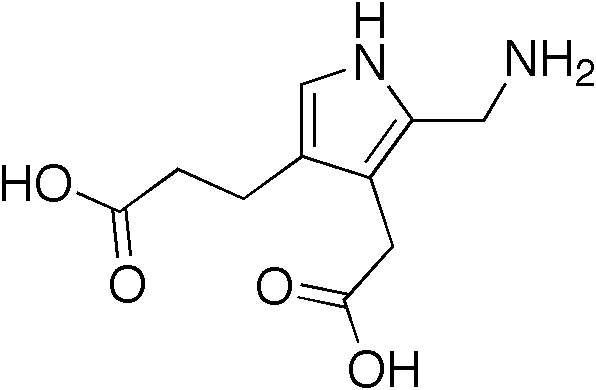
They show a uniform architecture of closely packed acinar or tubular structures of mature and bland appearance with scanty interposed stroma. [3] [4] [5] [6] [7] Cells are small with dark staining nuclei and inconspicuous nucleoli. ... Mitoses are conspicuously absent. [3] [4] [5] [6] [7] In the series reported by Jones et al. tumour cells were reactive for Leu7 in 3 cases of 5, to vimentine in 4 of 6, to cytocheratin in 2 of 6, to epithelial membrane antigen in 1 of 6 cases and muscle specific antigen in 1 of 6. [8] Olgac et al. found that intense and diffuse immunoreactivity for alpha-methylacyl-CoA racemase (AMACR) is useful in differentiating renal cell carcinoma from MA but a panel including AMACR, CK7 and CD57 is better in this differential diagnosis. [9] Differential diagnosis may be quite difficult indeed as exemplified by the three malignancies initially diagnosed as MA that later metastasized, in the report by Pins et al. [10] Cytogenetic characteristics [ edit ] Brunelli et al. stated that genetic analysis of chromosome 7, 17, and Y may facilitate discrimination of MA from papillary renal cell carcinoma in difficult cases. ... "Metanephric adenoma". Histol Histopathol . 7 (4): 689–92. PMID 1333853 . ^ a b Jones, E. ... Arch Pathol Lab Med. 123:415-420. ^ Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L (2003) Metanephric adenoma lacks the gains of chromosomes 7 and 17 and loss of Y that are typical of papillary renal cell carcinoma and papillary adenoma. ... "[Diagnosis of renal metanephric adenoma: relevance of immunohistochemistry and biopsy]". Ann Pathol (in French). 27 (5): 365–8. PMID 18185471 . ^ Nagashima Y, Arai N, Tanaka Y, Yoshida S, Sumino K, Ohaki Y, Matsushita K, Morita T, Misugi K 81991) Case record: two cases of renal epithelial tumour resembling immature nephron.
Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Treatment 5 Prognosis 6 Epidemiology 7 See also 8 References 9 External links Signs and symptoms [ edit ] AL amyloidosis can affect a wide range of organs, and consequently present with a range of symptoms. ... However, the diagnosis requires a sample of an affected organ. [4] [7] Treatment [ edit ] The most effective treatment is autologous bone marrow transplants with stem cell rescues . ... British Journal of Haematology . 140 (4): 365–377. doi : 10.1111/j.1365-2141.2007.06936.x .
Multiple myeloma is a cancer that develops in the bone marrow , the spongy tissue found in the center of most bones. The bone marrow produces red blood cells, which carry oxygen throughout the body; white blood cells, which form the body's defenses (immune system); and platelets, which are necessary for blood clotting . Multiple myeloma is characterized by abnormalities in plasma cells, a type of white blood cell. These abnormal cells multiply out of control, increasing from about one percent of cells in the bone marrow to the majority of bone marrow cells. The abnormal cells form tumors within the bone, causing bone pain and an increased risk of fractures.
A plasma cell disorder characterized by the aggregation and deposition of insoluble amyloid fibrils derived from misfolding of monoclonal immunoglobulin light chains usually produced by a plasma cell tumor. It usually presents as primary systemic amyloidosis (PSA) with multiple organ involvement and less frequently as primary localized amyloidosis (PLA) restricted to a single organ.
