Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Pyruvate Kinase Deficiency Of Red Cells
OMIM
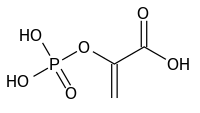
Biochemical Features Koler et al. (1964) found that patients with PK deficiency in red blood cells had normal PK activity in white blood cells, suggesting that the 2 enzymes were encoded by different loci. ... Bigley and Koler (1968) found decreased liver PK activity in a patient with hemolytic anemia due to red cell PK deficiency. ... Shinohara et al. (1976) described a new pyruvate kinase variant, PK Osaka, and discussed the various PK isozymes and their nomenclature. The variant was ascertained through a patient with PK deficiency hemolytic anemia. A genetic compound for 2 different PK mutations was studied by Zanella et al. (1978). ... The variants were called PK Sendai and PK Shinshu. Valentine et al. (1988) characterized the PK Greensboro variant.
-
Pallister-Killian Syndrome
OMIM
A number sign (#) is used with this entry because Pallister-Killian syndrome (PKS) is a dysmorphic condition caused by mosaicism for tetrasomy of chromosome 12p. ... Zakowski et al. (1992) described absence of the pericardium and focal aplasia cutis in the axillary area in PKS. Mauceri et al. (2000) reported a 15-year-old girl with Pallister-Killian syndrome and pineal tumor. ... Yeung et al. (2009) reported a girl with PKS who was referred at age 7 months for developmental delay and dysmorphic features. ... Pallister-Killian Syndrome Due to Hexasomy of Chromosome 12p Vogel et al. (2009) reported a 5-year-old girl with PKS resulting from mosaicism for 2 supernumerary isochromosomes, or hexasomy 12p. ... Vogel et al. (2009) suggested that phenotypic variation in PKS is most likely a result of which tissue types carry the mosaic cell line more than the percentage of mosaic cells or gene-dosage effects.
-
Adenosine Triphosphate, Elevated, Of Erythrocytes
OMIM
They showed low 2,3-diphosphoglycerate (2,3-DPG) and high adenosine triphosphate (ATP) levels. The PK electrophoretic patterns in these persons were abnormal by the presence of several additional bands. ... Inheritance - Autosomal dominant Lab - High erythrocyte adenosine triphosphate - Pyruvate kinase hyperactivity - Low 2,3-diphosphoglycerate (2,3-DPG) - Additional PK electrophoretic bands Heme - Polycythemia ▲ Close
-
Injection Fibrosis
Wikipedia
Orthopedic surgery is the typical treatment. [1] See also [ edit ] Fibrosis References [ edit ] ^ Mukherjee PK, Das AK (1980). "Injection fibrosis in the quadriceps femoris muscle in children".
-
Posterior Amorphous Corneal Dystrophy
Orphanet
Visual acuity is usually only minimally affected but in some more severe cases, penetrating keratoplasty (PK) may be warranted. Unlike other corneal dystrophies, non-corneal manifestations have been observed and include abnormalities of the iris (iridocorneal adhesions, corectopia, and pseudopolycoria).
-
Gelatinous Drop-Like Corneal Dystrophy
Orphanet
Management and treatment An unsatisfactory response has been observed to both lamellar keratoplasty (LKP) and penetrating keratoplasty (PK), as well as to a superficial keratectomy, since amyloid recurs in the graft within about 5 years.
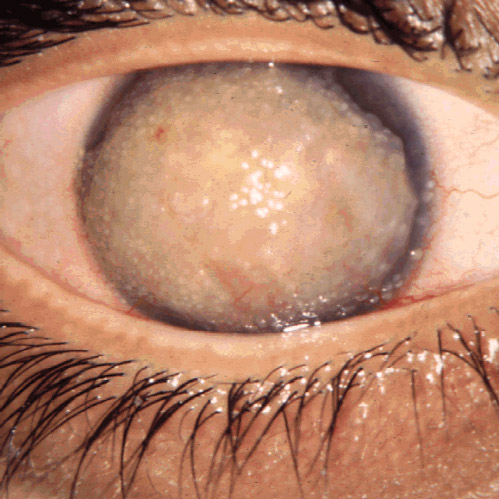
- Lattice Corneal Dystrophy Type I Orphanet
-
Reis-Bücklers Corneal Dystrophy
Orphanet
Management and treatment In advanced cases of RBCD, a superficial keratectomy, phototherapeutic keratectomy (PTK) or lamellar keratoplasty (LKP) may improve vision, but a penetrating keratoplasty (PK) is rarely necessary because the pathologic changes only involve the superficial cornea.

