Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... According to the WHO and UNICEF, deaths attributable to malaria in 2015 were reduced by 60% [77] from a 2000 estimate of 985,000, largely due to the widespread use of insecticide-treated nets and artemisinin-based combination therapies. [74] In 2012, there were 207 million cases of malaria.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
-
Neuroendocrine Tumor
Wikipedia
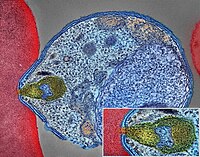
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Party And Play
Wikipedia
A key self-confidence issue for study participants was "body image", a concern that was heightened by the focus on social networking apps on appearance, because on these apps, there is a focus on idealized male bodies that are "toned and muscular". ... "Crystal Ball" . NYMag.com . Retrieved 2015-12-11 . ^ a b McCall, Hannah; Adams, Naomi; Mason, David; Willis, Jamie (2015-11-03). ... Deviant Behavior . 35 (11): 859–884. doi : 10.1080/01639625.2014.897116 . ... Archived from the original on February 6, 2008 . Retrieved 2015-12-11 . ^ "Up to 20 per cent of gay men have tried crystal meth" . ... California State University, Long Beach" . www.academia.edu . Retrieved 2015-12-11 . "The Internet and Drug Markets -Study" (PDF) .

-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Uric Acid Concentration, Serum, Quantitative Trait Locus 1
OMIM
Evidence for both an increased rate of uric acid synthesis and an impaired net elimination of uric acid by the kidney has been advanced. ... The Q141K allele was associated with a significantly increased risk of hyperuricemia (OR of 2.06, p = 1.53 x 10(-11)) and gout (OR of 2.23; p = 5.54 x 10(-11)). ... Autosomal dominant form Lab - Increased rate of uric acid synthesis - Impaired net elimination of uric acid by the kidney - Hyperuricemia Skin - Urate tophi ▲ Close
-
Pancreatic Neuroendocrine Tumor
Wikipedia
PanNETs are a type of neuroendocrine tumor , representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign , while some are malignant . ... Up to 60% [ medical citation needed ] of PanNETs are nonsecretory or nonfunctional, in which there is no secretion, or the quantity or type of products, such as pancreatic polypeptide (PPoma), chromogranin A, and neurotensin , do not cause a clinical syndrome although blood levels may be elevated. [10] In total, 85% of PanNETs have an elevated blood marker. [2] Functional tumors are often classified by the hormone most strongly secreted, for example: gastrinoma : the excessive gastrin causes Zollinger–Ellison syndrome (ZES) with peptic ulcers and diarrhea insulinoma : [11] hypoglycemia occurs with concurrent elevations of insulin , proinsulin and C peptide [12] glucagonoma : the symptoms are not all due to glucagon elevations, [12] and include a rash , sore mouth, altered bowel habits, venous thrombosis , and high blood glucose levels [12] VIPoma , producing excessive vasoactive intestinal peptide , which may cause profound chronic w atery d iarrhea and resultant dehydration , h ypokalemia , and a chlorhydria (WDHA or pancreatic cholera syndrome) somatostatinoma : these rare tumors are associated with elevated blood glucose levels, achlorhydria , cholelithiasis , and diarrhea [12] less common types include ACTHoma , CRHoma , calcitoninoma , GHRHoma , GRFoma , and parathyroid hormone–related peptide tumor In these various types of functional tumors, the frequency of malignancy and the survival prognosis have been estimated dissimilarly, but a pertinent accessible summary is available. [13] Diagnosis [ edit ] Because symptoms are non-specific, diagnosis is often delayed. [14] Measurement of hormones including pancreatic polypeptide , gastrin , proinsulin , insulin , glucagon , and vasoactive intestinal peptide can determine if a tumor is causing hypersecretion. [14] [15] Multiphase CT and MRI are the primary modalities for morphologic imaging of PNETs. ... However, morphological imaging alone is not sufficient for a definite diagnosis [14] [16] On biopsy , immunohistochemistry is generally positive for chromogranin and synaptophysin . [17] Genetic testing thereof typically shows altered MEN1 and DAXX / ATRX . [17] Staging [ edit ] The 2010 WHO classification of tumors of the digestive system grades all the neuroendocrine tumors into three categories, based on their degree of cellular differentiation (from well-differentiated "NET G1" through to poorly-differentiated "NET G3"). ... Combinations of several medicines have been used, such as doxorubicin with streptozocin and fluorouracil (5-FU) [12] and capecitabine with temozolomide. [ citation needed ] Although marginally effective in well-differentiated PETs, cisplatin with etoposide has some activity in poorly differentiated neuroendocrine cancers (PDNECs), [12] particularly if the PDNEC has an extremely high Ki-67 score of over 50%. [8] : 30 Several targeted therapy agents have been approved in PanNETs by the FDA based on improved progression-free survival (PFS): everolimus (Afinitor) is labeled for treatment of progressive neuroendocrine tumors of pancreatic origin in patients with unresectable, locally advanced or metastatic disease. [20] [21] The safety and effectiveness of everolimus