AL amyloidosisis the most common form of amyloidosis, a group of disorders in which an abnormal protein called amyloid builds up in tissues and organs. The signs and symptoms of AL amyloidosis vary among patients because the build up may occur in the tongue, intestines, muscles, joints, nerves, skin, ligaments, heart, liver, spleen, or kidneys. To diagnose AL amyloidosis, healthcare professionals use blood or urine tests to identify signs of amyloid protein and a biopsy to confirm the diagnosis. Treatment may include chemotherapy directed at the abnormal plasma cells, stem cell transplantation , or other treatments based on which symptoms have developed.
Primary systemic amyloidosis (PSA) is a form of AL amyloidosis (see this term) caused by the aggregation and deposition of insoluble amyloid fibrils derived from misfolded monoclonal immunoglobulin light chains usually produced by a plasma cell tumor (see this term) and characterized by multiple organ involvement.
It includes minor conditions such as heat cramps, heat syncope, and heat exhaustion as well as the more severe condition known as heat stroke. [1] Heat illness can relate to many of the organs and systems including: brain, heart, kidneys, liver, etc. [2] Contents 1 Classification 2 Prevention 3 Epidemiology 4 History 5 See also 6 References 7 External links Classification [ edit ] A number of heat illnesses exist including: [3] [4] Heat stroke - Defined by a body temperature of greater than 40 °C (104 °F) due to environmental heat exposure with lack of thermoregulation . ... Symptoms may include hyperventilation, respiratory problems, numbness or tingling, or muscle spasms. [5] Prevention [ edit ] Prevention includes avoiding medications that can increase the risk of heat illness (e.g. antihypertensives , diuretics , and anticholinergics ), gradual adjustment to heat, and sufficient fluids and electrolytes. [6] [7] Epidemiology [ edit ] A 2016 U.S. government report said that climate change could result in "tens of thousands of additional premature deaths per year across the United States by the end of this century." [8] Indeed, between 2014 and 2017, heat exposure deaths tripled in Arizona (76 deaths in 2014; 235 deaths in 2017) and increased fivefold in Nevada (29 deaths in 2014; 139 deaths in 2017). [9] Between 1999 and 2003, the US had a total of 3442 deaths from heat illness. ... Between 1992 and 2006, 423 workers died from heat illness in the US. [7] Exposure to environmental heat led to 37 work-related deaths. ... Current Reviews in Musculoskeletal Medicine . 7 (4): 355–365. doi : 10.1007/s12178-014-9240-0 .
Heat cramps , a type of heat illness , are muscle spasms that result from loss of large amount of salt and water through exercise. Heat cramps are associated with cramping in the abdomen , arms and calves . This can be caused by inadequate consumption of fluids or electrolytes . [1] Heavy sweating causes heat cramps, especially when the water is replaced without also replacing salt or potassium . [2] Although heat cramps can be quite painful, they usually don't result in permanent damage, though they can be a symptom of heat stroke or heat exhaustion . Heat cramps can indicate a more severe problem in someone with heart disease or if they last for longer than an hour. [2] In order to prevent them, one may drink electrolyte solutions such as sports drinks during exercise or strenuous work or eat potassium-rich foods like bananas and apples . When heat cramps occur, the affected person should avoid strenuous work and exercise for several hours to allow for recovery. [2] See also [ edit ] Dehydration References [ edit ] ^ Auerbach Paul S Wilderness Medicine. 4th ed.
"Traumatic dislocations and instability of the trapeziometacarpal joint of the thumb" (PDF) . Hand Clinics . 22 (3): 365–92. doi : 10.1016/j.hcl.2006.05.001 .
Eosinophilic pustular folliculitis (EPF) affects the skin causing itchy, red or skin-colored bumps and pustules (bumps containing pus). The papules mostly appear on the face, scalp, neck and trunk and may last for weeks or months. EPF affects males more than females. There are several forms of EPF. Classic eosinophilic pustular folliculitis mainly occurs in Japan. Immunosuppression-associated EPF is mainly associated with HIV infection, but has also been associated with certain cancers and medications.[16046] The infantile form of EPF is seen in infants from birth or within the first year of life. The underlying cause of EFP is unknown. All of these forms have similar skin findings.