-
Glycogen Storage Disease Ixb
OMIM
Clinical Features In an Israeli Arab family reported by Bashan et al. (1981), a 4-year-old brother and 2 sisters had marked hepatomegaly and marked accumulation of glycogen in both liver and muscle, without clinical symptoms. Liver phosphorylase kinase (PK) activity was 20% of normal, resulting in undetectable activity of phosphorylase a. Muscle PK was about 25% of normal, resulting in a marked decrease of phosphorylase a activity.
-
Fryns Syndrome
GeneReviews
Chromosome Anomalies Associated With Congenital Diaphragmatic Hernia and Additional Major Malformations/Dysmorphology View in own window Chromosome Abnormality Critical Genes Included Facial Phenotype Other Clinical Characteristics (in addition to CDH) Pallister-Killian syndrome (PKS) 1 (mosaic tetrasomy 12p) (OMIM 601803) Coarse w/wide-set eyes, prominent cheeks, & eversion of vermilion of lower lip Considered similar to Fryns syndrome 2, 3 Sparse hair, 4 syndactyly, & streaky skin pigmentation Hypotonia, seizures, & ID common 3 Short distal phalanges of fingers & toes, small nails, cloudy corneas, CHD, & internal malformations may be seen but are typically less frequent in PKS than in Fryns syndrome. ... In some persons, only chromosome analysis and/or the inheritance pattern can distinguish between PKS and Fryns syndrome [Veldman et al 2002]. To evaluate for PKS, skin fibroblasts, chorionic villus cells, or amniocytes should be karyotyped because of the phenomenon of tissue-specific mosaicism in which the isochromosome 12p can be present in some cells (e.g., fibroblasts), but not others (e.g., lymphocytes). It is important to note that a normal karyotype or CMA on peripheral blood lymphocytes does not exclude PKS, although CMA may detect PKS when the percentage of tetrasomic cells is relatively high. 3. Izumi & Krantz [2014] 4. Sparse hair is characteristic of PKS, in contrast to Fryns syndrome, in which the sisters originally described by Fryns had low hairlines and hypertrichosis. 5.

-
Samoyed Hereditary Glomerulopathy
Wikipedia
. ^ Jansen, B; Tryphonas L; Wong J; Thorner P; Maxie MG; Valli VE; Baumal R; Basrur PK. (June 1986). "Mode of inheritance of Samoyed hereditary glomerulopathy: an animal model for hereditary nephritis in humans". ... PMID 3711721 . ^ Jansen, B; Tryphonas, L; Wong, J; Thorner, P; Maxie, MG; Valli, VE; Baumal, R; Basrur, PK (1986). "Mode of inheritance of Samoyed hereditary glomerulopathy: an animal model for hereditary nephritis in humans".

-
Progressive Transformation Of Germinal Centres
Wikipedia
PMID 12145465 . ^ Hansmann ML, Fellbaum C, Hui PK, Moubayed P (February 1990). "Progressive transformation of germinal centers with and without association to Hodgkin's disease".

-
Marfanoid–progeroid–lipodystrophy Syndrome
Wikipedia
. ^ a b c Romere C, Duerrschmid C, Bournat J, Constable P, Jain M, Xia F, Saha PK, Del Solar M, Zhu B, York B, Sarkar P, Rendon DA, Gaber MW, LeMaire SA, Coselli JS, Milewicz DM, Sutton VR, Butte NF, Moore DD, Chopra AR (2016). ... Despite consuming between 5,000 and 8,000 calories daily, the communications student, has never tipped over 4st 3lbs. ^ Duerrschmid C, He Y, Wang C, Li C, Bournat J, Romere C, Saha PK, Lee M, Phillips KJ, Jain M, Jia P, Zhao Z, Farias M, Wu Q, Milewicz DM, Sutton VR, Moore DM, Butte NF, Krashes MJ, Xu Y, and Chopra AR (2017) - "Asprosin Activates the Hypothalamic Hunger-Circuitry" - article under consideration for publication as of 8/30/2017.

-
Frontotemporal Dementia And Parkinsonism Linked To Chromosome 17
Wikipedia
References [ edit ] ^ Mitra K, Gangopadhaya PK, Das SK. (Jun 2003). "Parkinsonism plus syndrome--a review".

