–Eaton myasthenic syndrome Anti-NMDA receptor encephalitis Musculoskeletal Dermatomyositis Hypertrophic osteopathy Mucocutaneous reactive erythema Erythema gyratum repens Necrolytic migratory erythema papulosquamous Acanthosis nigricans Ichthyosis acquisita Acrokeratosis paraneoplastica of Bazex Extramammary Paget's disease Florid cutaneous papillomatosis Leser-Trélat sign Pityriasis rotunda Tripe palms Other Febrile neutrophilic dermatosis Pyoderma gangrenosum Paraneoplastic pemphigus
-
Hypotransferrinemia
Wikipedia
Retrieved 20 February 2017 . ^ Nemeth E, Ganz T (2006). "Regulation of iron metabolism by hepcidin". ... Springer Science & Business Media. v t e Metal deficiency and toxicity disorders Iron excess: Iron overload Hemochromatosis Hemochromatosis/HFE1 Juvenile/HFE2 HFE3 African iron overload/HFE4 Aceruloplasminemia Atransferrinemia Hemosiderosis deficiency: Iron deficiency Copper excess: Copper toxicity Wilson's disease deficiency: Copper deficiency Menkes disease / Occipital horn syndrome Zinc excess: Zinc toxicity deficiency: Acrodermatitis enteropathica Other Inborn errors of metabolism
-
Diastrophic Dysplasia
Wikipedia
National Library of Medicine External links [ edit ] Classification D ICD - 10 : Q77.5 OMIM : 222600 MeSH : C536170 C536170, C536170 DiseasesDB : 30759 External resources eMedicine : orthoped/632 Orphanet : 628 GeneReviews/NCBI/NIH/UW entry on Diastrophic Dysplasia v t e Osteochondrodysplasia Osteodysplasia/ / osteodystrophy Diaphysis Camurati–Engelmann disease Metaphysis Metaphyseal dysplasia Jansen's metaphyseal chondrodysplasia Schmid metaphyseal chondrodysplasia Epiphysis Spondyloepiphyseal dysplasia congenita Multiple epiphyseal dysplasia Otospondylomegaepiphyseal dysplasia Osteosclerosis Raine syndrome Osteopoikilosis Osteopetrosis Other/ungrouped FLNB Boomerang dysplasia Opsismodysplasia Polyostotic fibrous dysplasia McCune–Albright syndrome Chondrodysplasia / chondrodystrophy (including dwarfism ) Osteochondroma osteochondromatosis Hereditary multiple exostoses Chondroma / enchondroma enchondromatosis Ollier disease Maffucci syndrome Growth factor receptor FGFR2 : Antley–Bixler syndrome FGFR3 : Achondroplasia Hypochondroplasia Thanatophoric dysplasia COL2A1 collagen disease Achondrogenesis type 2 Hypochondrogenesis SLC26A2 sulfation defect Achondrogenesis type 1B Autosomal recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia Chondrodysplasia punctata Rhizomelic chondrodysplasia punctata Conradi–Hünermann syndrome Other dwarfism Fibrochondrogenesis Short rib – polydactyly syndrome Majewski's polydactyly syndrome Léri–Weill dyschondrosteosis v t e Genetic disorder , membrane: Solute carrier disorders 1-10 SLC1A3 Episodic ataxia 6 SLC2A1 De Vivo disease SLC2A5 Fructose malabsorption SLC2A10 Arterial tortuosity syndrome SLC3A1 Cystinuria SLC4A1 Hereditary spherocytosis 4 / Hereditary elliptocytosis 4 SLC4A11 Congenital endothelial dystrophy type 2 Fuchs' dystrophy 4 SLC5A1 Glucose-galactose malabsorption SLC5A2 Renal glycosuria SLC5A5 Thyroid dyshormonogenesis type 1 SLC6A19 Hartnup disease SLC7A7 Lysinuric protein intolerance SLC7A9 Cystinuria 11-20 SLC11A1 Crohn's disease SLC12A3 Gitelman syndrome SLC16A1 HHF7 SLC16A2 Allan–Herndon–Dudley syndrome SLC17A5 Salla disease SLC17A8 DFNA25 21-40 SLC26A2 Multiple epiphyseal dysplasia 4 Achondrogenesis type 1B Recessive multiple epiphyseal dysplasia Atelosteogenesis, type II Diastrophic dysplasia SLC26A4 Pendred syndrome SLC35C1 CDOG 2C SLC39A4 Acrodermatitis enteropathica SLC40A1 African iron overload see also solute carrier family
-
Dementia With Lewy Bodies
MedlinePlus
The APOE gene provides instructions for making a protein, called apolipoprotein E, which packages cholesterol and other fats and carries them through the bloodstream. ... It is thought that the apolipoprotein E produced from the e4 allele of the APOE gene may disrupt the transport of alpha-synuclein into and out of cells, leading to its accumulation.
