The study noted the “hallmarks of epidemic typhus and relapsing fever”. [27] History of vaccines [ edit ] Major developments for typhus vaccines started during World War I , as typhus caused high mortality, and threatened the health and readiness for soldiers on the battlefield. [28] Vaccines for typhus, like other vaccines of the time, were classified as either living or killed vaccines. [28] Live vaccines were typically an injection of live agent, and killed vaccines are live cultures of an agent that are chemically inactivated prior to use. [28] Attempts to create a living vaccine of classical, louse -borne, typhus were attempted by French researchers but these proved unsuccessful. [28] Researchers turned to murine typhus to develop a live vaccine. [28] At the time, murine vaccine was viewed as a less severe alternative to classical typhus. Four versions of a live vaccine cultivated from murine typhus were tested, on a large scale, in 1934. [28] While the French were making advancements with live vaccines, other European countries were working to develop killed vaccines. [28] During World War II , there were three kinds of potentially useful killed vaccines. [28] All three killed vaccines relied on the cultivation of Rickettsia prowazekii , the organism responsible for typhus. [28] The first attempt at a killed vaccine was developed by Germany , using the Rickettsia prowazekii found in louse feces. [28] The vaccine was tested extensively in Poland between the two world wars and used by the Germans for their troops during their attacks on the Soviet Union . [28] A second method of growing Rickettsia prowazekii was discovered using the yolk sac of chick embryos . Germans tried several times to use this technique of growing Rickettsia prowazekii but no effort was pushed very far. [28] The last technique was an extended development of the previously known method of growing murine typhus in rodents. [28] It was discovered that rabbits could be infected, by a similar process, and contract classical typhus instead of murine typhus. [28] Again, while proven to produce suitable Rickettsia prowazekii for vaccine development, this method was not used to produce wartime vaccines. [28] During WWII, the two major vaccines available were the killed vaccine grown in lice and the live vaccine from France . [28] Neither was used much during the war. [28] The killed, louse-grown vaccine was difficult to manufacture in large enough quantities, and the French vaccine was not believed to be safe enough for use. [28] The Germans worked to develop their own live vaccine from the urine of typhus victims. [28] While developing a live vaccine, Germany used live Rickettsia prowazekii to test multiple possible vaccines' capabilities. [28] They gave live Rickettsia prowazekii to concentration camp prisoners, using them as a control group for the vaccine tests. [28] The use of DDT as an effective means of killing lice, the main carrier of typhus, was discovered in Naples . [28] Society and culture [ edit ] Biological weapon [ edit ] Typhus was one of more than a dozen agents that the United States researched as potential biological weapons before President Richard Nixon suspended all non-defensive aspects of the U.S. biological weapons program in 1969. [29] Poverty and displacement [ edit ] The CDC lists the following areas as active foci of human epidemic typhus: Andean regions of South America, some parts of Africa; on the other hand, the CDC only recognizes an active enzootic cycle in the United States involving flying squirrels (CDC).
A Rickettsial disease characterized by malaise and vague symptoms before the onset of high fever, headache, severe myalgias and less commonly petechial rash on the trunk and limbs, nausea, vomiting, coughing and pneumonia. Most patients also have some central nervous system disturbances, such as meningeal irritation, confusion, drowsiness, seizures, coma, and hearing loss.
A number sign (#) is used with this entry because immunodeficiency-28 (IMD28) is caused by homozygous or compound heterozygous mutation in the IFNGR2 gene (147569) on chromosome 21q22.
Mendelian susceptibily to mycobacterial diseases (MSMD) due to complete interferon gamma receptor 2 (IFN-gammaR2) deficiency is a genetic variant of MSMD (see this term) characterized by a complete deficiency in IFN-gammaR2, leading to an undetectable response to IFN-gamma, and consequently, to severe and often fatal infections with bacillus Calmette-Guérin (BCG) and other environmental mycobacteria (EM). Epidemiology The prevalence is unknown. Only ten children have been identified to date. Clinical description Severe and often fatal BCG and EM infections begin in early childhood (before the age of 3). The most common pathogens seen in patients include Mycobacterium fortuitum , Mycobacterium bovis BCG, Mycobacterium abscessus and Mycobacterium avium . Infections are disseminated and can involve soft tissue, bone marrow, lungs, skin, bones and lymph nodes.
