-
Retiform Parapsoriasis
Wikipedia
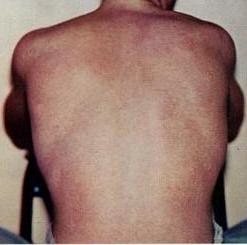
Retiform parapsoriasis Specialty Dermatology Retiform parapsoriasis is a cutaneous condition, considered to be a type of large-plaque parapsoriasis . [1] It is characterized by widespread, ill-defined plaques on the skin, that have a net-like or zebra-striped pattern. [2] Skin atrophy , a wasting away of the cutaneous tissue , usually occurs within the area of these plaques. [1] See also [ edit ] Parapsoriasis Poikiloderma vasculare atrophicans List of cutaneous conditions References [ edit ] ^ a b Lambert WC, Everett MA (Oct 1981).

-
Epileptic Encephalopathy, Early Infantile, 45
OMIM
In vitro functional studies in HEK293 cells showed that the mutation altered the kinetic properties of the channel, resulting in the net loss of GABAergic inhibition. In a boy with EIEE45, Lien et al. (2016) identified a de novo heterozygous missense mutation in the GABRB1 gene (T287I; 137190.0002).
-
Maple Syrup Urine Disease
GeneReviews
Acute metabolic decompensation is corrected by treating the precipitating stress while delivering sufficient calories, insulin, free amino acids, isoleucine, and valine to achieve sustained net protein synthesis in tissues. Some centers use hemodialysis/hemofiltration to remove BCAAs from the extracellular compartment, but this intervention does not alone establish net protein accretion. ... Thus, leucine tolerance reflects a balance between unmeasured protein losses (e.g., sloughed skin, hair, and nails) and the net accretion of body protein, which in turn is linked to growth rate [Strauss et al 2010]. ... The risk for metabolic crisis in any ill person with MSUD depends on residual in vivo BCKD enzyme activity in relation to the net liberation of free leucine from protein catabolism. ... Following the neonatal period, acute metabolic intoxication (leucinosis) and neurologic deterioration can develop rapidly at any age as a result of net protein degradation precipitated by infection, surgery, injury, or psychological stress (see Figure 1). ... Plasma leucine levels rise predictably as a result of net protein catabolism provoked by a variety of physiologic stresses, including (more...)DBT, BCKDHB, BCKDHA, BCAT2, PPM1K, DLD, ARID4B, BDNF, CTSD, SERPINE1, TNS3, CACNA2D2, MKRN3, UMOD, SPN, NME1, PAH, NBN, MEA1, IL1B, GPR4, GLI2, F2, MECP2

-
Afterdepolarization
Wikipedia
They are due to elevated cytosolic calcium concentrations, classically seen with digoxin toxicity. [3] [4] The overload of the sarcoplasmic reticulum may cause spontaneous Ca 2+ release after repolarization, causing the released Ca 2+ to exit the cell through the 3Na + /Ca 2+ -exchanger. This results in a net depolarizing current. The classical feature is Bidirectional ventricular tachycardia .
-
Nephrolithiasis, Calcium Oxalate
OMIM
In vitro flux studies indicated that mice lacking Slc26a6 have a defect in intestinal oxalate secretion resulting in enhanced net absorption of oxalate. Jiang et al. (2006) concluded that the anion exchanger, SLC26A6, has a major constitutive role in limiting net intestinal absorption of oxalate, thereby preventing hyperoxaluria and calcium oxalate urolithiasis.
-
Steroid Diabetes
Wikipedia
Mechanism [ edit ] Glucocorticoids oppose insulin action and stimulate gluconeogenesis , especially in the liver , resulting in a net increase in hepatic glucose output.