-
Hypokalemic Sensory Overstimulation
Wikipedia
PMID 18426576 . Medicine portal v t e Nervous system Central nervous system Meninges Spinal cord Brain Hindbrain Medulla Pons Cerebellum Midbrain Forebrain Diencephalon Retina Optic nerve Cerebrum Limbic system Peripheral nervous system Somatic Sensory nerve Motor nerve Cranial nerve Spinal nerve Autonomic Sympathetic Parasympathetic Enteric v t e Anesthesia and anesthesiology Types General Sedation Twilight anesthesia Local Topical Intercostal nerve block Neuraxial blockade Spinal Epidural Dental Inferior alveolar nerve Techniques Airway management Anesthesia provision in the US Arterial catheter Bronchoscopy Capnography Dogliotti's principle Drug-induced amnesia Intraoperative neurophysiological monitoring Nerve block Penthrox inhaler Tracheal intubation Scientific principles Blood–gas partition coefficient Concentration effect Fink effect Minimum alveolar concentration Second gas effect Measurements ASA physical status classification system Baricity Bispectral index Entropy monitoring Fick principle Goldman index Guedel's classification Mallampati score Neuromuscular monitoring Thyromental distance Instruments Anaesthetic machine Anesthesia cart Boyle's machine Gas cylinder Laryngeal mask airway Laryngeal tube Medical monitor Odom's indicator Relative analgesia machine Vaporiser Double-lumen endotracheal tube Endobronchial blocker Complications Emergence delirium Allergic reactions Anesthesia awareness Local anesthetic toxicity Malignant hyperthermia Perioperative mortality Postanesthetic shivering Postoperative nausea and vomiting Postoperative residual curarization Subspecialties Cardiothoracic Critical emergency medicine Geriatric Intensive care medicine Obstetric Oral sedation dentistry Pain medicine Professions Anesthesiologist Anesthesiologist assistant Nurse anesthetist Operating department practitioners Certified Anesthesia Technician Certified Anesthesia Technologist Anaesthetic technician Physicians' assistant (anaesthesia) History ACE mixture Helsinki Declaration for Patient Safety in Anaesthesiology History of general anesthesia History of neuraxial anesthesia History of tracheal intubation Organizations American Association of Nurse Anesthetists American Society of Anesthesia Technologists & Technicians American Society of Anesthesiologists Anaesthesia Trauma and Critical Care Association of Anaesthetists of Great Britain and Ireland Royal College of Anaesthetists Association of Veterinary Anaesthetists Australian and New Zealand College of Anaesthetists Australian Society of Anaesthetists International Anesthesia Research Society Category Outline
-
Alcohol Intolerance
Wikipedia
. ^ Potential risks to human health and the environment from the use of calcium cyanamide as fertiliser , page 29, Scientific Committee on Health and Environmental Risks , Retrieved 14 November 2016 v t e Alcohol and health Alcohol use Alcohol-related crimes Drunk drivers Alcohol-related traffic crashes in the United States Driving under the influence (DUI) Drunk driving in the United States Public intoxication Rum-running Adulterated moonshine / Denatured alcohol List of methanol poisoning incidents Alcoholism Alcohol and Native Americans Alcoholism in adolescence Alcoholism in family systems Collaborative Study on the Genetics of Alcoholism College student alcoholism Disease theory of alcoholism High-functioning alcoholic (HFA) Seeing pink elephants Chemistry Beer chemistry Congener Alcohol congener analysis Ethanol Blood alcohol content Breathalyzer Fusel alcohol Wine chemistry Effects Short-term effects of alcohol consumption Long-term effects of alcohol On memory Subjective response to alcohol Interactions Aging Brain Cancer breast cancer Cortisol Pregnancy Sleep Tolerance / intolerance Weight Beverage-specific Beer: Potomania Red wine: Red wine headache Social issues Alcohol advertising on college campuses Sex Alcohol myopia Alcohol abuse among college students Binge drinking Epidemiology Blackout (alcohol-related amnesia) Blackout Wednesday Drinking game list pregaming Drinking in public Drunk dialing Drunk walking Drunkorexia Dry drunk French paradox Hair of the dog Nightcap Pantsdrunk Passive drinking Binge drinking devices Beer bong Yard of ale Routes of administration Alcohol enema Alcohol inhalation Sconcing Surrogate alcohol Related issues Balconing Suicide History Dionysian Mysteries Dipsomania Gin Craze List of deaths through alcohol Rum ration Speakeasy General Beer day Drinking culture Apéritif and digestif Hangover remedies Health effects of wine Wine and food matching Long-distance race involving alcohol List of countries by alcohol consumption per capita Alcohol consumption by youth in the United States Nip joint Alcohol control Alcohol law Administrative license suspension (ALS) Alcohol packaging warning messages Drunk driving law by country DWI court Field sobriety testing Hip flask defence Ignition interlock device Legal drinking age Age controversy in US Underage drinking in US List of alcohol laws of US Alcohol prohibition List of countries with alcohol prohibition Neo-prohibitionism Temperance movement Sobriety Alcohol detoxification Alcohol-free zone Dry campus United States open-container laws Designated driver Alcohol rehabilitation Drunk tank Managed alcohol program Non-alcoholic drink List of cocktails List of mixed drinks Spritzer Malt drinks Teetotalism Temperance bar Twelve-step groups Al-Anon/Alateen Alcoholics Anonymous (AA): Adult Children of Alcoholics (ACA) Alcohol limitation 0-0-1-3 Alcohol education Alcohol server training FRAMES Dry January Foundation for Advancing Alcohol Responsibility Campaigns Get Your Sexy Back Liquor license Low-alcohol drinks Fermented tea Low-alcohol beer Low-alcoholic malt drinks Small beer Measurement Alcoholic spirits measure Standard drink Recommended maximum intake of alcoholic beverages Addiction medicine Disulfiram-like drugs : disulfiram , calcium carbimide , cyanamide . ... You can help Wikipedia by expanding it . v t e
-
Dissociated Sensory Loss
Wikipedia
. ^ Peres Serra, J; Martínez Yélamos, S; Ballabriga Planas, J; Basart Tarrats, E; Arbizu Urdiain, T (1994). "[Dissociated sensory loss syndrome in multiple sclerosis] (translated)". ... Neurological surgery . 12 (10): 1219–23. PMID 6504260 . v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline








