Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Neuroendocrine Tumor
Wikipedia
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... According to the WHO and UNICEF, deaths attributable to malaria in 2015 were reduced by 60% [77] from a 2000 estimate of 985,000, largely due to the widespread use of insecticide-treated nets and artemisinin-based combination therapies. [74] In 2012, there were 207 million cases of malaria.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
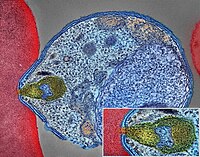
-
Pancreatic Neuroendocrine Tumor
Wikipedia
PanNETs are a type of neuroendocrine tumor , representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign , while some are malignant . ... However, morphological imaging alone is not sufficient for a definite diagnosis [14] [16] On biopsy , immunohistochemistry is generally positive for chromogranin and synaptophysin . [17] Genetic testing thereof typically shows altered MEN1 and DAXX / ATRX . [17] Staging [ edit ] The 2010 WHO classification of tumors of the digestive system grades all the neuroendocrine tumors into three categories, based on their degree of cellular differentiation (from well-differentiated "NET G1" through to poorly-differentiated "NET G3"). ... Combinations of several medicines have been used, such as doxorubicin with streptozocin and fluorouracil (5-FU) [12] and capecitabine with temozolomide. [ citation needed ] Although marginally effective in well-differentiated PETs, cisplatin with etoposide has some activity in poorly differentiated neuroendocrine cancers (PDNECs), [12] particularly if the PDNEC has an extremely high Ki-67 score of over 50%. [8] : 30 Several targeted therapy agents have been approved in PanNETs by the FDA based on improved progression-free survival (PFS): everolimus (Afinitor) is labeled for treatment of progressive neuroendocrine tumors of pancreatic origin in patients with unresectable, locally advanced or metastatic disease. [20] [21] The safety and effectiveness of everolimus in carcinoid tumors have not been established. [20] [21] sunitinib (Sutent) is labeled for treatment of progressive, well-differentiated pancreatic neuroendocrine tumors in patients with unresectable locally advanced or metastatic disease. [22] [23] Sutent also has approval from the European Commission for the treatment of 'unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors with disease progression in adults'. [24] A phase III study of sunitinib treatment in well differentiated pNET that had worsened within the past 12 months (either advanced or metastatic disease) showed that sunitinib treatment improved progression-free survival (11.4 months vs. 5.5 months), overall survival , and the objective response rate (9.3% vs. 0.0%) when compared with placebo. [25] Genetics [ edit ] Pancreatic neuroendocrine tumors may arise in the context of multiple endocrine neoplasia type 1 , Von Hippel–Lindau disease , neurofibromatosis type 1 (NF-1) or tuberose sclerosis (TSC) [26] [27] Analysis of somatic DNA mutations in well-differentiated pancreatic neuroendocrine tumors identified four important findings: [28] [6] as expected, the genes mutated in NETs, MEN1 , ATRX , DAXX , TSC2 , PTEN and PIK3CA , [28] are different from the mutated genes previously found in pancreatic adenocarcinoma . [29] [30] one in six well-differentiated pancreatic NETs have mutations in mTOR pathway genes, such as TSC2 , PTEN and PIK3CA . [28] The sequencing discovery might allow selection of which NETs would benefit from mTOR inhibition such as with everolimus , but this awaits validation in a clinical trial . mutations affecting a new cancer pathway involving ATRX and DAXX genes were found in about 40% of pancreatic NETs. [28] The proteins encoded by ATRX and DAXX participate in chromatin remodeling of telomeres ; [31] these mutations are associated with a telomerase -independent maintenance mechanism termed ALT (alternative lengthening of telomeres) that results in abnormally long telomeric ends of chromosomes . [31] ATRX / DAXX and MEN1 mutations were associated with a better prognosis . [28] References [ edit ] ^ Burns WR, Edil BH (March 2012).MEN1, ATRX, DAXX, ELK3, TP53, EPHB1, SLC6A2, CEACAM5, CEACAM7, UQCRFS1, DHDDS, CHPT1, RALBP1, CIB1, SEMA4D, RIPK1, CXCR4, VEGFA, TTR, GNA12, TSC2, TFE3, CDKN1B, PSG2, POMC, MYCN, CEACAM3, GRN, MUC16

- Dowling-Degos Disease GARD
-
Azotemia, Familial
OMIM
Furthermore, urea is reabsorbed actively by the tubule; this process is apparently brought into play particularly in states of low protein intake. Net reabsorption might be due to exaggerated active reabsorption or to deficient secretion.
-
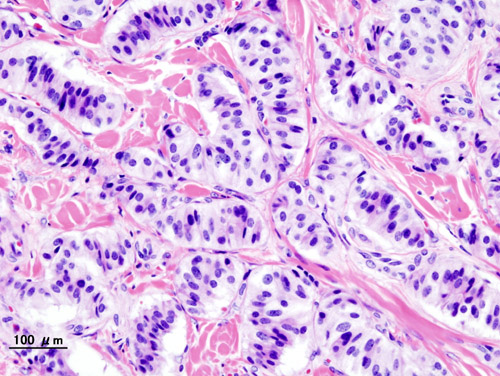
Insulinoma
GARD
Insulinoma is a type of pancreatic neuroendocrine tumor (pancreatic NET), which refers to a group of rare tumors that form in the hormone-making cells of the pancreas.MEN1, RPS15, CDKN2B, CDKN2C, IAPP, GCG, CDKN1B, CDKN1A, SST, FOXM1, GLP1R, PDX1, INS, IL1B, RIT2, PTPRN2, GAD1, EHMT1, IGF2, ZGLP1, CDKN2A, SLC30A8, SLC30A10, GCK, SSTR2, FFAR1, YY1, LEP, DPP4, INSM1, MNX1, HSPD1, GAD2, SLC2A2, CASR, RALBP1, RIPK1, PDHX, BTC, UQCRFS1, TP53, TGM2, SSTR5, CDKN1C, INSR, ABCC8, SLC6A2, SSTR4, SSTR3, WFS1, NIT1, SERPINA1, PTPRN, GIP, GCKR, CORO1A, H3P47, PRL, H3P10, ERBB2, GAST, EGR1, ELK3, CALCA, CASP3, EPHB1, G6PC, DLK1, CCN5, SQSTM1, PTTG1, GCM2, LHX2, KL, MAPK8IP1, INSL5, IRS2, ZNRD2, KHDRBS1, DCTN6, LILRB1, FASTK, CCND1, PDIA5, FAS, ATF6, KDM1A, PDZD2, BCL2, BRCA1, TNKS, PLA2G6, HNF1A, TCF19, TGFA, TGFB1, CASP8, THBD, TKT, TSPAN7, TPD52, TRP-AGG2-5, TRPC1, EIPR1, TXN, TYRP1, UCP2, VDR, CACNA1D, BRAF, STAB1, ERP44, NUP62, KCNH4, CAT, KCNH8, GPR119, STOML3, AKT1, HCAR2, GOLGA6A, TICAM2, HES3, MIR107, MIR144, MIR155, MIR204, MIR21, MIR375, INS-IGF2, ADSS2, TMED7-TICAM2, ECT, LINC02210-CRHR1, H3P23, ADM, SLC22A12, TXNDC5, TRABD, RCBTB1, FGF21, MCAT, MCTS1, TMED7, ADIPOR1, DCTN4, CDKAL1, SLC25A38, BANK1, MEG3, ZC3H12A, APOC2, SOX6, SELENOS, IGSF9, SEMA6A, HAMP, G6PC2, PDIA2, ANGPT2, SYP, STAT5A, STC1, STAT5B, KCNJ1, KCNJ6, KRT8, KRT16, KRT19, DECR1, LEPR, LGALS3, LMO2, EPCAM, SMAD2, SMAD3, SMAD4, MAPT, MC2R, MDK, RAB8A, CUX1, MET, CIITA, MLH1, EGF, EGFR, INPPL1, HK1, MTOR, FGF13, GNA12, GPD2, FBN1, GRN, GSK3B, GSR, GTF2H1, ESR2, ELK1, HLA-DQB1, HMGN2, HNF4A, EPHB2, IFI27, IGFBP1, IGFBP2, IL4, IL10, MRC1, NCAM1, NEDD4, SLC2A1, RAP1A, REG1A, CPE, CMA1, S100A8, SCT, CCL2, CXCL12, SDHD, CHGA, RAB3A, CDKN2D, SLC16A1, SNX1, CDC42, CDK1, CCND3, CCNC, CCK, STAT1, RANBP2, CR2, NF1, PIK3CG, NFE2L1, CTSB, NME1, OPA1, PAX4, PAX6, PCSK1, ENPP1, CTNNB1, PKD1, CRHR1, POLD1, MAPK1, MAPK3, MAPK8, ADCYAP1, PRSS1, PSEN2, PSMD9, PTEN, ACO2
- Sneddon Syndrome GARD
-
Mosquito Bites
Mayo Clinic
Avoid and exclude mosquitoes Limit exposure to mosquitoes by: Repairing any tears in the screens on windows, doors and camping gear Using mosquito netting over strollers and cribs Using mosquito netting when sleeping outdoors Selecting self-care products that don't have scents Use insect repellent Use insect repellent when mosquitoes are active. ... Some sporting goods stores sell clothing pretreated with permethrin. Don't wash bed nets or set them in sunlight, as this breaks down permethrin.
