-
Adult-Onset Still's Disease
Wikipedia
Medications that block the action of interleukin-1 , such as Anakinra , can be effective treatments when standard steroid treatments are insufficient. [3] Contents 1 Signs and symptoms 2 Pathophysiology 3 Diagnosis 3.1 Classification 4 Treatment 5 Epidemiology 6 History 7 Research directions 8 See also 9 References 10 External links Signs and symptoms [ edit ] The disease typically presents with joint pain , high fevers, a salmon-pink macular or maculopapular rash, enlargement of the liver and spleen , swollen lymph nodes , and a neutrophil-predominant increased white blood cell count in the blood. [1] Tests for rheumatoid factor and anti-nuclear antibodies are usually negative and serum ferritin is markedly elevated. ... Interleukin-18 is expressed at high levels. [2] [7] [8] Diagnosis [ edit ] The diagnosis is clinical, not based upon serology . [9] At least seven sets of diagnostic criteria have been devised, however the Yamaguchi criteria have the highest sensitivity. ... "Adult-onset Still's disease" . Autoimmunity Reviews . 13 (7): 708–722. doi : 10.1016/j.autrev.2014.01.058 . ... Expert Review of Clinical Immunology . 14 (5): 351–365. doi : 10.1080/1744666X.2018.1465821 . ... "Association between adult-onset Still's disease and interleukin-18 gene polymorphisms" . Genes and Immunity . 3 (7): 394–9. doi : 10.1038/sj.gene.6363922 .IL1B, IL18, TNF, IL1A, IL6, MEFV, IFNG, RBM45, NLRP3, HLA-DRB1, CXCL10, IL18BP, IL10, SMUG1, HMOX1, MIF, MAP4K3, GCK, TNFRSF1A, CRP, CD68, CASP1, CXCR4, SQSTM1, TP53, FCGR2C, TLR4, STAT3, AIM2, KHDRBS1, ATG5, RTN3, CXCL12, NUP62, IL37, DCTN4, PLAC8, IL23A, NOD2, CLEC7A, IL33, NLRP12, MIR134, MIR141, SELL, ACR, CCL2, SAFB, FAS, FASLG, BAG1, CLU, FCGR1A, FCGR2A, FCGR2B, GNAO1, CXCR3, GTF2H1, HLA-DPB1, HLA-DQB1, HLA-DQB2, ICAM1, IFI16, CCN1, IL1RN, IL4, CXCL8, IL15, IL17A, ITGAM, ALB, MPO, RARB, SAA1, SAA2, MIR29A
-
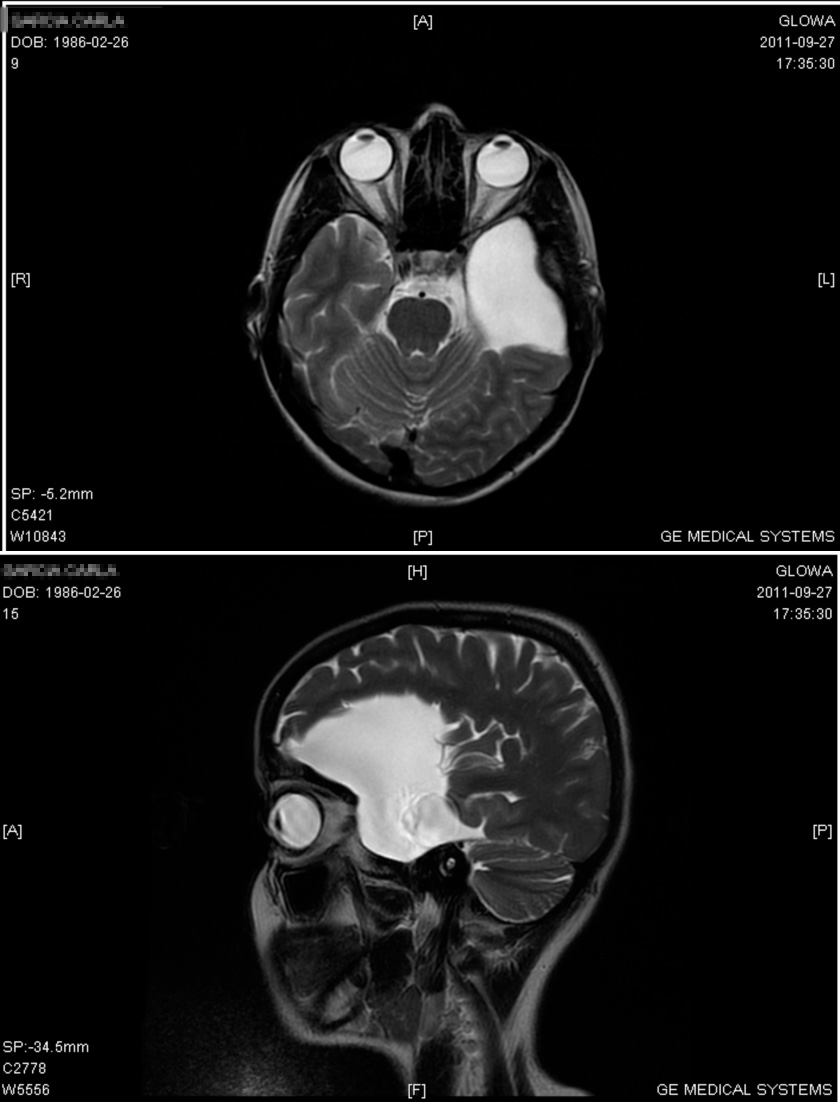
Porencephaly
Wikipedia
Cysts can develop in the frontal lobe , parietal lobe , forebrain , hindbrain , temporal lobe , or virtually anywhere in the cerebral hemisphere . [7] Genetics [ edit ] From recent studies, de novo and inherited mutations in the gene COL4A1 , suggesting genetic predisposition within the family, that encodes type IV collagen α1 chain has shown to be associated with and present in patients with porencephaly. ... In porencephaly patients, patients achieved good seizure control with appropriate drug therapy including valproate , carbamazepine , and clobazam . [6] [7] Also, anti-epileptic drugs served as another positive method of treatment. [7] Prognosis [ edit ] The severity of the symptoms associated with porencephaly varies significantly across the population of those affected, depending on the location of the cyst and damage of the brain. ... "The Differences in Epileptic Characteristics in Patients with Porencephaly and Schizencephaly". Brain Dev . 