in carcinoid tumors have not been established. [20] [21] sunitinib (Sutent) is labeled for treatment of progressive, well-differentiated pancreatic neuroendocrine tumors in patients with unresectable locally advanced or metastatic disease. [22] [23] Sutent also has approval from the European Commission for the treatment of 'unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors with disease progression in adults'. [24] A phase III study of sunitinib treatment in well differentiated pNET that had worsened within the past 12 months (either advanced or metastatic disease) showed that sunitinib treatment improved progression-free survival (11.4 months vs. 5.5 months), overall survival , and the objective response rate (9.3% vs. 0.0%) when compared with placebo. [25] Genetics [ edit ] Pancreatic neuroendocrine tumors may arise in the context of multiple endocrine neoplasia type 1 , Von Hippel–Lindau disease , neurofibromatosis type 1 (NF-1) or tuberose sclerosis (TSC) [26] [27] Analysis of somatic DNA mutations in well-differentiated pancreatic neuroendocrine tumors identified four important findings: [28] [6] as expected, the genes mutated in NETs, MEN1 , ATRX , DAXX , TSC2 , PTEN and PIK3CA , [28] are different from the mutated genes previously found in pancreatic adenocarcinoma . [29] [30] one in six well-differentiated pancreatic NETs have mutations in mTOR pathway genes, such as TSC2 , PTEN and PIK3CA . [28] The sequencing discovery might allow selection of which NETs would benefit from mTOR inhibition such as with everolimus , but this awaits validation in a clinical trial . mutations affecting a new cancer pathway involving ATRX and DAXX genes were found in about 40% of pancreatic NETs. [28] The proteins encoded by ATRX and DAXX participate in chromatin remodeling of telomeres ; [31] these mutations are associated with a telomerase -independent maintenance mechanism termed ALT (alternative lengthening of telomeres) that results in abnormally long telomeric ends of chromosomes . [31] ATRX / DAXX and MEN1 mutations were associated with a better prognosis . [28] References [ edit ] ^ Burns WR, Edil BH (March 2012). ... National Comprehensive Cancer Network, Inc. November 11, 2014 . Retrieved December 25, 2014 . ^ Pancreatic Neuroendocrine Tumors (Islet Cell Tumors) Treatment (PDQ®) National Cancer Institute [2] ^ Jensen RT, Berna MJ, Bingham DB, Norton JA (October 2008).MEN1, ATRX, DAXX, ELK3, TP53, EPHB1, SLC6A2, CEACAM5, CEACAM7, UQCRFS1, DHDDS, CHPT1, RALBP1, CIB1, SEMA4D, RIPK1, CXCR4, VEGFA, TTR, GNA12, TSC2, TFE3, CDKN1B, PSG2, POMC, MYCN, CEACAM3, GRN, MUC16

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Afterdepolarization
Wikipedia
They are due to elevated cytosolic calcium concentrations, classically seen with digoxin toxicity. [3] [4] The overload of the sarcoplasmic reticulum may cause spontaneous Ca 2+ release after repolarization, causing the released Ca 2+ to exit the cell through the 3Na + /Ca 2+ -exchanger. This results in a net depolarizing current. The classical feature is Bidirectional ventricular tachycardia . ... Neuroscience. ^ Katzung, B: Basic and Clinical Pharmacology (10th ed.), chapter 14: "Agents Used in Cardiac Arrhythmias", The McGraw-Hill Companies, 2007, ISBN 978-0-07-145153-6 ^ Lilly, L: "Pathophysiology of Heart Disease", chapter 11: "Mechanisms of Cardiac Arrhthmias", Lippencott, Williams and Wilkens, 2007 This article about a medical condition affecting the circulatory system is a stub .
-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Steroid Diabetes
Wikipedia
Mechanism [ edit ] Glucocorticoids oppose insulin action and stimulate gluconeogenesis , especially in the liver , resulting in a net increase in hepatic glucose output. ... Criteria [ edit ] The diagnostic criteria for steroid diabetes are those of diabetes (fasting glucoses persistently above 125 mg/dl (7 mM) or random levels above 200 mg/dl (11 mM)) occurring in the context of high-dose glucocorticoid therapy.
-
Esophageal Food Bolus Obstruction
Wikipedia
Rarely disorders of movement of the esophagus, such as nutcracker esophagus , can predispose to food bolus obstruction. [11] Treatment [ edit ] Conservative [ edit ] In an emergency room setting, someone with food bolus obstruction may be observed for a period to see if the food bolus passes spontaneously. ... ] and the use of large-bore tubes inserted into the esophagus to forcefully lavage it. [17] [ unreliable medical source? ] Endoscopic [ edit ] The Roth net can be inserted through the endoscope to remove pieces of the obstructed food. ... Traditional endoscopic techniques involved the use of an overtube, a plastic tube inserted into the esophagus prior to the removal of the food bolus, in order to reduce the risk of aspiration into the lungs at the time of endoscopy. [7] However, the "push technique", which involves insufflating air into the esophagus, and gently pushing the bolus toward the stomach instead, has emerged as a common and safe way of removing the obstruction. [7] [18] Other tools may be used to remove food boluses. The Roth Net is a mesh net that can be inserted through the endoscope, and opened and closed from the outside; it can be used to retrieve pieces of obstructed food.