-
Clanging
Wikipedia
. ^ Spitzer, Manfred (1999). The mind within the net: Models of learning, thinking, and acting .
-
Chronic Hallucinatory Psychosis
Wikipedia
Others, again, might be swept into the widespread net of dementia praecox . This state of affairs cannot be regarded as satisfactory, for they are not truly cases of melancholia, paranoia, dementia praecox or any other described affection.
-
Esophageal Food Bolus Obstruction
Wikipedia
] and the use of large-bore tubes inserted into the esophagus to forcefully lavage it. [17] [ unreliable medical source? ] Endoscopic [ edit ] The Roth net can be inserted through the endoscope to remove pieces of the obstructed food. ... Traditional endoscopic techniques involved the use of an overtube, a plastic tube inserted into the esophagus prior to the removal of the food bolus, in order to reduce the risk of aspiration into the lungs at the time of endoscopy. [7] However, the "push technique", which involves insufflating air into the esophagus, and gently pushing the bolus toward the stomach instead, has emerged as a common and safe way of removing the obstruction. [7] [18] Other tools may be used to remove food boluses. The Roth Net is a mesh net that can be inserted through the endoscope, and opened and closed from the outside; it can be used to retrieve pieces of obstructed food.

-
Mosquito-Borne Disease
Wikipedia
Depending on the mosquito vector, and the affected community, a variety of prevention methods may be deployed at one time. Insecticidal nets and indoor residual spraying [ edit ] The use of insecticide treated mosquito nets (ITNs) are at the forefront of preventing mosquito bites that cause malaria. ... Because the Anopheles gambiae feeds indoors (endophagic) and rests indoors after feeding (endophilic), insecticide treated nets (ITNs) interrupt the mosquito's feeding pattern. The ITNs continue to offer protection, even after there are holes in the nets, because of their excito-repellency properties which reduce the number of mosquitoes that enter the home. ... Infected individuals should avoid mosquito exposure by staying indoors or using a mosquito net . [34] Dengue fever [ edit ] Dengue infection's therapeutic management is simple, cost effective and successful in saving lives by adequately performing timely institutionalized interventions. ... "The Impact of Pyrethroid Resistance on the Efficacy of Insecticide-Treated Bed Nets against African Anopheline Mosquitoes: Systematic Review and Meta-Analysis" .