34 (7): 546–552. doi : 10.1016/j.braindev.2011.10.001 . ... J Matern Fetal Neonatal Med . 18 (6): 361–365. doi : 10.1080/14767050400029574 . ... "Genetic Mutation Predisposes to Porencephaly". Lancet Neurology . 4 (7): 400. doi : 10.1016/s1474-4422(05)70114-9 .

-
Video Game–related Health Problems
Wikipedia
"Video game epilepsy in the twentieth century: a review". Childs Nerv Syst . 23 (3): 265–7. doi : 10.1007/s00381-006-0285-2 . ... "The influence of violent media on children and adolescents:a public-health approach" (PDF) . The Lancet . 365 (9460): 702–10. doi : 10.1016/S0140-6736(05)17952-5 . ... Pediatr . 165 (6): 408–14. doi : 10.1007/s00431-005-0018-7 . PMID 16552547 . S2CID 25399018 . ^ Vaidya HJ (March 2004). ... "Overweight and obesity related to activities in Portuguese children, 7-9 years" . Eur J Public Health . 17 (1): 42–6. doi : 10.1093/eurpub/ckl093 . ... "Active video games to promote physical activity in children and youth: a systematic review" . Arch Pediatr Adolesc Med . 164 (7): 664–72. doi : 10.1001/archpediatrics.2010.104 .
-
Epstein–barr Virus Infection
Wikipedia
Current Topics in Microbiology and Immunology . 390 . pp. 365–85. doi : 10.1007/978-3-319-22822-8_15 . ... "Post-infectious acute cerebellar ataxia in children". Clinical Pediatrics . 42 (7): 581–4. doi : 10.1177/000992280304200702 . ... "Epstein–Barr virus-associated acute renal failure: diagnosis, treatment, and follow-up". Pediatric Nephrology . 18 (7): 667–674. doi : 10.1007/s00467-003-1152-y . ... Current Neurology and Neuroscience Reports . 7 (3): 253–8. doi : 10.1007/s11910-007-0038-y . ... PMID 12409644 . ^ "Nasopharyngeal Cancer" . HealthCommunities.com. 7 December 2011. ^ "Epstein-barr | Mononucleosis | About Virus | Mono | CDC" . www.cdc.gov . 2019-01-28 .
-
Progressive Vaccinia
Wikipedia
Although some vaccinia viruses commonly disseminate through the bloodstream, the NYCBOH strain reportedly causes only limited viremia in a small percentage of recipients during the period of pustule formation. [6] [7] The inflammatory process reaches its peak by days 10–12 after vaccination and begins to resolve by day 14, with the shedding of the scab and other pustules by day 21. ... Maryland Heights, Missouri, United States: jacionline.org. 110 (3): 357–365. doi : 10.1067/mai.2002.128052 .