- Naegeli-Franceschetti-Jadassohn Syndrome/dermatopathia Pigmentosa Reticularis MedlinePlus
-
Yellow Fever
Orphanet
Management and treatment As there is presently no antiviral drug available for YF, treatment is supportive, following the guidelines for treatment of severe septicemia. Insecticide-treated bed nets and/or room screens should be used in open-air settings to prevent further transmission.ERVK-32, ERVK-6, DDOST, PARTICL, TRIM56, CYP20A1, EMC3, TRAV12-2, BACE1, AMACR, TRPA1, TFPI, RPL19, RAF1, CRP, MRC1, IL10, IL6, IL1RN, IFNG, IFNAR1, GYPC, GPT, GLS, FCGR3B, FCGR3A, F10, MAP2K1

-
Dengue Fever
Orphanet
Management and treatment As there is presently no antiviral drug available for DF/DHF, treatment is supportive, following the guidelines for treatment of severe septicemia. Insecticide-treated bed nets, room screens and elimination of larval development sites should be used in open-air settings to prevent further transmission.CD209, PTPN11, SENP8, TNF, ERVK-6, IFNG, IVNS1ABP, IL10, ERVK-32, IFNA1, IFNA13, IL1B, PLAAT4, LSAMP, HLA-A, LAMP3, IFIH1, DDX58, HLA-DRB1, ROBO3, ERVW-1, IL6, PDR, ERVK-20, CLEC5A, CXCL10, CXCL8, IL4, CRP, GPT, RAF1, CCL5, MICB, VEGFA, CDK9, KRAS, RSAD2, MBL2, THBD, GP1BA, OAS3, RHD, ALB, FCGR2A, SARS1, PLCE1, HLA-C, FBXW7, SARS2, CCR5, SOAT1, RBM45, TNFSF10, TLR8, OAS1, MPZ, IFITM3, IFNAR2, CSPG5, IGHG3, CCL4L2, KDR, HPSE, EIF2AK3, KIR2DL1, PF4, SLC17A5, SPHK1, IL18, HAVCR1, MYOM2, KIR3DL1, KIR3DS1, TIMELESS, PMP22, IL1RN, ADAMTS13, PADI4, LGALS9, HSPA5, HLA-B, NLRP3, VDR, TAP2, MIR21, IFNL1, DHX9, ATN1, EGFR, CCL4, CCL3, CCL4L1, CCL2, ARHGEF5, FASN, THPO, RNASEL, RNASE1, CD40LG, STING1, TLR3, TAP1, GEM, PTGS2, MAVS, GZMB, TLR4, CFH, NR0B1, MBTPS1, CHST10, S1PR2, TXNRD1, TNFRSF10B, TTN, TSC2, HSP90B1, TPM4, IKBKE, TPI1, NUP153, SH2B3, VCAM1, P2RX6, PIWIL1, AURKB, PTPRU, HSD17B6, SQSTM1, USO1, MKNK1, APOL1, STAM, MGAM, HAP1, MOGS, SOCS3, CNBP, CLDN1, XBP1, VWF, VIM, IL1RL1, DNM1L, SERPINA3, RCL1, TIRAP, PIWIL4, NRSN1, TRIM69, EGFLAM, LRG1, CGAS, IL17F, GORASP1, IL33, HELZ2, MAGT1, DHX40, MAPKAP1, DDX50, RNF168, STT3B, MARCHF8, MIF-AS1, PRSS57, MIRLET7E, MIR146A, MIR378A, MIR484, NCF1, MIR744, KIR2DS2, LINC01672, MICA, P2RX5-TAX1BP3, PERCC1, MTCO2P12, WNK1, SMOC1, NOD1, TMED2, ARC, P2RX2, RAB18, MLXIP, CHP1, TPPP, LILRB1, GOLPH3, CERS1, OLFM4, AGPAT1, PRDX4, PROCR, NXF1, EXOC7, GRIP1, SEC14L2, SMUG1, NAAA, APEX2, MYLIP, NPC1L1, TRAT1, UBE2J1, SIRT6, RETREG1, SPHK2, AICDA, RPTOR, TRPV4, HPSE2, TP53, REL, TM7SF2, ILF2, GLS, GDF1, GC, GATA3, GAS6, GAPDH, GABPA, G6PD, IFI6, FLNB, FCGR3B, FCGR3A, FCGR2B, FCGR1A, F5, CXCR3, HGF, HLA-DQA1, IFNB1, IL6R, IL4R, IL3RA, IL2RA, IL2, IL1A, IFNAR1, HLA-E, ICAM3, HSP90AA1, HSF1, HP, HMGCR, HMGB1, ESR2, ESR1, ERN1, KLK3, CAT, BST2, BID, BCL2, BAK1, ARNTL, APRT, CD14, ANXA5, ANXA2, ANGPT1, AKT1, AHR, ADAR, CD9, CD33, ERBB4, CTLA4, ERBB2, ELAVL1, EIF4E, DDX6, DAXX, CYMD, CSF2, CD38, CLDN7, CP, CCR1, CEACAM8, CEBPB, CD69, IL11, ILF3, TLR2, INSR, PRNP, MAP2K1, PRKAB1, PRKAA2, PRKAA1, PPP2R5E, PLAT, PIK3CG, PIK3CD, PIK3CB, PIK3CA, SLC25A3, PAH, P4HB, P2RY2, RAB6A, ABO, REG1A, STAT3, TLR1, TGFB2, TGFB1, TCF7L2, SYT1, SYK, SSB, RELA, SPP1, SMARCB1, SI, SELE, CXCL11, SCD, P2RY1, P2RX7, P2RX5, KLRC1, MATK, CAPRIN1, LTA, TNPO1, KPNA2, KPNB1, KIR2DS5, CXCL9, KIR2DS1, KIR2DL3, ITGAX, IRF7, IRF1, INSRR, MFAP1, MMP2, P2RX4, NFKB1, P2RX3, P2RX1, OAS2, CNOT2, NOS3, NFYA, NFE2L2, MMP9, NCL, MYD88, COX2, COX1, MPL, MNT, H3P19