-
Cutaneous Actinomycosis
Wikipedia
PMID 19126014 . ^ Roy D, Roy PG, Misra PK (2003). "An interesting case of primary cutaneous actinomycosis" .
-
Macrotia
Wikipedia
Retrieved December 28, 2011 . ^ Brucker MJ, Patel J, Sullivan PK (August 2003). "A morphometric study of the external ear: age- and sex-related differences".ABL1, NIPBL, RAB18, IQSEC2, KDM6B, RHOBTB2, DENND5A, MED13L, SUZ12, FTSJ1, RAB3GAP2, POLR1A, MAN1B1, SIN3A, MMACHC, KIFBP, ZBTB20, PHGDH, BSCL2, TBL2, RBMX, PCLO, RAB3GAP1, CIT, AEBP1, PTDSS1, HERC1, AP3D1, BAZ1B, AIP, SMC3, FIBP, ZNHIT3, SNAP29, GTF2IRD1, SLC12A6, SRCAP, PQBP1, ALG3, ZMPSTE24, CWC27, APC2, SLC9A6, AGPAT2, YME1L1, WDR4, SETD2, ANKRD11, PSAT1, TP53RK, TTI2, SLC2A10, GPR101, FAR1, PHF6, WDR73, SHANK3, ALKBH8, ADAMTSL1, TOE1, TBX22, TBC1D20, MPLKIP, CEP120, DZIP1L, BRWD3, NALCN, KANSL1, NEXMIF, CTU2, PGAP1, NSD1, CXorf56, PIEZO2, TPRKB, MBTPS2, ACTL6B, OTUD6B, NDE1, PHIP, SETD5, OSGEP, PACS1, TENM3, HHAT, NUP133, TBC1D23, HDAC8, KMT2E, NUP107, THOC2, HECW2, PRUNE1, AP1S2, SYNGAP1, EED, KRAS, GRIA3, GRIA4, GRIN2B, GTF2I, HNRNPK, INSR, ITGA3, ITGB6, KCNJ1, LIMK1, OPHN1, LMNA, MAN2B1, MECP2, MGAT2, MGP, KMT2A, NAGLU, NBN, NDP, GLI2, FMR1, FLNA, FGFR2, JAG1, AHSG, AKT1, ASNS, RERE, ATP6V1A, KIF1A, BMP4, BRAF, CBL, CHRNA1, CHRNA7, CLIC2, DDX3X, DYRK1A, EIF2S3, ELN, EYA1, EZH2, NHS, PRDX1, MBTPS1, UBE2A, SSR4, CDKL5, ABCC8, TAF1, TAZ, TBCD, TRIO, TRPS1, UBA1, KDM6A, PAK3, CLIP2, ZNF711, SHOC2, KMT2D, KDM5C, SMC1A, NAA10, LAGE3, CASK, SLC16A2, SLC2A1, SIM1, SET, PAX1, PIK3R1, PKHD1, PMM2, PPP1CB, MAP2K1, MAP2K2, PSMB8, PTCH1, PTEN, NECTIN1, ALDH18A1, RAD21, RAP1A, RAP1B, DPF2, RFC2, RMRP, SALL1, SLC6A17
-
Hopkins Syndrome
Wikipedia
PMID 10826222 . ^ a b Acharya, AB; Lakhani, PK (1997). "Hopkins syndrome associated with Mycoplasma infection".