-
Neuromuscular Junction Disease
Wikipedia
The mechanism currently known that operates via the synaptic cleft causing impairment of normal functioning is another congenital myasthenia gravis.(reference 7) This mechanism is the only currently known disease that acts on the synapse. ... In other words, it is the most susceptible to negative intervention.(reference 7) The targets of these postsynaptic diseases can be multiple different proteins. ... (reference 14) Acquired myasthenia gravis is the most common neuromuscular junction disease.(reference 7) Important observations were made by Patrick and Lindstrom in 1973 when they found that antibodies attacking the acetylcholine receptors were present in around 85% of cases of myasthenia gravis. ... "Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies". Nat Med . 7 (3): 365–8. doi : 10.1038/85520 . ... "Myasthenia gravis and Lambert-Eaton myasthenic syndrome in the same patient". Muscle Nerve . 36 (1): 115–7. doi : 10.1002/mus.20735 . PMID 17206662 .
-
Generalized Pustular Psoriasis
Wikipedia
Contents 1 Signs and symptoms 2 Causes 3 Genetic factors 4 Diagnosis 4.1 Classification 4.1.1 von Zumbusch acute generalized pustular psoriasis 4.1.2 Generalized pustular psoriasis of pregnancy (Impetigo herpetiformis) 4.1.2.1 Infantile and juvenile 4.1.3 Circinate and annular 5 Treatments 6 Prognosis 7 Case reports 7.1 Case report 1 7.2 Case report 2 7.3 Case report 3 7.4 Case report 4 7.5 Case report 5 8 See also 9 References 10 External links Signs and symptoms [ edit ] GPP presents as pustules and plaques over a wide area of the body. ... Provocative Factors Influencing Pustular Psoriasis Drugs: lithium , aspirin , salicylates , methotrexate , corticosteroids , progesterone , phenylbutazone , trazodone , penicillin , hydrochloroquine Irritation from topical therapy: coal tar , anthralin Infections: dental, upper respiratory Pregnancy Solar irradiation Source: "Table II", "Pustular Psoriasis" Farber and Nall, 1993 [5] Genetic factors [ edit ] Although there are likely to be multiple genetic factors and environmental triggers, mutations causing defects in the IL-36RN , CARD14 and AP1S3 genes have been shown to cause GPP. [6] [7] [8] Diagnosis [ edit ] Classification [ edit ] It is important to note that while there are different forms of GPP, they are not exclusive of each other. ... "Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis". N. Engl. J. Med . 365 (7): 620–8. doi : 10.1056/NEJMoa1013068 . ... Journal of Dermatological Treatment . 19 (3): 185–7. doi : 10.1080/09546630701759587 . ... Journal of the American Academy of Dermatology . 25 (2 Pt 1): 336–7. doi : 10.1016/s0190-9622(08)80478-1 .
-
Opsoclonus Myoclonus Syndrome
Wikipedia
It affects 2 to 3% of children with neuroblastoma and has been reported to occur with celiac disease and diseases of neurologic and autonomic dysfunction. [2] [3] Contents 1 Signs and symptoms 1.1 Disease course and clinical subtypes 2 Cause 3 Diagnosis 4 Treatment 5 Prognosis 6 Research 7 Nomenclature 8 References 9 Further reading 10 External links Signs and symptoms [ edit ] Symptoms include: [ citation needed ] opsoclonus (rapid, involuntary, multivectorial (horizontal and vertical), unpredictable, conjugate fast eye movements without inter saccadic [quick rotation of the eyes] intervals) myoclonus (brief, involuntary twitching of a muscle or a group of muscles) cerebellar ataxia , both truncal and appendicular aphasia (a language disorder in which there is an impairment of speech and of comprehension of speech, caused by brain damage) mutism (a language disorder in which a person does not speak despite evidence of speech ability in the past, often part of a larger neurological or psychiatric disorder) lethargy irritability or malaise drooling strabismus (a condition in which the eyes are not properly aligned with each other) vomiting sleep disturbances emotional disturbances (including fits of rage [4] ) About half of all OMS cases occur in association with neuroblastoma (a cancer of the sympathetic nervous system usually occurring in infants and children). [ citation needed ] Disease course and clinical subtypes [ edit ] In most cases OMS starts with an acute flare-up of physical symptoms within days or weeks, but some less obvious symptoms such as irritability and malaise may begin weeks or months earlier. [ citation needed ] Cause [ edit ] In children, most cases are associated with neuroblastoma and most of the others are suspected to be associated with a low-grade neuroblastoma that spontaneously regressed before detection. ... Louis encephalitis , Chikungunya , Epstein-Barr , Coxsackie B , enterovirus , or just a flu) causes the remaining cases, though a direct connection has not been proven, [6] or in some cases Lyme disease . [7] OMS is not generally considered an infectious disease . ... "Longitudinal neurodevelopmental evaluation of children with opsoclonus-ataxia". Pediatrics . 116 (4): 901–7. doi : 10.1542/peds.2004-2377 . PMID 16199699 . ... "Delayed, recurrent opsoclonus-myoclonus syndrome responding to plasmapheresis". Pediatr. Neurol . 33 (5): 365–7. doi : 10.1016/j.pediatrneurol.2005.05.018 . ... "The association between neuroblastoma and opsoclonus-myoclonus syndrome: a historical review". Pediatr Radiol . 39 (7): 723–6. doi : 10.1007/s00247-009-1282-x .