-
Antiphospholipid Syndrome
Mayo Clinic
Some people develop a red rash with a lacy, net-like pattern. Less common signs and symptoms include: Neurological symptoms. ... Do you have frequent headaches? Have you noticed a red, net-like rash on your wrists or knees?APOH, PPARG, FRMD4A, TSHR, F5, F3, SYCP2L, F2, PTPRO, GPI, ANXA5, TLR4, SH2B2, KLK3, ANXA2, TNF, AGER, CPB2, MTOR, MTHFR, F10, PLG, HT, TFPI, PLAT, SELPLG, ACR, SERPINE1, MOK, LRP8, VWF, HMGB1, VIM, SELP, RAB4A, CCL2, CXCL12, ATXN2, RO60, S100A10, TRIM21, ABCA1, SSB, THBD, TNFRSF1B, NR1I2, ADIPOQ, SH2B3, PROCR, ADAMTS13, TREX1, PTPN22, FOXP3, SLC52A1, IL21, ANXA8, ANXA8L1, PROS1, NOS3, PON1, PLSCR1, HLA-DPB1, GP1BA, GCY, FGA, FCGR2A, F2RL1, EMD, EDN1, DECR1, CRP, CD36, CD1D, CALR, B2M, SERPINC1, AQP4, AMH, HLA-DRB1, HRES1, IDS, MBL2, PF4, PC, SERPINB2, TNFRSF11B, MYD88, MSN, MPL, LPA, IFNG, LGALS9, LCT, CXCL10, CXCL8, IL1B, IGFBP1, IGF1, C20orf181

-
Gerontophobia
Wikipedia
Part of a series on Discrimination General forms Age Class ( Caste ) Physical Disability Education Economic Employment Genetics Hair texture Height Housing Language Looks Race / Ethnicity / Nationality Rank Religion Sanity Sex Sexual orientation Size Skin color Specific forms Social Acephobia Adultism Amatonormativity Anti-albinism Anti-autism Anti-homelessness Anti-intellectualism Anti-intersex Anti-left handedness Anti-Masonry Antisemitism (Judeophobia) Aporophobia Audism Biphobia Clannism Cronyism Drug use Elitism Ephebiphobia Fatism Gerontophobia Heteronormativity Heterosexism HIV/AIDS stigma Homophobia Leprosy stigma Lesbophobia Misandry Misogyny Nepotism Pedophobia Perpetual foreigner Pregnancy Reverse Sectarianism Supremacism Black White Transphobia Non-binary Transmisogyny Vegaphobia Xenophobia Religious Ahmadiyya Atheism Baháʼí Faith Buddhism Catholicism Christianity post–Cold War era Druze Falun Gong Hinduism Persecution Islam Persecution Jehovah's Witnesses Judaism Persecution LDS or Mormon Neopaganism Eastern Orthodox Oriental Orthodox Copts Protestantism Rastafarianism Shi'ism Sufism Sunnism Zoroastrianism Ethnic/national African Albanian American Arab Armenian Australian Austrian Azerbaijani British Canadian Catalan Chechen Chilean Chinese Croat Dutch English Estonian European Filipino Finnish French Georgian German Greek Haitian Hazara Hispanic Hungarian Igbo Indian Indonesian Iranian Irish Israeli Italian Japanese Jewish Khmer Korean Kurdish Malay Manchu Mexican Middle Eastern Mongolian Montenegrin Pakistani Pashtun Polish Portuguese Quebec Romani Romanian Russian Scottish Serb Slavic Somali Soviet Tatar Thai Tibetan Turkish Ukrainian Venezuelan Vietnamese Western Manifestations Blood libel Bullying Compulsory sterilization Counter-jihad Cultural genocide Defamation Democide Disability hate crime Dog-whistle politics Eliminationism Ethnic cleansing Ethnic conflict Ethnic hatred Ethnic joke Ethnocide Forced conversion Freak show Gay bashing Gendercide Genital modification and mutilation Genocide examples Glass ceiling Hate crime Hate group Hate speech online Homeless dumping Indian rolling Lavender