Autosomal recessive mendelian susceptibility to mycobacterial diseases (MSMD) due to partial IFNgammaR2 deficiency is a genetic variant of MSMD (see this term) characterized by a partial deficiency in IFN-gammaR2, leading to a residual response to IFN-gamma and consequently to recurrent, moderately severe infections with bacillus Calmette-Guérin (BCG) and other environmental mycobacteria (EM). Epidemiology The prevalence is unknown. Only one patient has been reported with this variant to date. Clinical description The patient presented with a mild infection caused by BCG and M. abscessus . Etiology Autosomal recessive MSMD due to partial IFNgammaR2 deficiency is caused by a homozygous mutation (R114C) in IFNGR2 on chromosome 21q22.1-22.2 that encodes the IFN-gamma receptor ligand binding chain. This mutation leads to a residual cellular response to IFN-gamma in terms of IL12p40 production.
"The Murrain Now Known As Rinderpest" . taa.org.uk. Archived from the original on 28 August 2008 . Retrieved 28 July 2008 . ^ "murrain - Dictionary.com" . dictionary.com . ... ISBN 0-691-05891-1 . ^ "Murrain (WebBible Encyclopedia) - ChristianAnswers.Net" . christiananswers.net . Retrieved 28 July 2008 . ^ Billingsley, John. "Northern Earth - Medical Care, Magical Cure" . northernearth.co.uk. Archived from the original on 25 September 2006 . Retrieved 28 July 2008 . ^ Mullett, Charles F. (1946).
Some sources suggest that Peter himself may have had ulnar nerve entrapment . [4] [5] [6] References [ edit ] ^ "Image of Hand of benediction" . Stanford Medicine . Retrieved 28 May 2017 . ^ "The Hand Examination | Stanford Medicine 25" . stanfordmedicine25.stanford.edu . Stanford Medicine . Retrieved 28 May 2017 . ^ Schreuders, T. A. R. (2011-12-14). ... "Analysis of the Papal Benediction Sign: The ulnar neuropathy of St. Peter" . Clinical Anatomy . 28 (6): 696–701. doi : 10.1002/ca.22584 .
Description Age-related cataracts are one of the leading causes of visual impairment and blindness among the elderly worldwide. Among age-related cataracts, cortical opacities rank as the second most common type (Iyengar et al., 2004). The preferred title/symbol of this entry was formerly 'Cataract, Age-Related Cortical, 1; ARCC1.' Mapping To identify susceptibility loci for cortical cataracts, Iyengar et al. (2004) genotyped a subset of families from the Beaver Dam Eye Study (performed in the population of Beaver Dam, Wisconsin) and did a model-free genomewide linkage analysis for markers linked to a quantitative measure of cortical opacity. They obtained evidence for linkage at marker D1S1622 on chromosome 1p35 (P less than 0.0002) and at marker D6S1053 on chromosome 6q12 (p less than 0.00008) in the initial scan.
A number sign (#) is used with this entry because of evidence that inflammatory bowel disease-28 (IBD28) is caused by homozygous or compound heterozygous mutation in the IL10RA gene (146933) on chromosome 11q23.