-
Occupational Heat Stress
Wikipedia
Harvard Health . Retrieved 2020-11-11 . ^ Kenny, Glen P.; Yardley, Jane; Brown, Candice; Sigal, Ronald J.; Jay, Ollie (2010-07-13). ... Retrieved 2020-11-11 . ^ Publishing, Harvard Health. ... Retrieved 2020-11-03 . ^ "OSHA-NIOSH Heat Safety Tool App | NIOSH | CDC" . www.cdc.gov . 2020-02-20 . ... Retrieved 2020-11-11 . ^ "OSHA's Campaign to Prevent Heat Illness in Outdoor Workers" . www.osha.gov . ... Retrieved 2020-11-03 . ^ Parsons, Ken (2013-01-01).
-
West Nile Fever
Wikipedia
Clinical Infectious Diseases . 42 (11): 1527–35. doi : 10.1086/503841 . ... "Epidemiology and transmission dynamics of West Nile virus disease" . Emerging Infect. Dis . 11 (8): 1167–73. doi : 10.3201/eid1108.050289a . ... "West Nile virus infection and conjunctive exposure" . Emerging Infect. Dis . 11 (10): 1648–9. doi : 10.3201/eid1110.040212 . ... Retrieved 2018-11-28 . ^ Tyler KL, Pape J, Goody RJ, Corkill M, Kleinschmidt-DeMasters BK (February 2006). ... Emerging Infectious Diseases . 19 (11): 1836–8. doi : 10.3201/eid1911.130768 .CCR5, ERVK-32, ROBO3, MAVS, DDX58, PLAAT4, IFIT2, ERVK-6, STAT1, SPP1, OAS1, IL1B, IFNB1, RNASEL, CASP8, HLA-DRB1, PELI1, SELENBP1, ARHGEF2, LRRFIP1, NAMPT, TRAIP, RIPK3, SEC14L2, CSF1R, LAMP3, ERVW-1, FOXP3, ZMYND10, DDX56, CCR7, VCP, CDKN2A, IFIH1, DHX58, ZBP1, HAVCR2, PIK3IP1, NLRP3, TNFRSF13C, TRIM6, RBM45, CCR2, ERVK-20, ERVK-18, VAMP8, TNFRSF1A, IFNA1, TNF, IFNA13, HLA-DQA1, IL1A, HLA-C, IL10, IL17A, IL18, IRF3, IRF5, KIR2DL2, KIR3DL1, KIR3DS1, LSAMP, CD180, SMAD4, MMP9, HLA-A, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PZP, GLS, CASP1, SNCA, GEM, DDX3X, TAP1, TLR3, ATF4

-
Quartan Fever
Wikipedia
Spraying is a method in multiple regions and to control epidemics. [10] Nets treated with insecticide are effective in preventing mosquito contact for three years. ... When removing any water-filled containers from the surrounding area the mosquito life cycle is halted and acts as a method to reduce mosquito population within the surrounding area. [11] Clothing can act as a physical barrier to prevent exposure of flesh for mosquitoes to feed on, treating beds and clothing with insecticides/repellents can further reduce chances of infected mosquitoes from biting and passing quartan fever to individuals. [10] Avoiding areas which have high mosquito populations, specifically for quartan fever the P. malariae strain. [10] Avoiding travelling to regions which have a sub-tropic climate to prevent infection and developing quartan fever. [10] Implementing the sugar baiting method aids in reducing the population of Anopheles mosquitoes, and ultimately reducing the likelihood of catching quartan fever. ... "Attractive toxic sugar bait (ATSB) methods decimate populations of Anopheles malaria vectors in arid environments regardless of the local availability of favoured sugar-source blossoms" . Malaria Journal . 11 : 31. doi : 10.1186/1475-2875-11-31 . ... PMID 22297155 . v t e Malaria Biology Malaria Cerebral Quartan fever Blackwater fever Pregnancy-associated Plasmodium biology life cycle vivax falciparum ovale malariae knowlesi Anopheles mosquito Lifecycle Schizont Merozoite Hypnozoite Gametocyte Control and prevention Public health DDT Mosquito net Malaria prophylaxis Mosquito control Sterile insect technique Genetic resistance Duffy antigen Sickle-cell anaemia Thalassemia G6PDH deficiency Malaria vaccine RTS,S Diagnosis and treatment Diagnosis of malaria Malaria culture Blood film Malaria antigen detection tests Antimalarials Artemisinin Mefloquine Proguanil Society and malaria Diseases of poverty Millennium Development Goals History of malaria Roman fever National Malaria Eradication Program World Malaria Day Epidemiology Malaria and the Caribbean Malaria Atlas Project Organisations Malaria Consortium Against Malaria Foundation Bill & Melinda Gates Foundation Imagine No Malaria Malaria No More Africa Fighting Malaria African Malaria Network Trust South African Malaria Initiative African Leaders Malaria Alliance Amazon Malaria Initiative The Global Fund to Fight AIDS, Tuberculosis and Malaria Medicines for Malaria Venture Category v t e Topics in epidemiology Branches of epidemiology by physiology/disease: Cognitive epidemiology Neuroepidemiology Psychiatric epidemiology Veterinary epidemiology by methodological approach: Biostatistics Disease informatics E-epidemiology Environmental epidemiology Economic epidemiology Clinical epidemiology Computational epidemiology Conflict epidemiology Genetic epidemiology Life course approach Meta-analysis Molecular epidemiology Molecular pathological epidemiology Nutritional epidemiology Paleoepidemiology Pharmacoepidemiology Social epidemiology Spatial epidemiology Tele-epidemiology Surveillance epidemiology ( Clinical surveillance ) Global epidemiology of: Asthma Attention deficit hyperactivity disorder Autism Bed bugs Binge drinking Cancer breast Depression Diabetes mellitus Domestic violence Hepatitis C Herpes HIV/AIDS Leprosy Malnutrition Mental disorders child Motor vehicle collisions Obesity childhood Periodontal diseases Pneumonia Representations Schizophrenia Snakebites Suicide Syphilis Teenage pregnancy Tuberculosis Epidemiology journals General American Journal of Epidemiology Canadian Journal of Epidemiology and Biostatistics Global Epidemiological News Epidemiologic Reviews Epidemiology International Journal of Epidemiology Annals of Epidemiology Journal of Epidemiology and Community Health European Journal of Epidemiology Emerging Themes in Epidemiology Epidemiologic Perspectives and Innovations Eurosurveillance Speciality Cancer Causes & Control Cancer Epidemiology Biomarkers and Prevention Epidemiology and Infection Genetic Epidemiology Infection Control and Hospital Epidemiology Journal of Clinical Epidemiology Paediatric Perinatal Epidemiology Pharmacoepidemiology and Drug Safety Preventive Medicine

-
Lymphatic Filariasis
Wikipedia
"Filariasis and lymphoedema" . Parasite Immunology . 31 (11): 664–72. doi : 10.1111/j.1365-3024.2009.01133.x . ... "Lymphatic Filariasis" . Archived from the original on 11 May 2012 . Retrieved 24 March 2012 . ^ Niwa S. ... Archived from the original on 15 February 2015 . Retrieved 11 February 2015 . ^ a b "Lymphatic filariasis. ... Archived (PDF) from the original on 2016-03-04 . Retrieved 2015-11-28 . ^ a b c d e f The Carter Center. ... International Journal of Environmental Research and Public Health . 11 (5): 5133–6. doi : 10.3390/ijerph110505133 .GPT2, THAS, IL10, HSPD1, TLR9, TLR2, LMLN, GGTLC1, TMPRSS13, NARS2, RBM33, TLR1, HSPA14, NBEAL2, HERPUD1, NR0B2, VEGFA, TXN, TNFRSF1B, TLR4, CISH, CTLA4, MSMB, EDN1, FOXC2, FLT4, HGF, IGHG3, IL3, MST1, TGFB1, NARS1, PRL, PTEN, PTPN6, SLA, SMS, LOC102724197

-
Leishmaniasis
Wikipedia
Prevention [ edit ] Using insecticide can reduce phlebotomine sandfly numbers which in turn reduces the risk of cutaneous leishmaniasis infection. [18] Leishmaniasis can be partly prevented by using nets treated with insecticide while sleeping. [2] To provide good protection against sandflies, fine mesh sizes of 0.6 mm or less are required, but a mosquito net with 1.2mm mesh will provide a limited reduction in the number of sandfly bites. [19] Finer mesh sizes have the downside of higher cost and reduced air circulation which can cause overheating. Many Phlebotomine sandfly attacks occur at sunset rather than at night, so it may also be useful to put nets over doors and windows or to use insect repellents . ... The Journal of Antimicrobial Chemotherapy . 