Contents 1 Signs and symptoms 2 Pathogenesis 3 Diagnosis 3.1 Macroscopic 3.2 Microscopic 3.3 Immunohistochemistry 3.4 Differential diagnoses 4 Management 5 Epidemiology 6 References 7 Further reading 8 External links Signs and symptoms [ edit ] Most patients present with a very rapidly growing mass that often gives a bluish appearance in the mouth. ... "Melanotic neuroectodermal tumor of infancy" . Ear Nose Throat J . 85 (6): 365. doi : 10.1177/014556130608500608 . ... JAMA Otolaryngol Head Neck Surg . 140 (7): 667–8. doi : 10.1001/jamaoto.2014.632 .
Garrod's pads (also known as violinist's pads [1] ) are a cutaneous condition characterized by calluses on the dorsal aspect of the interphalangeal joints , [2] i.e. the back side of the finger joints. They are often seen in violin , viola , and cello players, along with fiddler's neck and other dermatologic conditions peculiar to string musicians. [2] Although Garrod's pads are conventionally described as appearing on the proximal interphalangeal joint, distal interphalangeal joint involvement has also been described. [2] Garrod's pads are named after Archibald Garrod who first documented them in 1904 in association with Dupuytren's contracture . [3] H.A. Bird described them as an incidental finding in a professional violinist and proposed that they arise in such cases due to repeated extreme tension of the extensor tendons over the interphalangeal joints. [4] Bird noted that violin players use the left hand for a markedly different task than the right hand, with the extensor tendons in the left hand subjected to considerable tension, and that Garrod's pads only arise on the left hand in such cases. This unilateral finding differentiates the occupational hazard of Garrod's pads from more significant disorders. Among violinists and violists, Garrod's pads apparently arise as a protective mechanism for the skin and subcutaneous tissues above the tendons; Bird notes that they do not protect against external trauma unlike most calluses. [4] Patients with Dupuytren's contracture are four times more likely to have coexisting Garrod's pads. [5] [6] See also [ edit ] Knuckle pads Harpist's finger Fiddler's neck Cellist's chest Cello scrotum Paget-Schroetter syndrome List of cutaneous conditions List of eponymously named medical signs References [ edit ] ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).
A number sign (#) is used with this entry because knuckle pads are associated with certain genetic disorders such as epidermolytis palmoplantar keratoderma (144200) or Dupuytren contractures (126900), both of which are autosomal dominant. Knuckle pads are sometimes associated with Dupuytren contractures and it is not completely certain that a different gene is involved. Camptodactyly (114200) also has an uncertain relationship. Skoog (1948) defined knuckle pads as 'subcutaneous nodules on the dorsal aspect of the proximal interphalangeal joints.' Lu et al. (2003) reported association of knuckle pads with epidermolytic palmoplantar keratoderma in a Chinese family and identified a novel leu160-to-phe mutation in the keratin-9 gene (L160F; 607606.0012) as the presumed cause. They presented evidence that both the hyperkeratosis and the knuckle pads were friction-related.
However, in young puppies or immunocompromised animals, mixed or secondary infections can progress to lower respiratory infections such as pneumonia . [5] Contents 1 Symptoms 1.1 Types 2 Transmission 3 Treatment and prevention 4 Vaccines 5 Complications 6 See also 7 References 8 External links Symptoms [ edit ] The incubation period is 5–7 days (with a range of 3–10). [5] Symptoms can include a harsh, dry cough, retching, sneezing, snorting, gagging or vomiting in response to light pressing of the trachea or after excitement or exercise. ... To put the relative levels of shedding bacteria into perspective, a study analyzing the shedding kinetics of B. bronchiseptica presents the highest levels of bacterial shedding one week post-exposure, with an order of magnitude decrease in shedding observed every week. [7] This projection places negligible levels of shedding to be expected six weeks post-exposure (or approximately five weeks post-onset of symptoms). Dogs which had been administered intranasal vaccine four weeks prior to virulent B. bronchiseptica challenge displayed little to no bacterial shedding within three weeks of exposure to the virulent strain. [7] Treatment and prevention [ edit ] See also: DA2PPC Vaccine Antibiotics are given to treat any bacterial infection present. ... "Detection of Respiratory Viruses and Bordetella Bronchiseptica in Dogs with Acute Respiratory Tract Infections" . The Veterinary Journal . 201 (3): 365–369. doi : 10.1016/j.tvjl.2014.04.019 .