Archived from the original (PDF) on 2006-01-05 . Retrieved 2006-06-28 . Carta, A.; et al. (June 1999). "Neuro-ophthalmological presentation of pneumosinus dilatans" . Neuro-Ophthalmology . Retrieved 2006-06-28 . External links [ edit ] MR/CT scans of pneumosinus dilatans from MedPix
Foot deformities were first observed between ages two and ten years, were moderately or severely disabling, and required surgery in 6% (1/18) to 11% (3/28) of cases (Table 2). Table 2. Occurrence of Manifestations of CMT4C by Study View in own window Study Finding Study (Total Patients) Azzedine et al [2006] (28) Colomer et al [2006] (14) Senderek et al [2003] (18) Houlden et al [2009] (6) Baets et al [2011] (9) Laššuthová et al [2011] (16) Yger et al [2012] (14) Fischer et al [2012] (6) Cumulative Data Age at onset 1 st symptoms 2-10 4-39 Infancy-12 1-16 <1 1-12 1-12 ND 1-16 Neuropathy 2-10 Infancy-12 1-16 <1 2-50 2-50 2-25 1-50 Age at (last) exam (yrs) 5-45 8-45 11-56 8-42 ND ND 8-59 5-59 Foot deformity Pes cavus 20/28 14/14 1 8/18 Yes ND 13/15 12/14 ND Pes planus 7/28 4/18 Yes ND no no ND Pes valgus 1/28 ND ND ND no 3/14 ND Other No Hammer toes 8/18 Small feet ND Hammer toes no ND Total 28/28 14/14 13/18 2 6/6 1 7/9 14/15 14/14 ND 96/104 (92%) Age at onset (yrs) 2-10 No data 2-12 ND ND ND 1,12 3 ND 1-12 Surgery 3/28 None 1/13 No ND 9/14 4/14 ND 17/69 (24%) Spine deformity Total 27/28 5/14 4 11/18 4 6/6 6/9 10/12 12/12 5/6 82/105 (78%) Age at onset (yrs) 2-10 4 4-12 5 ND 2, 6, 7, 12 6 ND 7-15 ND 2-15 Surgery 7 7 + 6 8 = 13/27 1/14 1/11 3/6 3/6 ND 1/12 ND 22/76 (29%) ND = not done or not documented 1. ... Additional Clinical Findings in CMT4C by Study View in own window Clinical Finding Study (Total Patients) Azzedine et al [2006] (28) Colomer et al [2006] (14) Senderek et al [2003] (18) Houlden et al [2009] (6) Baets et al [2011] (9) Laššuthová et al [2011] (16) Yger et al [2012] (14) Cumulative Data Hypoacusis 5/28 0/14 2/18 0/6 0/9 0/15 8/13 15/103 Deafness 0/28 5/14 1/18 2/6 1/9 3/15 0/13 12/103 Nystagmus 0/28 0/14 2/18 0/6 2/9 0/15 0/13 4/103 Pupillary light reflexes 0/28 3/14 0/18 1/6 0/9 0/15 14/13 4/20 Other pupillary disturbances -- -- -- Asymmetric size 1/6 -- -- -- 1/6 Lingual fasciculation -- 3/14 -- -- -- -- -- 3/14 Tongue atrophy and/or weakness -- -- -- 1/6 -- -- 2/13 3/19 Facial paresis 1/28 -- -- 1/6 1/9 -- 4/13 7/56 Facial weakness -- -- -- 1/6 -- -- -- 1/6 Head tremor -- 2/14 -- -- -- -- -- 2/14 Vocal cord involvement -- -- -- -- -- -- 1/13 1/13 Total patients w/cranial nerve involvement 5/28 9/14 5/14 1 4/6 -- -- 10/13 33/73 Respiratory insufficiency or hypoventilation 7/28 2 -- 2/18 -- 1/9 -- -- 10/55 Sensory ataxia 1/28 2/14 -- -- -- -- -- >3/42 3 Diabetes mellitus -- -- 1/18 -- -- -- -- 1/18 Romberg sign -- 2/14 -- -- -- -- -- 2/14 1. 14 of 18 patients were examined for cranial nerve involvement. 2. ... Genotype-Phenotype Correlations Significant intrafamilial variability in the disease course makes it difficult to identify genotype-phenotype correlations [Kessali et al 1997, Gabreëls-Festen et al 1999, Senderek et al 2003, Azzedine et al 2005a, Azzedine et al 2005b, Azzedine et al 2006]. In 28 individuals with CMT4C, Azzedine et al [2006] found no correlation between the nature and the position of the pathogenic variant, disease duration, and the stage of disability.