-
Myotonic Dystrophy 1
OMIM
Using antisera developed against synthetic DM-PK peptide antigens for biochemical and histochemical studies, van der Ven et al. (1993) found lower levels of immunoreactive DM-kinase protein of 53 kD in skeletal and cardiac muscle extracts of DM patients than in normal controls. Immunohistochemical staining revealed that DM-PK is localized predominantly at sites of neuromuscular and myotendinous junctions of human and rodent skeletal muscles. ... By quantitative RT-PCR and by radioimmunoassay using antisera developed against both synthetic peptides and purified myotonin-protein kinase (Mt-PK) protein expressed in E. coli, Fu et al. (1993) demonstrated that decreased levels of the mRNA and protein expression are associated with the adult form of myotonic dystrophy. From this they suggested that the autosomal dominant nature of the disease is due to an Mt-PK dosage deficiency and that means of elevating Mt-PK level or activity should be explored for therapeutic intervention in adult patients.DMPK, MBNL1, CELF1, IFIH1, ERBB2, INSR, MBNL2, SIX5, ALB, IL6, SLC2A1, CCR5, NKX2-5, DMD, IL10, ERBB3, ESR1, TNNI3, LDB3, IGF1, IFNA13, IFNA1, CLCN1, CNBP, TNF, APOC2, MIR206, PTH, CAT, ADIPOQ, TRIM24, TAC3, KLF4, SOCS5, TRIM21, TNFRSF10A, SLPI, SLC22A1, MVP, SLC6A3, TRBV20OR9-2, TNFRSF25, VEGFA, TERT, TG, TGFB1, TGM2, TH, BECN1, FZD3, TIMP2, FZD5, CXCR4, DGCR2, TIMP3, TJP1, XPO1, HNRNPDL, ACTB, ABCB6, LILRB2, SIRT6, TRIM33, MPC1, ASB2, RBFOX1, NAT10, MYH14, ASRGL1, RSPH6A, PRRT2, RMI2, TADA1, TMEM119, MIR126, MIR130A, MIR27B, MIR29C, TLR7, CD274, REM1, SPEN, CCT2, CTCF, NEK6, CCL27, CCL21, MORF4L1, PUF60, ZNF609, PDLIM3, NBEAL2, SYNE1, SIRT1, CABIN1, DIANPH, ATRNL1, FGF21, LILRB1, PRKCA, CCL2, DDX6, CHI3L1, CKM, CRP, CST3, CTLA4, CTSD, ACE, FN1, CEBPA, GAD2, MSTN, GEM, GH1, GSK3B, HAS2, HCRT, CEBPB, CD19, HGF, APOE, AGER, AIC, ALPP, AMH, ANGPT2, APOA1, APOB, ATHS, CAV1, ATP2A1, ATP2A2, AVP, BRCA1, BUB1, CAD, CASP3, HFE, HLA-B, SCN5A, PRKAA2, MYC, NPPB, ROR1, TNFRSF11B, OXA1L, POU2F1, PRKAA1, PRKAB1, MSH3, ADA, PRKCB, PSMB8, PTCH1, MOK, BRD2, RYR1, MUC1, MMP9, MNX1, HSPD1, HLA-DQA1, HLA-DQB1, HLA-DRB4, HNRNPC, HNRNPH1, HNRNPH2, HP, IFNG, MMP2, IGFBP5, IL4, IL17A, KCNQ1, KIR3DL1, SMAD3, MAL, ZASP
-
Human Genetic Resistance To Malaria
Wikipedia
Contents 1 Development of genetic resistance to malaria 2 Innate resistance 2.1 Mechanisms of protection 3 Types of innate resistance 3.1 Hemoglobin abnormalities 3.1.1 Distribution of abnormal hemoglobins 3.1.2 Sickle-cell 3.1.3 Thalassemias 3.1.4 HbC and HbE erythroids 3.2 Other erythrocyte mutations 3.2.1 Glucose-6-phosphate dehydrogenase deficiency 3.2.2 PK deficiency 3.2.3 Elliptocytosis 3.2.4 Southeast Asian ovalocytosis 3.2.5 Duffy antigen receptor negativity 3.2.6 Gerbich antigen receptor negativity 3.2.7 Other rare erythrocyte mutations 3.3 Human leucocyte antigen polymorphisms 3.4 Hereditary persistence of fetal hemoglobin 4 Validating the malaria hypothesis 4.1 Fitnesses of different genotypes 5 See also 6 Notes 7 Glossary 8 References 9 Further reading 10 External links Development of genetic resistance to malaria [ edit ] Microscopic parasites , like viruses, protozoans that cause malaria, and others, cannot replicate on their own and rely on a host to continue their life cycles. ... Malaria parasites were significantly more often observed in normal red cells than in enzyme-deficient cells. [43] An evolutionary genetic analysis of malarial selection of G6PD deficiency genes has been published by Tishkoff and Verelli. [40] The enzyme deficiency is common in many countries that are, or were formerly, malarious, but not elsewhere. PK deficiency [ edit ] Main article: PK deficiency See also: Pyruvate kinase Pyruvate kinase (PK) deficiency, also called erythrocyte pyruvate kinase deficiency, is an inherited metabolic disorder of the enzyme pyruvate kinase. ... There is a significant correlation between severity of PK deficiency and extent of protection against malaria. [44] Elliptocytosis [ edit ] Main article: Elliptocytosis Elliptocytosis a blood disorder in which an abnormally large number of the patient's erythrocytes are elliptical.
- Xanthelasma Wikipedia