-
Escherichia Coli O104:h4
Wikipedia
Analysis of genomic sequences obtained by BGI Shenzhen shows that the O104:H4 outbreak strain is an enteroaggregative E. coli (EAEC or EAggEC) type that has acquired Shiga toxin genes, presumably by horizontal gene transfer . [2] [3] [4] Genome assembly and copy-number analysis both confirmed that two copies of the Shiga toxin stx2 prophage gene cluster are a distinctive characteristic of the genome of the O104:H4 outbreak strain. [5] [6] The O104:H4 strain is characterized by these genetic markers: [6] [7] Shiga toxin stx2 positive tellurite resistance gene cluster positive intimin adherence gene negative β-lactamases ampC, ampD, ampE, ampG, ampH are present. ... "Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology" . PLoS One . 6 (7): e22751. doi : 10.1371/journal.pone.0022751 . ... "Epidemic profile of Shiga-toxin–producing Escherichia coli O104:H4 outbreak in Germany". New England Journal of Medicine . 365 (19): 1771–1780. doi : 10.1056/NEJMoa1106483 . ... Science News, Articles and Information | Scientific American. Scientific American, 7 Aug. 2011. Web. 08 Nov. 2011. < http://www.scientificamerican.com/article.cfm?
-
Hereditary Pancreatitis
Wikipedia
The term "hereditary pancreatitis" is used when a genetic biomarker is identified, and "familial pancreatitis" otherwise. [3] Contents 1 Presentation 2 Genetics 3 Diagnosis 4 Management 5 Prognosis 6 References 7 External links Presentation [ edit ] HP is characterised by attacks of epigastric pain , which are often associated with nausea and vomiting. ... Lifetime risk of cancer has been variously calculated as 35–54% [4] [5] [6] to the age of 75 years and screening for early pancreatic cancer is being offered to HP sufferers on a scientific basis. [7] Some patients may choose to have their pancreas surgically removed to prevent pancreatic cancer from developing in the future. [8] The epidemiology of HP follows a similar pattern to alcohol-associated chronic pancreatitis, but there are important differences. For example, HP typically has an earlier age of pancreatitis onset; although malabsorption and diabetes mellitus occur at a later stage in the disease progression. [5] Genetics [ edit ] The vast majority of the cases of HP are caused by substitutions, at base 365 (c.365G>A) and base 86 of the cDNA (c.86A>T) on the PRSS1 gene. ... "A signal peptide cleavage site mutation in the cationic trypsinogen gene is strongly associated with chronic pancreatitis". Gastroenterology . 117 (1): 7–10. doi : 10.1016/s0016-5085(99)70543-3 .SPINK1, PRSS1, CTRC, CFTR, PRSS2, CPA1, CASR, HP, STAT6, OR10A4, NAB2, MSR1, ELAC2, TP53, PCAP, RNASEL, GAST, CBX4, NGFR, KLK3, CD274, LGR5, NPEPPS, TNF, PIK3CA, IL10, PROS1, CTSB, CRP, PIK3CB, PSAT1, PRSS3, PLAG1, BRCA2, PIK3CG, CDKN2A, COX2, PIK3CD, VEGFA, UBB, TSC2, SERPINA1, TGFB1, PTGS2, TLR4, SYP, SRD5A2, S100A8, SOX9, S100A9, MAPK3, ABL2, CLDN1, CRISPLD2, PINX1, SPHK2, SMURF1, EHMT1, CAMKMT, APOL3, PANX3, ADIPOQ, PRSS58, MIR204, HPC3, ZGLP1, MIR4270, MTCO2P12, PGPEP1, DLL4, FOXP3, MBL3P, BHLHE22, AMACR, HEY1, CELA3B, LZTS1, NCOA2, HOXB13, DLC1, HDAC6, MAGEC1, PECAM1, HDAC9, HPCX, PGR, MUC5AC, PDCD1, DSPP, CDKN1C, CP, CPB2, CLDN7, CYP1B1, CYP17A1, EEF1B2P2, CDK2, EPHB4, ERBB2, ERCC1, F13A1, FAP, FCGR2B, CDKN1B, CDH1, GCG, BDNF, APC, BIRC2, APP, RERE, CCND1, BCL2, BGN, CD40, BRAF, CALM1, CALM2, CALM3, CCKBR, KRIT1, FOXD1, GDNF, PCNA, MLH1, MAP2, MBL2, MGMT, MGST1, CD99, MIF, MMP9, LCN2, MPO, MUC1, ABO, MUC6, NGF, NOTCH1, EPCAM, KRT19, GLP1R, HDGF, GPI, GPT, GSTM1, GSTM3, GSTT1, HDAC1, HIF1A, IRF1, HLA-DQA1, HLA-DQB1, IL1A, IL1B, IL4, CXCL8, H3P10
-
Red Eye (Medicine)
Wikipedia
It may be more common in occupations such as farming and welding. inflamed pinguecula [7] – a yellow-white deposit close to the junction between the cornea and sclera, on the conjunctiva. ... "The science of pterygia". Br J Ophthalmol . 94 (7): 815–20. doi : 10.1136/bjo.2008.151852 . ... American Academy Ophthalmology. p. 365. ISBN 1-56055-814-8 . ^ "Keratoconjunctivitis, Sicca" . eMedicine .