It is the most common of the acute porphyrias . [1] [2] [3] Contents 1 Signs and symptoms 1.1 Acute attacks 2 Pathophysiology 3 Diagnosis 4 Treatment 5 Society 6 References 7 External links Signs and symptoms [ edit ] The clinical presentation of AIP is highly variable and non-specific. ... The syndrome marked by acute attacks affects only 10% of gene carriers. [4] The mean age at diagnosis is 33 years old. [5] Like other porphyrias, AIP is more likely to present in women. [6] A distinguishing feature of AIP that separates it from other porphyrias is the absence of photosensitive cutaneous symptoms that occur in addition to acute attacks. [7] Acute attacks [ edit ] AIP is one of the four porphyrias that presents as an acute attack. 90% of affected individuals never experience an acute attack and are asymptomatic, while an estimated 5% of affected individuals experience repeat attacks. [8] Attacks are most common in young adult women and are rare before puberty or after menopause. [9] Severe acute attacks may require hospitalization. ... PMID 26159905 . ^ Kauppinen, Raili (15–21 Jan 2005). "Porphyrias". Lancet . 365 (9455): 241–252. doi : 10.1016/s0140-6736(05)70154-9 . ... PMID 15652607 . ^ Kauppinen, Raili (15–21 Jan 2005). "Porphyrias". Lancet . 365 (9455): 241–252. doi : 10.1016/s0140-6736(05)70154-9 . ... Journal of Community Hospital Internal Medicine Perspectives . 7 (2): 100–102. doi : 10.1080/20009666.2017.1317535 .
Each parent was heterozygous for 1 of the mutations. Hessels et al. (2004) described a 7-year-old boy with homozygous AIP who demonstrated hepatosplenomegaly, mild anemia, mild mental retardation, yellow-brown teeth, and dark red urine and who had excessively elevated levels of urinary delta-aminolevulinic acid, porphobilinogen, and uroporphyrin. ... Although affected members from 21 families had absent CRM to the PBGD protein, all 7 AIP heterozygotes from 1 family of Basque origin had positive CRM results detected in red cell lysates. ... The findings indicated that the toxic accumulation of porphyrin precursors, rather than heme deficiency, is primarily responsible for acute neurologic attacks in heterozygous AIP. In their patient, a 7-year-old boy with excessively elevated levels of urinary delta-aminolevulinic acid, porphobilinogen, and uroporphyrin, Hessels et al. (2004) found a novel homozygous leu81-to-pro (L81P) mutation in exon 6 of the porphobilinogen deaminase gene (609806.0045). ... In affected members of 11 different families with either CRM-negative or CRM-positive AIP, Grandchamp et al. (1990) identified 7 different point mutations in the PBGD gene.
Genes of Interest in the Differential Diagnosis of Clinically Manifest Acute Intermittent Porphyria (AIP) View in own window Gene Disorder MOI Clinical Features Biochemical Characteristics 1 Urine Stool Plasma Erythrocytes CPOX Hereditary coproporphyria (HCP) AD Acute attack ± skin lesions 2 ↑ PBG, ALA 3 , porphyrins 4 ↑ copro- porphyrin III ↑ plasma porphyrins; fluorescence emission peak ~620 nm 5 PPOX Variegate porphyria (VP) AD Acute attack ± skin lesions 2 ↑ PBG, ALA 3 , porphyrins 4 ↑ proto- porphyrin 6 ↑ plasma porphyrins; fluorescence emission peak ~626 nm 7 ALAD ALAD deficiency porphyria (OMIM 612740) AR Acute attack ↑ ALA, copropor- phyrin III, normal PBG ↑ zinc-protoporphyrin; ↓ ALAD activity FAH Tyrosinemia type 1 AR Acute attack ↑ ALA ↓ ALAD activity AD = autosomal dominant; ALA = 5-aminolevulinic acid; ALAD = 5-aminolevulinate dehydratase; AR = autosomal recessive; MOI = mode of inheritance 1. ... Protoporphyrin is the main stool porphyrin, but a small increase in coproporphyrin III is also observed 7. Plasma porphyrin concentration is always increased and fluorescence emission spectroscopy distinguishes VP from all other porphyrias.