A number sign (#) is used with this entry because Charcot-Marie-Tooth (CMT) disease type 4C is caused by homozygous or compound heterozygous mutation in the SH3TC2 gene (608206). Mild mononeuropathy of the median nerve (MNMN; 613353) is a less severe allelic disorder caused by heterozygous mutation in the SH3TC2 gene. For a phenotypic description and a discussion of genetic heterogeneity of autosomal recessive demyelinating Charcot-Marie-Tooth disease, see CMT4A (214400). Clinical Features Kessali et al. (1997) reported 2 large consanguineous Algerian families with autosomal recessive demyelinating CMT. Mean age at onset was 5.2 years (range 2 to 10 years). All patients had foot deformities and scoliosis, often requiring surgery.
Charcot-Marie-Tooth disease type 4C (CMT4C) is a subtype of Charcot-Marie-Tooth type 4 characterized by childhood or adolescent-onset of a relatively mild, demyelinating sensorimotor neuropathy that contrasts with a severe, rapidly progressing, early-onset scoliosis, and the typical CMT phenotype (i.e. distal muscle weakness and atrophy, sensory loss, and often foot deformity). A wide spectrum of nerve conduction velocities are observed and cranial nerve involvement and kyphoscoliosis have also been reported.
A number sign (#) is used with this entry because DFNA28 is caused by heterozygous mutation in the GRHL2 gene (608576) on chromosome 8q22. Clinical Features Peters et al. (2002) reported a large American family in which 11 members spanning 5 generations were affected with sensorineural hearing loss. Affected individuals showed a mild to moderate hearing loss across most frequencies that progressed to severe loss in the higher frequencies by the fifth decade. Age at onset varied, with the earliest case documented at 7 years of age. Vona et al. (2013) studied a large 5-generation family segregating autosomal dominant postlingual hearing loss with a highly variable age of onset and progression.
Clinical Characteristics Clinical Description To date, 28 individuals from six families have been identified with a pathogenic variant in TTBK2 [Houlden et al 2007, Bauer et al 2010, Lindquist et al 2017, Deng et al 2019]. ... Clinical Features of Spinocerebellar Ataxia Type 11 View in own window Feature Number of Persons w/Feature Comment Cerebellar ataxia 28/28 Variable truncal &/or gait ataxia Limb ataxia 21/28 Dysarthria 22/28 Jerky pursuit 18/28 Nystagmus 20/28 Ophthalmoplegia 2/28 Diplopia 4/28 Hyperreflexia 18/28 Most prominent in the British family Lower > upper limbs Extrapyramidal features 1/28 Laterocollis "No-no" head tremor Onset. ... It is unclear if abnormal eye findings progress, but ocular symptoms were the only presenting feature for one individual out of 28 affected. Although abnormal eye findings may be seen at the time of presentation, they are rarely symptomatic. ... No other pyramidal signs apart from hyperreflexia were observed in the 28 individuals apart from one individual with upgoing plantar reflexes.
A number sign (#) is used with this entry because of evidence that this form of spinocerebellar ataxia, SCA11, is caused by mutations in the gene encoding tau tubulin kinase-2 (TTBK2; 611695). Pure autosomal dominant cerebellar ataxia is a relatively benign, late-onset, slowly progressive neurologic disorder characterized by an uncomplicated cerebellar syndrome (see SCA1; 164400). For a general discussion of autosomal dominant spinocerebellar ataxia, see SCA1 (164400). Clinical Features Worth et al. (1999) identified 2 British families with a relatively pure form of autosomal dominant cerebellar ataxia in which affected individuals did not have the CAG expansion in the CACNA1A gene (601011), excluding a diagnosis of SCA6 (183086), and in which the disease was not linked to the SCA5 (600224) or SCA10 (603516) loci. Houlden et al. (2007) described an affected family from Devon, on the southwest coast of England, which stretched over 8 generations.
A rare neurologic disease that is characterized by the early-onset of cerebellar signs, eye movement abnormalities and pyramidal signs. Epidemiology Spinocerebellar ataxia type 11 (SCA11) prevalence is unknown but SCA11 is thought to account for 2% of ADCA type III cases. More than sixty clinically affected members from six families (of British, Pakistani, Danish, Chinese, German and French descent) have been reported to date. Clinical description SCA11 presents between the ages of 11-70 years with a mean age of onset of 25 years. It presents with the cerebellar signs such as dysarthria and progressive ataxia, eventually leading to difficulty walking and loss of balance as well as eye movement abnormalities (jerky pursuit, horizontal and vertical nystagmus and ophthalmoplegia).