-
Hemolytic–uremic Syndrome
Wikipedia
"New insights into postrenal transplant hemolytic uremic syndrome". Nature Reviews Nephrology . 7 (1): 23–35. doi : 10.1038/nrneph.2010.155 . ... "Hemolytic Uremic Syndrome: pathogenesis and update of interventions". Expert Rev Anti Infect Ther . 7 (6): 697–707. doi : 10.1586/eri.09.49 . ... New York, NY: McGraw-Hill. ISBN 978-0-07-138875-7 . ^ Rivero, MA; Passucci, JA; Rodriguez, EM; Signorini, ML; Tarabla, HD; Parma, AE (2011). ... "Hemolytic–uremic syndrome". Pediatr Rev . 22 (11): 365–9. doi : 10.1542/pir.22-11-365 . ... "German outbreak of Escherichia coli O104:H4 associated with sprouts". N Engl J Med . 365 (19): 1763–1770. doi : 10.1056/NEJMoa1106482 .CD46, DGKE, C3, CFB, CFH, IL6, IL1B, TNF, NQO1, ALB, IL1A, EPO, CCL2, ADAMTS13, CFI, CFHR1, THBD, CFHR3, MMACHC, PRDX1, STX1A, TRIM25, STX2, VWF, CAPG, RNR2, ST8SIA2, IL10, PCSK5, PLG, SH3BP4, MTHFR, CXCR4, F3, MCHR2, CXCL12, ZFP36, TLR4, CCR2, LIPG, PLA2G7, ST11, MIR126, GRHPR, WT1, AIMP2, ACKR3, GADL1, SUMF2, A4GALT, CLEC1B, DBR1, DESI1, HPLH1, CFHR5, POLDIP2, RNF19A, IL33, GRAP2, CADM1, NT5C2, AZIN2, SLC35G1, IL24, RAB7B, NLE1, AHSA1, ABO, SAA2, TP53, FGF2, EDN1, SLC25A10, MAPK14, CRK, CPB2, CLU, CDA, CD40LG, CD22, CAT, CASP1, VPS51, C4BPA, C3AR1, APCS, ANGPT2, ANGPT1, FABP1, FHL1, TM7SF2, HLA-DRB1, TGFB1, SPARC, CX3CL1, CCL20, CCL4, AGT, SAA1, REN, MAPK1, COX1, MATK, IL18, CXCL8, IL4R, IL2RA, IFNB1, HMGB1, RN7SL263P

-
Bruce Effect
Wikipedia
"Pregnancy-Block in the Meadow Vole, Microtus Pennsylvanicus" . Reproduction . 24 (2): 275–7. doi : 10.1530/jrf.0.0240275 . PMID 5551417 . ^ MALLORY, F. ... Hormones and Behavior . 55 (1): 240–7. doi : 10.1016/j.yhbeh.2008.10.013 . ... Pennsylvanicus and Peromyscus maniculatus" . Reproduction . 49 (2): 365–7. doi : 10.1530/jrf.0.0490365 . ... Behavioral Ecology and Sociobiology . 52 (1): 31–7. doi : 10.1007/s00265-002-0484-0 . ... "Laboratory Studies with Rodents: Facts or Artifacts?" . BioScience . 53 (4): 421–7. doi : 10.1641/0006-3568(2003)053[0421:LSWRFO]2.0.CO;2 .