Acute intermittent porphyria (AIP) is one of the liver (hepatic) porphyrias . AIP is caused by low levels of porphobilinogen deaminase (PBGD), an enzyme also often called hydroxymethylbilane synthase. The low levels of PBGD are generally not sufficient to cause symptoms; however, activating factors such as hormones, drugs, and dietary changes may trigger symptoms. Although most individuals with AIP never develop symptoms, symptomatic individuals typically present with abdominal pain with nausea. Treatment is dependent on the symptoms.
A rare, severe form of the acute hepatic porphyrias characterized by the occurrence of neuro-visceral attacks without cutaneous manifestations. Clinical description Patients suffer intermittent neuro-visceral attacks that can persist for several days and that repeat over several weeks. These attacks manifest as intense abdominal pain (>95% of cases) and neurological and/or psychological symptoms. The abdominal pain is often associated with lumbago irradiating to the thighs, and with nausea, vomiting and relentless constipation. Psychological symptoms are variable: irritability, emotionality, depression, considerable anxiety and, more rarely, auditory and visual hallucinations, disorientation, mental confusion.
Contents 1 Signs and symptoms 2 Genetics 3 Diagnosis 4 Treatment 5 Prognosis 6 Epidemiology 7 See also 8 References 9 External links Signs and symptoms [ edit ] This syndrome is characterized by overgrowth and advanced bone age. ... If persons with this disorder have any increased cancer risk, their risk is only slightly greater than that of the general population. [7] Genetics [ edit ] Mutations in the NSD1 gene cause Sotos syndrome. [8] [9] The NSD1 gene provides instructions for making a protein (histone methyltransferase) that is involved in normal growth and development. ... Developmental delays may improve in the school-age years; however, coordination problems may persist into adulthood, along with any learning disabilities and/or other physical or mental issues. [ citation needed ] Epidemiology [ edit ] Incidence is approximately 1 in 14,000 births. [11] See also [ edit ] Proteus syndrome Beckwith-Wiedemann syndrome Perlman syndrome References [ edit ] ^ http://www.exploringautism.org/autism/evaluation.htm ^ "7-foot-tall Broc Brown: Facts" . Morning News USA . ... "Haploinsufficiency of NSD1 causes Sotos syndrome". Nat. Genet . 30 (4): 365–6. doi : 10.1038/ng863 . PMID 11896389 .
Molecular Genetic Testing Used in Sotos Syndrome View in own window Gene 1 Method Proportion of Probands with a Pathogenic Variant 2 Detectable by Method NSD1 Sequence analysis 3 45%-80% CMA 4 15%-50% 5, 6 Gene-targeted deletion/duplication analysis 7 20%-55% 7 , 8 1. See Table A. Genes and Databases for chromosome locus and protein. 2. ... The incidence of the common 1.9-Mb deletion varies by population [Kurotaki et al 2003, Tatton-Brown et al 2005a, Tatton-Brown et al 2005b, Visser et al 2005]. 7. Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. ... BWS should be clinically distinguishable from Sotos syndrome (molecular testing is indicated in individuals w/clinical overlap). 7 Macroglossia Anterior earlobe creases / helical pits Omphalocele Visceromegaly ↑ risk of embryonal tumors, esp Wilms tumor Simpson-Golabi-Behmel syndrome type 1 GPC3 GPC4 XL Pre- & postnatal overgrowth Variable ID Predominantly affects males Polydactyly Supernumerary nipples Diastasis recti Pectus excavatum Facial gestalt differs. 8 Bannayan-Riley-Ruvalcaba syndrome (see PTEN Hamartoma Tumor Syndrome) PTEN AD Macrocephaly Somewhat similar facial gestalt May be associated w/autistic spectrum disorder Vascular malformations Hamartomatous polyps of distal ileum & colon Pigmented macules on shaft of penis Lipomas ↑ risk of thyroid & breast cancer 8 Fragile X syndrome (see FMR1 -Related Disorders) FMR1 XL Macrocephaly ID Typical but subtle facial appearance may overlap w/Sotos syndrome: dolicocephalic head shape, prominent jaw & forehead. ... Approximately 85% of individuals with BWS have no family history of BWS while approximately 15% have a family history consistent with autosomal dominant transmission of BWS. 7. NSD1 pathogenic variants were reported in two individuals with BWS; however, the individuals do not fulfill diagnostic criteria for BWS and do fulfill diagnostic criteria for Sotos syndrome [Baujat et al 2004, Tatton-Brown & Rahman 2004]. 8.