Spinocerebellar ataxia type 11 (SCA11) is characterized by progressive cerebellar ataxia (difficulty walking and balance) and abnormal eye signs (jerky pursuit, horizontal and vertical movements (nystagmus), pyramidal features (increased muscular tonus, increased reflexes and an abnormal reflex known as Babinski sign and inability to make to perform fine movements), peripheral neuropathy with numbness, weakness or pain in the feet or hands or other places of the body and dystonia. It is a very rare disease and very few patients have been reported to date. Age of onset ranges from the early teens to the mid 20s and life span is normal. Diagnosis is based on signs and symptoms and is confirmed by genetic testing finding a change (mutation) in the TTBK2 gene . It is inherited in an autosomal dominant manner. Treatment may include speech and language therapy for talking and swallowing problems, occupational therapy, including home adaptations, physiotherapy and use of assistive walking devices and ankle-foot orthotics (AFOs) for those with neuropathy.
February 13, 2014 . Retrieved 28 December 2014 . ^ a b c d e "Oral Candidiasis Statistics" . cdc.gov . ... Archived from the original on 29 December 2014 . Retrieved 28 December 2014 . ^ "Symptoms of Oral Candidiasis" . cdc.gov . ... Archived from the original on 29 December 2014 . Retrieved 28 December 2014 . ^ "Candidiasis" . cdc.gov . ... Archived from the original on 29 December 2014 . Retrieved 28 December 2014 . ^ a b c Walker M (2008). ... Archived from the original on 29 December 2014 . Retrieved 28 December 2014 . ^ a b Dolin GL, Mandell JE, Bennett R (2010).
A number sign (#) is used with this entry because of evidence that childhood-onset dystonia-28 (DYT28) is caused by heterozygous mutation in the KMT2B gene (606834) on chromosome 19p13. Description Dystonia-28 is an autosomal dominant neurologic disorder characterized by onset of progressive dystonia in the first decade of life.
Presenting manifestations (documented for 37/39 individuals) include the following: Lower-limb dystonia characterized by foot posturing, toe walking, and gait disturbance (in 28 individuals) Upper-limb dystonia leading to abnormal hand/arm posturing, dystonic tremor, and difficulties in handwriting and hand dexterity (6) Cervical dystonia (1) Truncal/axial dystonia (1) Dystonic tremor and (action) myoclonus (1) Over time, the majority of individuals developed progressive cranial, cervical, and laryngeal dystonia with features of retrocollis, torticollis, dysarthria/anarthria, dysphonia, and difficulties in swallowing and chewing.
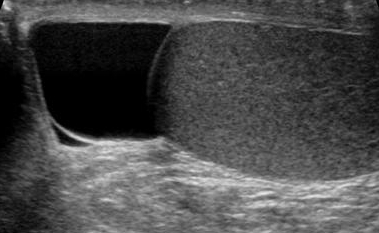
Overview A spermatocele (SPUR-muh-toe-seel) is an abnormal sac (cyst) that develops in the epididymis — the small, coiled tube located on the upper testicle that collects and transports sperm. Noncancerous and generally painless, a spermatocele usually is filled with milky or clear fluid that might contain sperm. Spermatocele A spermatocele, also known as a spermatic cyst, is a typically painless, noncancerous (benign), fluid-filled sac that grows near the top of a testicle. The exact cause of spermatoceles isn't clear, but they might be due to a blockage in one of the tubes that transport sperm. Spermatoceles, sometimes called spermatic cysts, are common. They typically don't reduce fertility or require treatment.
Epididymal cyst Specialty Urology Causes Congenital but manifests in adults. Diagnostic method By clinical examination it appears as a mass which resembles a bunch of grapes. It may also be detected using a transillumination test, where a mass will appear as the 'Chinese Lantern' pattern. Treatment Surgical excision can be performed but it is not preferred in young adults as it can cause infertility as well as psychological effects on the patient. Epididymal cyst An epididymal cyst is a cyst of the epididymis containing serous liquid.