-
Large Granular Lymphocytic Leukemia
Wikipedia
As the name suggests, T-cell large granular lymphocyte leukemia is characterized by involvement of cytotoxic- T cells ). [2] In a study based in the US, the average age of diagnosis was 66.5 years [3] whereas in a French study the median age at diagnosis was 59 years (with an age range of 12-87 years old). [4] In the French study, only 26% of patients were younger than 50 years which suggests that this disorder is associated with older age at diagnosis. [4] Due to lack of presenting symptoms, the disorder is likely to be underdiagnosed in the general population. [5] Contents 1 Signs and symptoms 1.1 Sites of involvement 2 Cause 3 Diagnosis 3.1 Laboratory findings 3.2 Peripheral blood 3.3 Bone marrow 3.4 Immunophenotype 3.5 Genetic findings 4 Treatment 5 Prognosis 6 Epidemiology 7 History 8 References 9 External links Signs and symptoms [ edit ] This disease is known for an indolent clinical course and incidental discovery. [1] The most common physical finding is moderate splenomegaly . B symptoms are seen in a third of cases, and recurrent infections due to anaemia and/or neutropenia [6] are seen in almost half of cases. [7] [8] [9] [10] Rheumatoid arthritis is commonly observed in people with T-LGLL, leading to a clinical presentation similar to Felty's syndrome . [11] Signs and symptoms of anemia are commonly found, due to the association between T-LGLL and erythroid hypoplasia . [12] Sites of involvement [ edit ] The leukemic cells of T-LGLL can be found in peripheral blood , bone marrow , spleen , and liver . Nodal involvement is rare. [1] [7] Cause [ edit ] The postulated cells of origin of T-LGLL leukemia are transformed CD8+ T-cell with clonal rearrangements of β chain T-cell receptor genes for the majority of cases and a CD8- T-cell with clonal rearrangements of γ chain T-cell receptor genes for a minority of cases. [1] Diagnosis [ edit ] Laboratory findings [ edit ] The requisite lymphocytosis of this disease is typically 2-20x10 9 /L. [12] Immunoglobulin derangements including hypergammaglobulinemia, autoantibodies, and circulating immune complexes are commonly seen. [10] [13] [14] [15] Peripheral blood [ edit ] The neoplastic lymphocytes seen in this disease are large in size with azurophilic granules that contains proteins involved in cell lysis such as perforin and granzyme B . [16] Flow cytometry is also commonly used. [17] Bone marrow [ edit ] Bone marrow involvement in this disease is often present, but to a variable extent. ... Acta Clinica Croatica . 57 (2): 362–365. doi : 10.20471/acc.2018.57.02.18 . ... "Laboratory findings and clinical courses of 33 patients with granular lymphocyte-proliferative disorders". Leukemia . 7 (6): 782–8. PMID 8388971 . ^ a b Loughran TP, Starkebaum G, Aprile JA (March 1988).
-
Primary Hyperparathyroidism
Wikipedia
Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Treatment 4.1 Surgery 4.2 Medications 5 Epidemiology 6 Children 7 Research directions 8 See also 9 References 10 External links Signs and symptoms [ edit ] The signs and symptoms of primary hyperparathyroidism are those of hypercalcemia. ... Recently, it was demonstrated that liquidators of the Chernobyl power plant are faced with a substantial risk of primary hyperparathyroidism, possibly caused by radioactive strontium isotopes . [7] Diagnosis [ edit ] The diagnosis of primary hyperparathyroidism is made by blood tests. ... "The parathyroid as a target for radiation damage". N. Engl. J. Med . 365 (7): 676–8. doi : 10.1056/NEJMc1104982 . ... "Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial" . J. Clin. Endocrinol. Metab . 89 (7): 3319–25. doi : 10.1210/jc.2003-030908 .CDC73, PTH, CASR, MEN1, CDKN1B, GCM2, GNA11, AP2S1, VDR, RET, HPT, CTNNB1, CALCA, FGF23, ALB, PTHLH, KL, DMD, BEST1, IL6, NAT10, CCL27, PDLIM3, GRN, GNA12, TNFRSF11B, ASRGL1, NR3C2, CHGA, SLPI, ATRNL1, SPINK1, ALPP, ATHS, CCND1, CFTR, DCTN6, TNFSF10, LPAR3, RAPGEF5, TNFRSF11A, BCAR1, AIP, ZNRD2, ADGRG2, ADAMTS4, ADM, LGR6, HAVCR1, OXER1, TMX2-CTNND1, TMED7-TICAM2, GPR166P, VN1R17P, MIR30E, MIR28, TICAM2, MRGPRX1, GPRC6A, GPR151, DERL2, MRGPRX4, MRGPRX3, CRISPLD2, SLC26A6, RGS5, ZNF410, ADAMTS9, SDHAF2, TMED7, TNFSF11, SDHC, FZD4, ACE, IGFBP3, IGF1, IFI27, GCG, GC, ESR1, EMP1, DMP1, DBP, IL17A, CYP19A1, CTNND1, CSE1L, CD38, BTF3P11, BGLAP, APRT, APC, IL11, JAK2, FZD1, RASA1, FZD5, WNT10B, TNF, TAT, ADAM17, SPP1, SLC12A1, AGT, PTMS, KCNQ1, PSMD9, PRSS2, PRLR, NOS3, MIP, MFAP1, LRP5, LRP2, H3P23

-
Filariasis
Wikipedia