scare LGBT hate crimes Lynching Mortgage Murder music Occupational segregation Persecution Pogrom Purge Red Scare Religious persecution Religious terrorism Religious violence Religious war Scapegoating Segregation academy Sex-selective abortion Slavery Slut-shaming Trans bashing Victimisation Violence against women White flight White power music Wife selling Witch-hunt Policies Age of candidacy Blood purity Blood quantum Crime of apartheid Disabilities Catholic Jewish Ethnocracy Ethnopluralism Gender pay gap Gender roles Gerontocracy Gerrymandering Ghetto benches Internment Jewish quota Jim Crow laws Law for Protection of the Nation McCarthyism MSM blood donation restrictions Nonpersons Numerus clausus (as religious or racial quota) Nuremberg Laws One-drop rule Racial quota Racial steering Redlining Same-sex marriage (laws and issues prohibiting) Segregation age racial religious sexual Sodomy law State atheism State religion Ugly law Voter suppression Countermeasures Affirmative action Anti-discrimination law Cultural assimilation Cultural pluralism Diversity training Empowerment Feminism Fighting Discrimination Hate speech laws by country Human rights Intersex rights LGBT rights Masculism Multiculturalism Nonviolence Racial integration Reappropriation Self-determination Social integration Toleration Related topics Allophilia Anti-cultural, anti-national, and anti-ethnic terms Bias Christian privilege Civil liberties Cultural assimilation Dehumanization Diversity Ethnic penalty Eugenics Internalized oppression Intersectionality Male privilege Masculism Medical model of disability autism Multiculturalism Net bias Neurodiversity Oikophobia Oppression Police brutality Political correctness Polyculturalism Power distance Prejudice Prisoner abuse Racial bias in criminal news Racism by country Religious intolerance Second-generation gender bias Snobbery Social exclusion Social model of disability Social stigma Stereotype threat The talk White privilege v t e Gerontophobia is the fear of age-related self-degeneration (similar to Gerascophobia ), or a hatred or fear of the elderly due to memento mori . ... External links [ edit ] AGEISM AND AGING UP: A Q and A with Mariah MedFriendly Age Wave v t e Discrimination General forms Age Caste Class Disability Education Economic Employment Genetic Hair texture Height Housing Language Looks Race / Ethnicity / Nationality Rank Sanity Sex Sexual orientation Size Skin color Social Acephobia Adultism Amatonormativity Anti-albinism Anti-autism Anti-homelessness Anti-intellectualism Anti-intersex Anti-left handedness Anti-Masonry Antisemitism (Judeophobia) Aporophobia Audism Biphobia Clannism Cronyism Drug use Elitism Ephebiphobia Fatism Gerontophobia Heteronormativity Heterosexism HIV/AIDS stigma Homophobia Leprosy stigma Lesbophobia Misandry Misogyny Nepotism Pedophobia Perpetual foreigner Pregnancy Reverse Sectarianism Supremacism Black White Transphobia Non-binary Transmisogyny Vegaphobia Xenophobia Religious Ahmadiyya Atheism Baháʼí Faith Buddhism Catholicism Christianity post–Cold War era Falun Gong Hinduism Persecution Islam Persecution Jehovah's Witnesses Judaism Persecution LDS or Mormon Neopaganism Eastern Orthodox Oriental Orthodox Protestantism Rastafarianism Shi'ism