Psychomotor development of the sister, who also had advanced osseous maturation, improved significantly at the age of 7 years. Accelerated growth with normal bone age, optic atrophy, renal agenesis with contralateral double kidney, and significant mental retardation (IQ, 45) were shown in the brother at 3.5 years of age. ... Before molecular analyses, the patients were phenotypically scored into 4 groups: 37 patients comprising group 1 had a phenotype typical of Sotos syndrome; 13 patients comprising group 2 had a Sotos-like phenotype but with some atypical features; 7 patients comprising group 3 had been diagnosed with Weaver syndrome (277590); and 18 patients comprising group 4 had an overgrowth condition that was neither Sotos nor Weaver syndrome. There was a strong correlation between presence of an NSD1 alteration and clinical phenotype, as 28 of 37 patients (76%) in group 1 had NSD1 mutations or deletions, whereas none of the patients in group 4 had alterations in NSD1. Three of the 7 patients who had been diagnosed with Weaver syndrome had NSD1 mutations (see 606681.0006). ... Compared with controls, 14 deletions and 7 duplications were significantly overrepresented in cases, providing a clinical diagnosis as pathogenic.
A rare genetic overgrowth syndrome characterized by a typical facial appearance, overgrowth with macrocephaly and variable intellectual impairment. Epidemiology Prevalence at birth is estimated at 1/14,000. Clinical description Excessive growth is evident across life, especially in childhood, and can manifest since fetal life., with final adult height beyond or in the upper part of the normal ranges. Macrocephaly is usually striking and disproportioned with respect to height. The distinct facial appearance is most easily recognized in early childhood (long narrow face, flushed cheeks, prominent forehead with frontotemporal hair scarcity, down-slanting palpebral fissures, hypertelorism, a high arched palate, and pointed chin). Sotos syndrome is associated with mild to severe intellectual disability as well as a wide spectrum of behavioral disorders.
A number sign (#) is used with this entry because of evidence that Sotos syndrome-3 (SOTOS3) is caused by homozygous mutation in the APC2 gene (612034) on chromosome 19p13. One such family has been reported. For a discussion of genetic heterogeneity of Sotos syndrome, see SOTOS1 (117550). Clinical Features Almuriekhi et al. (2015) reported 2 sibs, born of consanguineous Egyptian parents, with intellectual disability (IQs of 60 and 56), a severe receptive and expressive language disorder, learning disabilities, and hyperactive behavior associated with relative macrocephaly, long face, and prominent chin and nose. Brain imaging in 1 patient showed dilated lateral ventricles. Neither patient had advanced bone age, hypotonia, seizures, or autism. The findings were reminiscent of Sotos syndrome, but genetic analysis excluded mutations in the NSD1 gene (606681).
Sotos syndrome is a condition characterized mainly by distinctive facial features; overgrowth in childhood; and learning disabilities or delayed development. Facial features may include a long, narrow face; a high forehead; flushed (reddened) cheeks; a small, pointed chin; and down-slanting palpebral fissures . Affected infants and children tend to grow quickly; they are significantly taller than their siblings and peers and have a large head. Other signs and symptoms may include intellectual disability; behavioral problems; problems with speech and language; and/or weak muscle tone (hypotonia). Sotos syndrome is usually caused by a mutation in the NSD1 gene and is inherited in an autosomal dominant manner.
Instead, they experience a thickening of the skin being repeatedly bitten. [5] Contemporary research suggests a link between impulse control disorders and obsessive–compulsive disorders , [6] and this was addressed in the DSM-5 when dermatophagia and other related disorders were classified as 'other specified obsessive-compulsive related disorders' and are given the specification of body focused repetitive behavior . [7] Further information on OCD, other anxiety disorders, dermatophagia and other related body focused repetitive behaviors can be found in the DSM-5 [8] Contents 1 Behavior 2 Management 2.1 Management in children with disabilities 3 In popular culture 4 See also 5 Notes Behavior [ edit ] People with dermatophagia chew their skin out of compulsion, and can do so on a variety of places on their body. [9] Those with dermatophagia typically chew the skin surrounding their fingernails and joints. ... Journal of the American Academy of Dermatology . 53 (2): 365. doi : 10.1016/j.jaad.2005.04.021 .