67 (11): 2576–97. doi : 10.1093/jac/dks275 . ... Expert Review of Anti-Infective Therapy . 11 (1): 79–98. doi : 10.1586/eri.12.148 . ... External links [ edit ] Wikipedia's health care articles can be viewed offline with the Medical Wikipedia app . Classification D ICD - 10 : B55 ICD - 9-CM : 085 MeSH : D007896 DiseasesDB : 3266 External resources MedlinePlus : 001386 eMedicine : emerg/296 Patient UK : Leishmaniasis Wikimedia Commons has media related to Leishmaniasis .TNF, IFNG, IL10, IL6, ARG1, IL18, CRP, TNFRSF18, MCL1, HSPA4, IL1B, SLC11A1, CXCL10, NLRP3, IL17A, TLR2, CCR5, TLR4, IL32, PRDX2, LEP, TGFB1, CD274, FCN2, CD163, MTOR, HM13, IL4, BCL2, BAX, LMLN, IGF1, HIF1A, ANXA1, VDR, UNG, TAM, NR0B2, EZR, ADA, TLR3, STAT1, MAPK3, MAPK4, EIF2AK2, PSG5, PSMD7, PTHLH, PTPN1, PTPN2, PTPN6, RPA1, RPS6, CCL2, CCL8, CXCL11, SLC1A5, SLC1A7, SNAP25, SOAT1, SPP1, TP63, EIF2S2, CDK5R1, GOPC, FOXP3, HSPA14, CD244, TOLLIP, FBLIM1, MSTO1, FBXW7, ACSS2, PDXP, SLC52A2, ALDH1A2, TMPRSS13, DCLK3, IL33, CDCA5, PWAR1, ARMH1, HNP1, CCR2, UPK3B, DLL1, SGSM3, NOX1, PABPC1, NR1I2, SPHK1, EIF2B4, EIF2B2, PRKAB1, HSPB3, SLC7A6, ARHGEF2, AIM2, H6PD, RABEPK, LANCL1, TNFSF13B, EBNA1BP2, CD160, GABARAPL2, GABARAPL1, PRDX5, POLR1A, MAPK1, NOS2, PRKAA2, PRKAA1, CST3, CTLA4, CTSB, CTSL, CYP51A1, DDT, DHFR, DPAGT1, DPP4, DSPP, DUSP4, EEF1B2, EEF2, EGFR, EIF2B1, F2R, FCGR2A, FECH, FLI1, CPB1, CCR7, LRBA, ATR, AKT1, ALDH1A1, APEX1, APRT, AQP1, ATM, ATP2A3, ATP2B4, PRDM1, CD69, BRCA1, CAPN1, CD1A, CD28, CD86, CD40, CD40LG, CD44, FPR2, G6PD, GAPDH, CYTB, MNAT1, CD200, MPG, MPL, MPST, MRC1, MSMB, MST1, AHR, MFAP1, PAEP, PHB, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PLP1, PNOC, MAP3K10, MBL2, GCHFR, IFNB1, GCK, GTF3C1, HLA-C, HMOX1, HSPD1, IFN1@, IFNA1, IFNA13, IL1A, LTA, IL9, IL12A, IL12RB1, IL13, ITGA4, ITGAL, JAK2, RPSA, H3P28

-
Pancreatic Cancer
Wikipedia
These cancerous cells have the ability to invade other parts of the body. [9] A number of types of pancreatic cancer are known. [10] The most common, pancreatic adenocarcinoma , accounts for about 90% of cases, [11] and the term "pancreatic cancer" is sometimes used to refer only to that type. [10] These adenocarcinomas start within the part of the pancreas that makes digestive enzymes . [10] Several other types of cancer, which collectively represent the majority of the nonadenocarcinomas, can also arise from these cells. [10] About 1–2% of cases of pancreatic cancer are neuroendocrine tumors , which arise from the hormone-producing cells of the pancreas. [10] These are generally less aggressive than pancreatic adenocarcinoma. [10] Signs and symptoms of the most-common form of pancreatic cancer may include yellow skin , abdominal or back pain , unexplained weight loss , light-colored stools , dark urine, and loss of appetite . [1] Usually, no symptoms are seen in the disease's early stages, and symptoms that are specific enough to suggest pancreatic cancer typically do not develop until the disease has reached an advanced stage. [1] [2] By the time of diagnosis, pancreatic cancer has often spread to other parts of the body. [10] [12] Pancreatic cancer rarely occurs before the age of 40, and more than half of cases of pancreatic adenocarcinoma occur in those over 70. [2] Risk factors for pancreatic cancer include tobacco smoking , obesity , diabetes , and certain rare genetic conditions. [2] About 25% of cases are linked to smoking, [3] and 5–10% are linked to inherited genes . [2] Pancreatic cancer is usually diagnosed by a combination of medical imaging techniques such as ultrasound or computed tomography , blood tests, and examination of tissue samples ( biopsy ). [3] [4] The disease is divided into stages , from early (stage I) to late (stage IV). [12] Screening the general population has not been found to be