Sufism Zoroastrianism Ethnic/National African Albanian American Arab Armenian Australian Austrian British Canadian Catalan Chilean Chinese Croat Dutch English Estonian European Filipino Finnish French Georgian German Greek Haitian Hazara Hindu Hispanic Hungarian Igbo Indian Indonesian Iranian Irish Israeli Italian Japanese Jewish Khmer Korean Kurdish Malay Manchu Mexican Middle Eastern Mongolian Pakistani Pashtun Polish Portuguese Quebec Romani Romanian Russian Scottish Serb Slavic Somali Soviet Tatar Thai Turkish Ukrainian Venezuelan Vietnamese Western Manifestations Blood libel Bullying Compulsory sterilization Counter-jihad Cultural genocide Defamation Democide Disability hate crime Dog-whistle politics Eliminationism Enemy of the people Ethnic cleansing Ethnic conflict Ethnic hatred Ethnic joke Ethnocide Forced conversion Freak show Gay bashing Gendercide Genital modification and mutilation Genocide examples Glass ceiling Hate crime Hate group Hate speech Homeless dumping Indian rolling Lavender scare LGBT hate crimes Lynching Mortgage Murder music Native American sports mascots Occupational segregation Persecution Pogrom Purge Red Scare Religious persecution Religious terrorism Religious violence Religious war Scapegoating Segregation academy Sex-selective abortion Slavery Slut-shaming Trans bashing Victimisation Violence against women White flight White power music Wife selling Witch-hunt Discriminatory policies Age of candidacy Blood purity Blood quantum Crime of apartheid Disabilities Catholic Jewish Ethnocracy Ethnopluralism Gender pay gap Gender roles Gerontocracy Gerrymandering Ghetto benches Internment Jewish quota Jim Crow laws Law for Protection of the Nation McCarthyism MSM blood donation restrictions Nonpersons Numerus clausus (as religious or racial quota) Nuremberg Laws One-drop rule Racial quota Racial steering Redlining Same-sex marriage (laws and issues prohibiting) Segregation age racial religious sexual Sodomy law State atheism State religion Ugly law Voter suppression Countermeasures Affirmative action Anti-discrimination law Cultural assimilation Cultural pluralism Diversity training Empowerment Feminism Fighting Discrimination Hate speech laws by country Human rights Intersex rights LGBT rights Masculism Multiculturalism Nonviolence Racial integration Reappropriation Self-determination Social integration Toleration Related topics Allophilia Anti-cultural, anti-national, and anti-ethnic terms Bias Christian privilege Civil liberties Cultural assimilation Dehumanization Diversity Ethnic penalty Eugenics Internalized oppression Intersectionality Male privilege Masculism Medical model of disability autism Multiculturalism Net bias Neurodiversity Oikophobia Oppression Police brutality Political correctness Polyculturalism Power distance Prejudice Prisoner abuse Racial bias in criminal news Racism by country Religious intolerance Second-generation gender bias Snobbery Social exclusion Social model of disability Social stigma Stereotype threat The talk White privilege Category
-
Rahman Syndrome
OMIM
The truncated proteins were predicted to have a reduced net charge compared to the wildtype protein, rendering them likely to be less effective in neutralizing negatively charged linker DNA.