It is often classified under the umbrella entity microscopic colitis , that it shares with a related condition, lymphocytic colitis . [1] Contents 1 Signs and symptoms 2 Causes 3 Diagnosis 4 Treatment 5 See also 6 References 7 External links Signs and symptoms [ edit ] Microscopic colitis causes chronic watery diarrhea with greater than 10 bowel movements per day. ... Zeitschrift für Gastroenterologie . 42 (5): 365–9. doi : 10.1055/s-2004-812709 .
Recently, several studies found that a single course of prenatal steroids (betamethasone) may increase survival in hydropic fetuses with microcystic CPAMs to 75-100%. [6] [7] These studies indicate that large microcystic lesions may be treated prenatally without surgical intervention. ... American Journal of Obstetrics and Gynecology . 208 (2): 151.e1–7. doi : 10.1016/j.ajog.2012.11.012 . ... "Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival" . Fetal Diagn Ther . 22 (5): 365–371. doi : 10.1159/000103298 . PMID 17556826 .
A rare respiratory malformation characterized by a hamartomatous mass of non-functioning lung tissue of variable extent and with variable degrees of cystic or adenomatoid change. Clinical presentation, prognosis, and presence of associated abnormalities depend on the subtype of the lesion. Based on histopathological findings, five subtypes (types 0 to 4) can be differentiated.
It was first described by Verhoeven et al. in 2001. [3] Contents 1 Signs and Symptoms 2 Causes 3 Diagnosis 3.1 Metabolite Analyses 3.2 Mutation Analysis 4 Treatment 5 Epidemiology 6 See also 7 References 8 External links Signs and Symptoms [ edit ] The leading symptoms of Transaldolase Deficiency are coagulopathy , thrombocytopenia , hepatosplenomegaly , hepatic fibrosis and dysmorphic features. [4] The dysmorphic features can include antimongoloid slant , low-set ears, and cutis laxa . [5] Those affected by this disease have abnormal polyol concentrations in urine and other bodily fluids, this can determined by an abnormal liver function tests. [4] [5] With transaldolase deficiency there is a buildup of sedoheptulose 7-phosphate (it is increased six to sevenfold in the blood compared to normal), which decreases the change of ribose 5-phosphate into glucose 6-phosphate . [6] [7] This reaction is important in releasing NADPH. ... Nephrol Dial Transplant . 27 (8): 3224–7. doi : 10.1093/ndt/gfs061 . PMID 22510381 . ^ a b c d Balasubramaniam S, Wamelink MM, Ngu LH, Talib A, Salomons GS, Jakobs C, Keng WT (Jan 2011). ... "The pathogenesis of transaldolase deficiency" . IUBMB Life . 59 (6): 365–73. doi : 10.1080/15216540701387188 . ... "Transaldolase: from biochemistry to human disease". Int J Biochem Cell Biol . 41 (7): 1482–94. doi : 10.1016/j.biocel.2009.02.001 . ... JIMD Reports. 12 . pp. 47–50. doi : 10.1007/8904_2013_243 . ISBN 978-3-319-03460-7 . PMC 3897798 . PMID 23846909 . ^ Wamelink MM, Struys EA, Salomons GS, Fowler D, Jakobs C, Clayton PT (Jun 2008).
A number sign (#) is used with this entry because of evidence that transaldolase deficiency is caused by homozygous or compound heterozygous mutation in the TALDO1 gene (602063) on chromosome 11p15. Description Transaldolase deficiency is a rare inborn error of pentose metabolism. Typical features include intrauterine growth restriction, triangular vase, loose wrinkly skin at birth, and development of progressive liver failure (summary by Lee-Barber et al., 2019). Clinical Features Verhoeven et al. (2001) described deficiency of transaldolase in the first child of healthy, consanguineous Turkish parents. Soon after birth, the patient had undergone surgical correction of aortic coarctation.