effective. [13] The risk of developing pancreatic cancer is lower among nonsmokers, and people who maintain a healthy weight and limit their consumption of red or processed meat . [5] Smokers' chances of developing the disease decrease if they stop smoking and almost return to that of the rest of the population after 20 years. [10] Pancreatic cancer can be treated with surgery, radiotherapy , chemotherapy , palliative care , or a combination of these. [1] Treatment options are partly based on the cancer stage. [1] Surgery is the only treatment that can cure pancreatic adenocarcinoma, [12] and may also be done to improve quality of life without the potential for cure. [1] [12] Pain management and medications to improve digestion are sometimes needed. [12] Early palliative care is recommended even for those receiving treatment that aims for a cure. [14] In 2015, pancreatic cancers of all types resulted in 411,600 deaths globally. [8] Pancreatic cancer is the fifth most-common cause of death from cancer in the United Kingdom, [15] and the third most-common in the United States. [16] The disease occurs most often in the developed world, where about 70% of the new cases in 2012 originated. [10] Pancreatic adenocarcinoma typically has a very poor prognosis; after diagnosis, 25% of people survive one year and 5% live for five years. [6] [10] For cancers diagnosed early, the five-year survival rate rises to about 20%. [17] Neuroendocrine cancers have better outcomes; at five years from diagnosis, 65% of those diagnosed are living, though survival varies considerably depending on the type of tumor. [10] Contents 1 Types 1.1 Exocrine cancers 1.2 Neuroendocrine 2 Signs and symptoms 2.1 Other findings 2.2 Symptoms of spread 3 Risk factors 3.1 Alcohol 4 Pathophysiology 4.1 Precancer 4.2 Invasive cancer 4.3 PanNETs 5 Diagnosis 5.1 Histopathology 5.2 Staging 5.2.1 Exocrine cancers 5.2.2 PanNETs 6 Prevention and screening 7 Management 7.1 Exocrine cancer 7.1.1 Surgery 7.1.2 Chemotherapy 7.1.3 Radiotherapy 7.2 PanNETs 7.3 Palliative care 8 Outcomes 9 Distribution 9.1 PanNETs 10 History 10.1 Recognition and diagnosis 10.2 Surgery 11 Research directions 12 See also 13 References 14 External links Types [ edit ] The pancreas has many functions, served by the endocrine cells in the islets of Langerhans and the exocrine acinar cells . ... They are being detected at a greatly increased rate as CT scans become more powerful and common, and discussion continues as how best to assess and treat them, given that many are benign. [24] Neuroendocrine [ edit ] Main article: Pancreatic neuroendocrine tumor The small minority of tumors that arise elsewhere in the pancreas are mainly pancreatic neuroendocrine tumors (PanNETs). [25] Neuroendocrine tumors (NETs) are a diverse group of benign or malignant tumors that arise from the body's neuroendocrine cells , which are responsible for integrating the nervous and endocrine systems. NETs can start in most organs of the body, including the pancreas, where the various malignant types are all considered to be rare . ... This creates a tumor microenvironment that is short of blood vessels (hypovascular) and so of oxygen ( tumor hypoxia ). [2] It is thought that this prevents many chemotherapy drugs from reaching the tumor, as one factor making the cancer especially hard to treat. [2] [3] Cancer type Relative incidence [11] Microscopy findings [11] Micrograph Immunohistochemistry markers [11] Genetic alterations [11] Pancreatic ductal adenocarcinoma (PDAC) 90% Glands and desmoplasia Aberrant p53 SMAD4 loss MUC1 MSLN CA19-9 p53 SMAD4 KRAS p16 Pancreatic acinar cell carcinoma (ACC) 1% to 2% Granular appearance Trypsin Chymotrypsin Lipase p53 SMAD4 APC ARID1A GNAS Adenosquamous carcinoma 1% to 4% [53] Combination of