Oldest conjoined twins Ronnie and Donnie Galyon Born Ronald Galyon Donald Galyon ( 1951-10-28 ) October 28, 1951 Dayton, Ohio Died July 4, 2020 (2020-07-04) (aged 68) Dayton, Ohio Occupation Sideshow attractions, reality TV personalities Known for Oldest living set of conjoined twins Ronnie and Donnie Galyon (October 28, 1951 – July 4, 2020) [1] were American conjoined twins . ... Elizabeth Hospital in Dayton, Ohio , on October 28, 1951, to Wesley and Eileen Galyon.
"Summary of the National Institute of Arthritis, Diabetes, Digestive and Kidney Diseases Workshop on Interstitial Cystitis, National Institutes of Health, Bethesda, Maryland, August 28-29, 1987" . Journal of Urology . 140 (1): 203–206. doi : 10.1016/S0022-5347(17)41529-1 . ^ "How is a finding of glomerulations characterized in interstitial cystitis/bladder pain syndrome (IC/BPS)?" . www.medscape.com . 2018 . Retrieved 2020-07-28 . ^ "Cystoscopy with Hydrodistention" . Interstitial Cystitis Association . 2015 . Retrieved 2020-07-28 . ^ Wennevik Gjertrud E.; Meijlink Jane M.; Hanno Philip; Nordling Jørgen (2016).
RightDiagnosis.com . Healthgrades . Retrieved 28 October 2015 . ^ Liebeskind, David S (8 January 2015). "Hemorrhagic Stroke Clinical Presentation" . Medscape . WebMD . Retrieved 28 October 2015 . This medical sign article is a stub .
Lucio and Simplicio Godina Born March 8, 1908 Samar , Philippines Died Lucio: 24 November 1936 (1936-11-24) (aged 28) Simplicio: 8 December 1936 (1936-12-08) (aged 28) New York City , U.S.
Limited mood effects were shown at 250mg/day and negative mood effects above. [3] Contents 1 Chocolate 1.1 In humans 1.2 In other species 2 Symptoms 3 See also 4 Footnotes 5 References 6 External links Chocolate [ edit ] In humans [ edit ] Cocoa beans contain about 1.2% theobromine by weight , [4] so 28 g (1 ounce ) of raw cocoa contains approximately 0.3 g theobromine. ... Archived from the original on January 1, 2009 . Retrieved July 28, 2009 . ^ "Greedy dog cheats chocolate death" . ... Archived from the original on April 6, 2009 . Retrieved July 28, 2009 . ^ "The Poisonous Chemistry of Chocolate" . ... Archived from the original on January 14, 2014 . Retrieved July 28, 2009 . ^ Gwaltney-Brant, Sharon (February 2001). ... Archived from the original on February 26, 2011 . Retrieved July 28, 2009 . References [ edit ] Theobromine in the ChemIDplus database (September 9, 2004) Merck Veterinary Manual (Toxicology/Food Hazards section), Merck & Co., Inc., Chocolate Poisoning .
., David. "Cryptotia" . Retrieved December 28, 2011 . CS1 maint: multiple names: authors list ( link ) ^ University of North Carolina, Chapel Hill, School of Medicine. "Cryptotia" . Retrieved December 28, 2011 . CS1 maint: multiple names: authors list ( link ) External links [ edit ] Classification D OMIM : 123557 MeSH : C565140 v t e Congenital malformations and deformations of ears Size Macrotia Microtia Anotia Position Low-set ears Other Accessory auricle Mondini dysplasia This article about a disease of the ear and mastoid process is a stub .
Cryptotia is a congenital auricular deformity in which the upper pole of the cartilage is buried beneath the scalp. The incidence of cryptotia is said to be higher among Japanese than Caucasians and to be approximately 1 in 400 (Hayashi et al., 1993). Six individuals in 2 generations, with 2 instances of male-to-male transmission, were affected in the family reported by Hayashi et al. (1993). Inheritance - Autosomal dominant Ears - Upper ear cartilage buried under scalp ▲ Close