Transaldolase deficiency is an inborn error of the pentose phosphate pathway that presents in the neonatal or antenatal period with hydrops fetalis, hepatosplenomegaly, hepatic dysfunction, thrombocytopenia, anemia, and renal and cardiac abnormalities. Epidemiology Less than ten cases have been reported in the literature so far, all involving children born to consanguineous parents of Turkish and Arabic origin. Clinical description Dysmorphic features (downward-slanting palpebral fissures, low-set ears, and cutis laxa) have also been described. The severity of the symptoms and outcome vary widely. Etiology The disorder is caused by mutations in the transaldolase gene ( TALDO1 , 11p15.5-p15.4).
This can result from the release of mediators from a specific site, such as the skin or mucosal tissue, or activation of mast cells around the vasculature. [10] Diagnosis [ edit ] MCAS is often difficult to identify due to the heterogeneity of symptoms and the "lack of flagrant acute presentation." [7] The condition can also be difficult to diagnose, especially since many of the numerous symptoms are non-specific in nature. ... World Journal of Hematology . 3 (1): 1–7. ^ a b c d e Frieri M (June 2018). ... Clinical Reviews in Allergy & Immunology . 54 (3): 353–365. doi : 10.1007/s12016-015-8487-6 . ... "Quercetin is more effective than cromolyn in blocking human mast cell cytokine release and inhibits contact dermatitis and photosensitivity in humans" . PloS One . 7 (3): e33805. Bibcode : 2012PLoSO...733805W . doi : 10.1371/journal.pone.0033805 . ... "Demonstration of an aberrant mast-cell population with clonal markers in a subset of patients with "idiopathic" anaphylaxis" . Blood . 110 (7): 2331–3. doi : 10.1182/blood-2006-06-028100 .
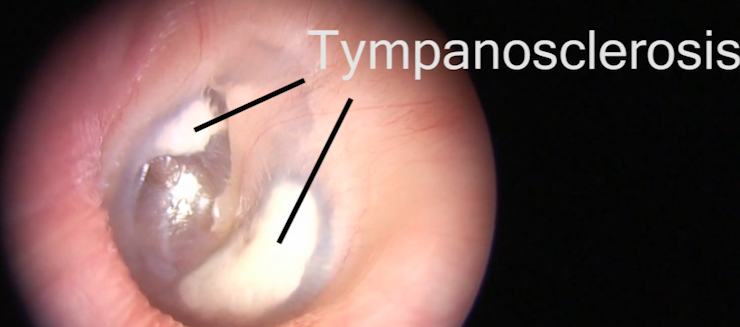
tympanosclerosis | name = | synonyms = Myringosclerosis, intratympanic tympanosclerosis | field = ENT surgery | symptoms = | complications = | onset = | duration = | types = | causes = | risks = | diagnosis = | differential = | prevention = | treatment = | medication = | prognosis = | frequency = | deaths = }} Tympanosclerosis is a condition caused by hyalinization and subsequent calcification of subepithelial connective tissue of TM and middle ear, sometimes resulting in a detrimental effect to hearing. [1] [2] Contents 1 Signs and symptoms 2 Causes 3 Pathophysiology 4 Diagnosis 4.1 Classification 5 Treatment 6 Prognosis 7 Epidemiology 8 References 9 External links Signs and symptoms [ edit ] Myringosclerosis rarely causes any symptoms. [3] Tympanosclerosis, on the other hand, can cause significant hearing loss [3] or chalky, white patches on the middle ear or tympanic membrane. [1] Causes [ edit ] The aetiology for tympanosclerosis is not extensively understood. There are several probable factors which could result in the condition appearing, including: Long term otitis media (or 'glue ear') [2] Insertion of a tympanostomy tube . [4] [5] [6] If aspiration is performed as part of the insertion, the risk of tympanosclerosis occurring increases. [7] Risk also increases if a larger tube is used, [8] or if the procedure is repeated. [9] Atherosclerosis [10] There is ongoing research as to whether or not cholesteatoma is associated with tympanosclerosis. ... The Journal of Laryngology and Otology . 105 (8): 614–7. doi : 10.1017/s0022215100116822 . ... The American Journal of Otology . 16 (3): 365–72. PMID 8588632 . External links [ edit ] Classification D ICD - 10 : H74.0 ICD - 9-CM : 385.0 MeSH : D063371 External resources Patient UK : Tympanosclerosis Wikimedia Commons has media related to Tympanosclerosis .