-
Felty Syndrome
Orphanet
Neutrophil extracellular chromatin traps (NETs) containing deiminated histones, in complex with bacterial adjuvants, are the most likely antigenic trigger for the production of autoantibodies to deiminated histones.
-
Retinal Dystrophy, Reticular Pigmentary, Of Posterior Pole
OMIM
Variation in the presence of autofluorescent chromophores was observed, with the older sister and the unrelated boy exhibiting a milder and more punctiform hyperautofluorescence of the net, whereas the younger sister showed a more intense hyperautofluorescent pattern.
-
Body Inflation
Wikipedia
ISBN 1-890451-03-7 . ^ More proof that the Net's a weird place: A new book assembles strange Web sites. , The Globe and Mail , 27 April 2000, page R7 ^ The Juicy, Round World of Blueberry Porn ^ a b Allerhand, Rhalou (2008).

-
Lichen Spinulosus
Wikipedia
PMID 2179296 . External links [ edit ] Derm Net NZ Emedicine Thehinhso v t e Diseases of the skin and appendages by morphology Growths Epidermal Wart Callus Seborrheic keratosis Acrochordon Molluscum contagiosum Actinic keratosis Squamous-cell carcinoma Basal-cell carcinoma Merkel-cell carcinoma Nevus sebaceous Trichoepithelioma Pigmented Freckles Lentigo Melasma Nevus Melanoma Dermal and subcutaneous Epidermal inclusion cyst Hemangioma Dermatofibroma (benign fibrous histiocytoma) Keloid Lipoma Neurofibroma Xanthoma Kaposi's sarcoma Infantile digital fibromatosis Granular cell tumor Leiomyoma Lymphangioma circumscriptum Myxoid cyst Rashes With epidermal involvement Eczematous Contact dermatitis Atopic dermatitis Seborrheic dermatitis Stasis dermatitis Lichen simplex chronicus Darier's disease Glucagonoma syndrome Langerhans cell histiocytosis Lichen sclerosus Pemphigus foliaceus Wiskott–Aldrich syndrome Zinc deficiency Scaling Psoriasis Tinea ( Corporis Cruris Pedis Manuum Faciei ) Pityriasis rosea Secondary syphilis Mycosis fungoides Systemic lupus erythematosus Pityriasis rubra pilaris Parapsoriasis Ichthyosis Blistering Herpes simplex Herpes zoster Varicella Bullous impetigo Acute contact dermatitis Pemphigus vulgaris Bullous pemphigoid Dermatitis herpetiformis Porphyria cutanea tarda Epidermolysis bullosa simplex Papular Scabies Insect bite reactions Lichen planus Miliaria Keratosis pilaris Lichen spinulosus Transient acantholytic dermatosis Lichen nitidus Pityriasis lichenoides et varioliformis acuta Pustular Acne vulgaris Acne rosacea Folliculitis Impetigo Candidiasis Gonococcemia Dermatophyte Coccidioidomycosis Subcorneal pustular dermatosis Hypopigmented Tinea versicolor Vitiligo Pityriasis alba Postinflammatory hyperpigmentation Tuberous sclerosis Idiopathic guttate hypomelanosis Leprosy Hypopigmented mycosis fungoides Without epidermal involvement Red Blanchable Erythema Generalized Drug eruptions Viral exanthems Toxic erythema Systemic lupus erythematosus Localized Cellulitis Abscess Boil Erythema nodosum Carcinoid syndrome Fixed drug eruption Specialized Urticaria Erythema ( Multiforme Migrans Gyratum repens Annulare centrifugum Ab igne ) Nonblanchable Purpura Macular Thrombocytopenic purpura Actinic/solar purpura Papular Disseminated intravascular coagulation Vasculitis Indurated Scleroderma / morphea Granuloma annulare Lichen sclerosis et atrophicus Necrobiosis lipoidica Miscellaneous disorders Ulcers Hair Telogen effluvium Androgenic alopecia Alopecia areata Systemic lupus erythematosus Tinea capitis Loose anagen syndrome Lichen planopilaris Folliculitis decalvans Acne keloidalis nuchae Nail Onychomycosis Psoriasis Paronychia Ingrown nail Mucous membrane Aphthous stomatitis Oral candidiasis Lichen planus Leukoplakia Pemphigus vulgaris Mucous membrane pemphigoid Cicatricial pemphigoid Herpesvirus Coxsackievirus Syphilis Systemic histoplasmosis Squamous-cell carcinoma This condition of the skin appendages article is a stub .