Contents 1 Signs and symptoms 2 Cause 3 Diagnosis 3.1 Concentration methods 4 Treatment 5 Society and culture 5.1 Research teams 5.2 Prospects for elimination 6 Other animals 6.1 Cattle 6.2 Horses 6.3 Dogs 7 See also 8 References 9 Further reading 10 External links Signs and symptoms [ edit ] The most spectacular symptom of lymphatic filariasis is elephantiasis – edema with thickening of the skin and underlying tissues—which was the first disease discovered to be transmitted by mosquito bites. [2] Elephantiasis results when the parasites lodge in the lymphatic system . ... Treatment [ edit ] The recommended treatment for people outside the United States is albendazole combined with ivermectin . [5] [6] A combination of diethylcarbamazine and albendazole is also effective. [5] [7] Side effects of the drugs include nausea, vomiting, and headaches. [8] All of these treatments are microfilaricides; they have no effect on the adult worms. ... This drug has shown signs of inhibiting the reproduction of the bacteria, further inducing sterility. [7] Clinical trials in June 2005 by the Liverpool School of Tropical Medicine reported an eight-week course almost completely eliminated microfilaraemia. [14] [ non-primary source needed ] [15] Society and culture [ edit ] Research teams [ edit ] In 2015 William C. ... Lancet . 376 (9747): 1175–85. doi : 10.1016/s0140-6736(10)60586-7 . PMID 20739055 . S2CID 29589578 . ^ Turkington CA. ... PMID 17078904 . ^ Hoerauf A, Mand S, Fischer K, Kruppa T, Marfo-Debrekyei Y, Debrah AY, Pfarr KM, Adjei O, Buttner DW (2003), "Doxycycline as a novel strategy against bancroftian filariasis-depletion of Wolbachia endosymbionts from Wuchereria bancrofti and stop of microfilaria production", Med Microbiol Immunol (Berl) , 192 (4): 211–6, doi : 10.1007/s00430-002-0174-6 , PMID 12684759 , S2CID 23349595 ^ Taylor MJ, Makunde WH, McGarry HF, Turner JD, Mand S, Hoerauf A (2005), "Macrofilaricidal activity after doxycycline treatment of Wuchereria bancrofti: a double-blind, randomised placebo-controlled trial", Lancet , 365 (9477): 2116–21, doi : 10.1016/S0140-6736(05)66591-9 , PMID 15964448 , S2CID 21382828 ^ a b Andersson J, Forssberg H, Zierath JR (5 October 2015), "Avermectin and Artemisinin - Revolutionary Therapies against Parasitic Diseases" (PDF) , The Nobel Assembly at Karolinska Institutet , retrieved 5 October 2015 ^ Ndeffo-Mbah ML, Galvani AP (April 2017).IL10, IFNG, SLCO6A1, GPT2, MBL2, IL5, IL4, IGHG3, GSTK1, CHIT1, EDN1, HLA-A, TLR2, DTYMK, PRDX5, DLEU2, ALKBH1, TNFRSF18, TTR, TNF, TLR4, TGM1, HSPA8, LGALS2, IL13, CALU, F13A1, F13B, IL2, GLB1, GPT, ABO

-
Hypertensive Heart Disease
Wikipedia
Contents 1 Signs and symptoms 2 Diagnosis 2.1 Differential diagnosis 3 Prevention 3.1 Blood pressure goals 4 Treatment 5 Epidemiology 5.1 Sex differences 5.2 Ethnic differences 6 References 7 External links Signs and symptoms [ edit ] The symptoms and signs of hypertensive heart disease will depend on whether or not it is accompanied by heart failure . ... "Hypertensive heart disease: a new clinical classification (VIA)" . e-Journal of the European Society of Cardiology Council for Cardiology Practice . 7 (20): ePub. ^ GBD 2013 Mortality and Causes of Death, Collaborators (17 December 2014). ... "The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report" (PDF) . JAMA . 289 (19): 2560–2572. doi : 10.1001/jama.289.19.2560 . ... "Global burden of hypertension: analysis of worldwide data" . The Lancet . 365 (9455): 217–223. doi : 10.1016/S0140-6736(05)17741-1 . ... American Journal of Cardiology . 101 (7): 1016–1022. doi : 10.1016/j.amjcard.2007.11.061 .NPPB, ACE, MMP2, AGTR1, AGT, NPR1, CAPN9, FGF21, ITGB1BP2, GDE1, ADRA1A, FENDRR, SOCS3, MIR155, MIR19B1, MIR25, CARMN, PGR-AS1, SLC33A1, SLC8A1, BCL10, TIMP2, REN, PPARA, OGN, MMP9, NR3C2, DPP4, CYP11B2, CTF1, ATM, MHRT
-
Paternal Depression
Wikipedia
However, the recent increase of research into paternal depression shows society's views on increasing gender equality in social roles and the changing culture on masculine and feminine concepts. [7] Stigma of men with mental illness [ edit ] How the stigma of men with mental illness influence the prevalence of seeking treatment [27] There is often stigma around mental illness, especially for men. ... Paired with gender roles and the concepts of masculinity and femininity, society views men with mental impairments as weak and vulnerable and not the stereotypical alpha male . [7] This then affects how men view their own mental disability, influencing the seeking of treatment and acceptance of the illness. [16] This cause and effect relationship can create a cycle, leading men to be disheartened and ashamed of reaching out. ... "Paternal depression in the postnatal period and child development: a prospective population study" . Lancet . 365 (9478): 2201–5. doi : 10.1016/S0140-6736(05)66778-5 . ... S2CID 13093367 . ^ Nierenberg, Cari; October 27, Contributing writer; ET, 2016 03:22am. "7 Ways Depression Differs in Men and Women" . ... Complementary Therapies in Medicine . 7 (3): 191–192. 1990–1999. doi : 10.1016/s0965-2299(99)80132-0 .