-
Dogger Bank Itch
Wikipedia
In Dogger Bank itch, sensitivity is acquired after repeated handling of the sea chervils that become entangled in fishing nets. [ citation needed ] The specific toxin responsible for the rash was determined to be the sulfur -bearing salt (2-hydroxyethyl) dimethylsulfoxonium chloride. [3] This salt is also found in some sea sponges and has potent in vitro activity against leukemia cells. [4] Treatment [ edit ] A study of two cases in 2001 suggests that the rash responds to oral ciclosporin . ... The sea chervil, abundant in the area, frequently came up with the fishing nets and had to be thrown back into the water.
-
Uric Acid Concentration, Serum, Quantitative Trait Locus 1
OMIM
Evidence for both an increased rate of uric acid synthesis and an impaired net elimination of uric acid by the kidney has been advanced. ... Autosomal dominant form Lab - Increased rate of uric acid synthesis - Impaired net elimination of uric acid by the kidney - Hyperuricemia Skin - Urate tophi ▲ Close
-
West Nile Fever
Wikipedia
Prevention [ edit ] Low-cost, ceiling hung mosquito netting for a bed Many of the guidelines for preventing occupational West Nile virus exposure are common to all mosquito-borne diseases . [57] Public health measures include taking steps to reduce mosquito populations. ... DEET formulations as high as 30% are recommended for children over two months of age. [58] The CDC also recommends the use of: IR3535, oil of lemon eucalyptus, para-menthane-diol, or 2-undecanone. [59] Protect infants less than two months of age by using a carrier draped with mosquito netting with an elastic edge for a tight fit. ... Repellents containing permethrin ( e.g. , Permanone) or other insect repellents may be applied to clothing, shoes, tents, mosquito nets, and other gear. (Permethrin is not suitable for use directly on skin.) ... They are now widespread in the United States, and in Florida they have been found in all 67 counties. [60] In an at-risk area, staying in air-conditioned or well- screened room, or sleeping under an insecticide-treated bed net is recommended. Bed nets should be tucked under mattresses, and can be sprayed with a repellent if not already treated with an insecticide. [57] Monitoring and control [ edit ] West Nile virus can be sampled from the environment by the pooling of trapped mosquitoes via ovitraps , carbon dioxide -baited light traps, and gravid traps, testing blood samples drawn from wild birds, dogs, and sentinel monkeys, and testing brains of dead birds found by various animal control agencies and the public. ... Retrieved 28 October 2017 . ^ Gompf, Sandra. "West Nile Virus" . Medicine Net . MedicineNet Inc . Retrieved 15 January 2019 . ^ "Symptoms, Diagnosis, & Treatment" .CCR5, ERVK-32, ROBO3, MAVS, DDX58, PLAAT4, IFIT2, ERVK-6, STAT1, SPP1, OAS1, IL1B, IFNB1, RNASEL, CASP8, HLA-DRB1, PELI1, SELENBP1, ARHGEF2, LRRFIP1, NAMPT, TRAIP, RIPK3, SEC14L2, CSF1R, LAMP3, ERVW-1, FOXP3, ZMYND10, DDX56, CCR7, VCP, CDKN2A, IFIH1, DHX58, ZBP1, HAVCR2, PIK3IP1, NLRP3, TNFRSF13C, TRIM6, RBM45, CCR2, ERVK-20, ERVK-18, VAMP8, TNFRSF1A, IFNA1, TNF, IFNA13, HLA-DQA1, IL1A, HLA-C, IL10, IL17A, IL18, IRF3, IRF5, KIR2DL2, KIR3DL1, KIR3DS1, LSAMP, CD180, SMAD4, MMP9, HLA-A, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PZP, GLS, CASP1, SNCA, GEM, DDX3X, TAP1, TLR3, ATF4