-
Diabetic Angiopathy
Wikipedia
Pathophysiology [ edit ] As insulin is required for glucose uptake, hyperglycemia in diabetes mellitus does not result in a net increase in intracellular glucose in most cells.NOS3, AGER, HP, THBS1, SERPINF1, MTHFR, ADCY3, HMOX1, CREM, CASP3, ASS1, FASLG, ADCY8, THBS2, ALB, RELA, VEGFA, FN1, PTK2B, NOS2, CCL2, SPARC, ICAM1, BDKRB1, MOK, EDN1, MPO, ADM, RENBP, AKR1B1, PPARG, PON1, SERPINE1, SMPD1, DPP4, SOD1, TFRC, STIP1, TLR2, ADAMTS13, MIR185, TGFBI, CIP2A, JTB, MIR200C, TCF7L2, SERP1, MIR216B, NOX4, SOD2, MIR146A, TNF, RNF10, MIR130A, TNFRSF1A, TRIB3, MIR126, NOP53, TRAF6, HPSE, VDR, CCDC8, FZD5, ARHGEF5, AOC3, SQSTM1, KLF4, OR10A4, PON2, SGK1, IAPP, GLP1R, GCG, FGF2, EGFR, E2F1, DUSP6, ACE, CMA1, CAT, CANX, ATF3, APOE, AKT1, AHR, AGT, HMGB1, IFIT3, SELL, IGF1, REN, PTEN, MAPK7, MAPK3, PRKCB, PPIA, PLG, TNFRSF11B, MSX2, LGALS3, KLK1, KDR, ITGAM, IRAK1, IGFBP7, H3P7
- Megalencephaly-Capillary Malformation Syndrome MedlinePlus
-
Distal Renal Tubular Acidosis
Orphanet
Distal renal tubular acidosis (dRTA) is a disorder of impaired net acid secretion by the distal tubule characterized by hyperchloremic metabolic acidosis.SLC4A1, ATP6V0A4, ATP6V1B1, WDR72, SLC4A4, CA2, GATA3, OSGEP, EPHA3, NCOA7, MRGPRF, SLC26A7, VAX2, RBFOX2, ALB, SLC26A4, AMH, GYPB, GYPA, FOXI1, CANX, ATP6AP1, GYPE

-
Mutyh Polyposis
GeneReviews
A heterozygous MUTYH pathogenic variant was identified in two of 45 individuals with neuroendocrine tumors (NET) of the pancreas and eight of 160 individuals with adrenocortical carcinomas (ACC) [Pilati et al 2017, Scarpa et al 2017]. Two of 15 probands with familial NET of the small intestine and four of 215 individuals with nonfamilial NET of the small intestine were heterozygous for MUTYH pathogenic variant p.Gly396Asp [Dumanski et al 2017]. It is unclear if a heterozygous MUTYH pathogenic variant is a risk factor for NET or ACC, as the risk of NET or ACC in individuals with biallelic MUTYH pathogenic variants appears to be quite low.