-
Aromatase Deficiency
Wikipedia
As of 2016, only 35 cases have been described in medical literature. [3] Contents 1 Signs and symptoms 1.1 Female 1.2 Male 1.3 During pregnancy 1.4 Comorbidity 1.5 Complications 1.5.1 Pregnant mother 1.5.2 Female 1.5.3 Male 2 Cause 2.1 Gene Mutation 3 Diagnosis 4 Treatment 5 History 6 See also 7 References 8 Further reading 9 External links Signs and symptoms [ edit ] The deficiency causes the virilization of XX fetuses. ... Thus, RORA deficiency is linked to aromatase deficiency, which in turn can lead to elevated testosterone levels, a proposed risk factor for autism. [6] Complications [ edit ] Pregnant mother [ edit ] Aromatase is an estrogen synthase that synthesize estrone (E1) and estradiol (E2) from Androstenedione and Testosterone respectively. [7] During pregnancy, the placenta , which is fetal tissue, synthesizes large amounts of the intermediates in the biosynthesis of the estrogens, androstenedione and testosterone , but cannot convert them to estrogens due to the absence of aromatase. [7] The levels of accumulated androgens in the mother can elevate 100-fold higher than normal cycling levels which subsequently virilise both the mother and the fetus. The mother will experience cystic acne, deepening of the voice and hirsutism. [2] However, these symptoms are normally resolved following parturition. [2] If the fetus is a male, it will develop a normal male genitalia and will proceed to grow normally and exhibit secondary male sex characteristics. [8] If the fetus is a female, it will be born with ambiguous genitalia including labioscrotal fusion and a greatly enlarged phallus . [7] Female [ edit ] Aromatase deficient female cannot synthesize estrone or estradiol in the absence of aromatase. ... With a very low level of circulating estrogen (<7pg/mL), resulting in a higher level of FSH and LH in the blood. [2] Elevated level of androgens do not contribute to harmonic skeletal muscle growth like estrogen, thus, patients exhibits eunuchoid body habitus. [4] Patients are generally tall in stature and have a pattern of persistent linear bone growth into adulthood. [2] [7] Without estrogen, the epiphyseal plates cannot fuse together properly, resulting in continuous height growth. ... Molecular Autism, May 2015 DOI: 10.1186/2040-2392-6-7 ^ a b c d Blakemore J, Naftolin F (July 2016).

-
Arachnoid Cyst
Wikipedia
S2CID 34344350 . ^ a b c d e f g h i j k l m "Arachnoid Cysts Information Page" . NINDS . Retrieved April 7, 2017 . ^ Gelabert-González M (2004). ... Acta Neurochir (Wien) . 113 (1–2): 42–7. doi : 10.1007/bf01402113 . PMID 1799142 . ... "Intracranial cysts in autosomal dominant polycystic kidney disease". J. Neurosurg . 83 (6): 1004–7. doi : 10.3171/jns.1995.83.6.1004 . ... "Haemorrhage into an arachnoid cyst: a serious complication of minor head trauma" . Emerg Med J . 19 (4): 365–6. doi : 10.1136/emj.19.4.365 . PMC 1725893 . ... "Arachnoid cysts do not contain cerebrospinal fluid: A comparative chemical analysis of arachnoid cyst fluid and cerebrospinal fluid in adults" . Cerebrospinal Fluid Res . 7 : 8. doi : 10.1186/1743-8454-7-8 .ACO2, OFD1, ARID2, POLR3H, DNAJC19, TICRR, SHANK3, PUS3, HDAC8, PHIP, GPSM2, SACS, RTTN, KIF7, FGFR1, FGFR2, SON, NOTCH3, NEK1, LAMC2, CSF2, FOXC2, SPAST, HOXD4, NID1, COL1A2, SLC12A2, SOX2, ZIC2, WNT1, TYMS