gland-like cells and squamous epithelial cells. ... Typical sites for metastatic spread (stage IV disease) are the liver, peritoneal cavity and lungs , all of which occur in 50% or more of fully advanced cases. [58] PanNETs [ edit ] The 2010 WHO classification of tumors of the digestive system grades all the pancreatic neuroendocrine tumors (PanNETs) into three categories, based on their degree of cellular differentiation (from "NET G1" through to the poorly differentiated "NET G3"). [19] The U.S.BRCA2, TP53, CDKN2A, STK11, KRAS, BRCA1, SMAD4, EP300, PALB2, ABO, NR5A2, MYC, CTNNB1, TNF, MMP2, AKT2, APC, PDX1, PTEN, PTGS2, STAT3, ATM, KDR, CXCL8, TYMS, SST, TNFSF10, TERT, EGFR, TGFB1, HIF1A, MSLN, CD44, SPINK1, MMP9, PPARG, CD24, LDHA, PLAU, SOD2, PALLD, SSTR2, SLC29A1, MAP2K4, DPYD, TGM2, AXL, IL24, IFNA1, PTCH1, EPCAM, NR4A1, TFPI2, CDH1, LINC00673, NRP1, RAP1GAP, MEN1, RB1, VHL, WT1, KLF5, TYMP, PTHLH, GADD45A, FGF13, CCL20, EFEMP1, CFLAR, AHR, TNFRSF10B, SSTR1, FXYD3, TP63, BAP1, FOXP1, ANXA10, DAXX, NFKBIA, TSC2, RNF43, BUB1, VEGFB, MIR1179, SSTR3, IFNA2, RABL3, SUGCT, CNR1, IFNA5, CNR2, ETAA1, CLPTM1, SYCP1, RBBP8, GAGE1, CCAT1, BCL2L1, BCAR1, LY6E, ZNRF3, ATRX, COL18A1, CLPTM1L, DAB2, SBF2, HSPB2, ERBB2, CCKBR, HRAS, HSPB1, HSP90AA1, LINC01829, PPFIBP1, EPHB2, PIK3CA, CCK, HGF, ERBB3, MIA, PDHX, PSCA, SIRT1, HSPB3, H3P10, PIM3, NOTCH1, CEACAM5, EGF, SMUG1, PIK3CB, PARP1, MAPK1, MAPK8, MTCO2P12, GPC1, CDK4, HDAC9, CEACAM3, ABCG2, ACTB, CEACAM7, LINC01088, MAPK3, NDRG1, PSG2, YAP1, PIK3CG, TAFA5, SLC19A1, SPARC, PRSS1, PROM1, PIK3CD, HP, GLI1, MAP2K7, F3, NGF, ZEB1, FBXW7, NFE2L2, TEX11, ITGB1, VEGFA, GABPA, BICD1, ANXA2, CD82, GAST, DCLK1, COX2, CCND1, XIAP, MET, BACH1, S100A4, LGALS1, MDM2, DPP6, MUC16, FOXM1, MIR21, SLC39A4, MIR221, IFNG, EZH2, LINC00964, AKT1, ALB, LDLRAD4, C9, MUC5AC, SETD2, CXCR4, VIM, MUC4, CD274, LEPQTL1, XRCC1, IGF1, MUC1, IGF1R, CXCL12, IL1B, IL6, BCL2, HNF1A, SCT, TWIST1, CCN2, TLR4, DNMT1, AURKA, CAV1, PLK1, CASP3, PKM, MTHFR, LGALS3, MTOR, BRAF, PRKAA1, MIR301A, AGER, HOTAIR, EPHA2, MCL1, VTCN1, COL11A2, RAC1, DUSP6, CDKN2B, CRP, MAPK14, RUNX3, MALAT1, FN1, MIR155, MIR210, ERCC2, DCK, AGR2, CFTR, GDF15, CDKN1A, ADIPOQ, PRKN, MIR200C, TIMP1, KIF20A, PTF1A, PSMD9, TGFBR2, TICAM2, RRM1, MIR137, MIR142, ALOX5, MLH1, MIR29A, SOCS3, IFI27, SRC, GLP1R, PRKAA2, SP1, SOX2, TMED7-TICAM2, HMGA1, SOAT1, H3P23, PAK4, CHEK1, ZNRD2, BMI1, HMGA2, RALBP1, PALD1, TMED7, TLR7, BSG, DCTN6, WNT5A, EPAS1, WEE1, POSTN, BNIP3, MST1, FGFR1, ATN1, MST1R, GSK3B, PRKAB1, CD47, NFATC2, HSPA5, FANCG, PVT1, SHH, MBD1, ETS1, NPTX2, ABCB1, SLC2A1, MUC2, IFNA13, MMP7, MRC1, IGF2, CCN1, NQO1, LUM, MDK, IL10, PTPA, ELAVL2, POU5F1, FLT1, CXCR2, GSTT1, AIMP2, DKK3, MIR23A, KLF10, CRK, MIR214, MUL1, MIR203A, MIR200B, MIR195, APEX1, GRAP2, MIR145, CDKN1B, MIR141, RIPK1, TNFRSF6B, NR1I2, LINC01194, TGFA, THBS1, ALDH1A1, POLDIP2, MICA, NES, HOTTIP, UCA1, POU5F1P4, MIR34A, POU5F1P3, RNF19A, CASP8, CBLL2, CTLA4, AHSA1, RUNX2, ERCC1, OGG1, FANCC, ZG16B, DNER, NTRK1, FGF2, F2RL1, MMP14, FOXO3, CD276, NUPR1, RMC1, MSH2, AFAP1-AS1, DCC, LIF, MIR10B, HDAC1, HDAC2, HMGB1, ACTN4, HMOX1, ADM, ADRB2, IAPP, ICAM1, GDNF, MIR96, MIR25, IL1A, IL2, MIR223, MIR212, MIR205, ANXA1, MIR148A, MMRN1, MIR143, LCN2, MIR132, REG3A, TMEM97, UQCRFS1, STAT1, RET, TGFBI, CDC42, DPP4, CDK2, KLF4, CTNND1, IL32, DSPP, BAG3, CUX1, VDR, TAZ, PIM1, PCNA, SOX9, SEMA5A, XPO1, MAP2K1, SUB1, PRKD1, SPP1, JUN, TOP2A, MLRL, XIST, RAF1, IL4, IL4R, VEGFC, MIR222, MIR216A, NAT2, IL13, MOK, RAD51, TP73, JAK2, MIR23B, MIR29C, IGFBP3, MIR506, SMARCA1, SLC5A5, H3P9, COMMD3-BMI1, SNAI1, HK2, HLA-A, SKP2, SSTR5, FOXA2, SDC1, HSF1, CCL5, MIR27A, MIR494, MIR429, TNC, RRM2, MIR335, MIR17HG, TRA, RREB1, ROCK1, MIR30A, L1CAM, TEAD1, NCOA3, IKZF3, STMN1, BHLHE40, ESRP1, NT5C3A, IL22, PLEC, NXT1, SLC22A3, IL17B, ZEB2, NF2, NFATC1, TSPAN1, CIB1, CACYBP, MRPL28, PDPN, NFKB1, WWTR1, IGF2BP3, BRD4, HPSE, NRAS, NTSR1, COPS5, SERPINE1, PAK1, SIRT3, CDK14, PARPBP, PACC1, MSX2, TP53INP1, SOCS1, LEP, TBC1D9, MIR107, LGALS4, DUSP28, MTA1, PRKCA, UPRT, TM4SF1, LPAR2, SMAD2, MAGT1, MUC13, REG4, VMP1, LONP1, EHMT1, MMP1, FTO, TRPM8, GAS5, NDRG2, KEAP1, ACKR3, SLC12A9, ADAM9, NAT1, CDK6, GEM, GCG, CALR, CDKN3, FGFR2, ADH1C, CCR6, GLS, FHIT, CSF2, FOS, ALDH2, ACVR1B, FKBP5, FOXO1, ALPP, ALPI, GATA6, CETN1, ASS1, TIMM8A, CLDN7, F2R, TSACC, GPRC5A, ADH1B, HULC, FOSB, CASR, PCLAF, SMARCA4, DECR1, CDX2, CDK5, EPHX2, RCBTB1, FSCN1, SOX4, FLT3, MIR663A, LGALS9, UGT1A7, MIR217, CEL, CYP17A1, CLDN1, TRPM7, ISG15, CLOCK, MIR345, SET, CDK7, MIR200A, MIR216B, JUNB, JUND, ADAMTS1, KL, ITGAM, SERPINF1, SFTPD, SHC1, SELE, ITGA2, HDAC3, ARNTL, PBK, TUG1, SLC16A1, PDGFRB, DCN, RGN, KISS1, CHI3L1, SREBF1, IQGAP1, BSND, FGF7, ZNF35, VASH2, YY1, ESR1, AGT, PEAK1, TFF1, TFRC, NOTCH3, EZR, COX8A, FBP1, CLDN4, HAVCR2, VAV1, CRYZ, FASN, TIMP2, MUC6, TSPAN8, MYB, MIR196B, TXN, MIR330, ATF2, MIR31, IL33, AGTR1, VSIR, CES2, LOXL2, TNFRSF10C, PGK1, SSTR4, ABCC3, PEBP1, CEMIP, CLIC1, EGLN3, ADRA1A, PRDM14, HHIP, SLC52A2, MIR29B1, CDCP1, ADRA2B, MIR29B2, TAM, KLK7, MAP3K1, CCR7, ALCAM, MAP3K7, MGMT, MIF, CXCL10, EDNRA, NEK2, RASA1, TPX2, NEAT1, EEF1B2P2, PRMT1, GJB2, KDM1A, APLP2, CXCR6, RBM14-RBM4, MIR146A, GJA1, KANK2, NOX4, REG1A, USP22, MIR150, CA9, HTC2, MAP2K2, CD40, GGT1, GPR42, DKK1, MIR15B, E2F1, PDCD4, FAS, AREG, HHLA2, GOLGA6A, HPGDS, BHLHE22, HNRNPA2B1, MCAT, GOT1, SLCO1B3, RGCC, EIF4E, LILRB1, MIR106B, NRG1, PTK2, PHGDH, PWAR1, CCKAR, ARF6, GSTP1, MSH6, MARK2, EBI3, IGFBP1, S100A8, CDK1, S100A9, RBPJ, S100A11, CAT, GATA4, MIR191, SATB1, CDA, MIR1247, ENO1, CBX7, DNM1L, ABCC5, SERPINA1, SCO2, ARL2BP, BRS3, CHST15, CHEK2, GRP, AKR1A1, GSTM1, PLG, ECT2, PRNP, RPL10, SDF4, MIR17, RBM14, MIRLET7B, PLAC8, ADH1A, TRIM29, THY1, NAF1, C17orf97, ASPH, PTPN1, TCF7L2, TWF1, TPD52, PTPN11, TP53BP1, LPAR1, MTDH, TK1, LMLN, TLR1, TNS4, TGFBR1, TFF3, CRY2, CRMP1, AR, FAM83A, PTK6, PTBP1, TFAP2A, E2F3, KISS1R, TGFB2, MAK16, TIAM1, CCL21, TBX2, IDO2, CYP24A1, RFXAP, CASC2, ROBO1, DNM2, ROS1, SMARCA2, RBM45, RPS6KA2, RPS6KB1, RPS27A, ABHD11-AS1, S100A2, RELA, SIX1, TRIM69, SFRP4, ARHGAP4, LDLRAD3, SEL1L, SARDH, RMDN2, CCL2, PDIK1L, TICAM1, CCL18, DNMT3A, LRG1, TAT, DLX6-AS1, TACR1, PTPN14, TAC1, PTPRC, SOX2-OT, RAB5A, SYT1, STING1, GADL1, PRRT2, CDCA5, DUSP1, STK4, CYP2B6, STAT5B, SNHG15, CYP1A1, DPYSL3, DNMT3B, CYP1A2, CYP1B1, OSBPL5, SSAV1, RECQL, SRY, SRF, AZIN2, BHLHE23, TPM3, CD68, IL23A, PRMT3, PSME3, ABCC4, B3GALT5, GOLM1, TUBB3, ING4, PRMT5, WASF2, VAV3, RMDN1, SEMA4D, DESI2, ATG7, ANP32B, CD40LG, FOXP3, GDE1, ZFR, SLIT2, DLL4, MEG3, COX5A, RMDN3, CDH17, HEATR1, SLC52A1, CDH3, PAF1, IQSEC1, NAMPT, MFN2, KRT20, TLR9, BCL2L11, PTPRU, CDC25B, WWOX, CDC20, ASAP1, TXNIP, BTK, PRPF31, KLF8, DDR1, CTRC, BBC3, CILK1, MYOF, ZHX2, NOC2L, SMG1, CD36, SULF1, CASP9, TFIP11, SLC7A8, ANGPTL2, SNHG1, SRRM2, DAPK2, RASSF1, MAP4K1, FSTL1, SRPX2, DELEC1, CELF2, CTCF, CD86, ERO1A, SLC39A3, AP4B1, EEF2K, GIPC1, CD80, UHRF1, TBK1, MCTS1, CD14, SGSM3, RBMS3, WDR5, CDK8, CDK9, TPT1, COL4A2, COL11A1, WNT10B, DHDDS, XPC, XRCC3, XRCC4, BAX, ZFP36, COL1A2, ULBP2, LIN28A, MANF, MCPH1, TUSC3, TCTN1, BHLHE41, CMM, FOSL1, LAT2, VIP, SYBU, UBC, NR2C2, TRAF2, TRAF6, TMPRSS13, CPOX, TTK, ITCH, TYRO3, NUF2, BAG1, ARHGAP24, ATR, AVP, CLDN3, UTRN, KDM6A, UVRAG, VCL, USP5, LTB4R, WNK1, HAP1, KSR1, CDK5R1, PER3, PER2, PNO1, SQSTM1, BNC1, DIABLO, METTL3, USP9X, TM4SF5, ARTN, AIP, DMAP1, SLC16A4, SLC16A3, DYRK1B, BUB3, CES1, CCN5, CFL1, CHPT1, CMA1, GORASP1, ROBO3, CUL4B, IL21, CIP2A, DENR, CLU, DANCR, TP73-AS1, SMURF1, TNFSF13, AKR1B10, CEACAM1, TNFRSF10D, TNFRSF10A, IL18R1, CCR5, PADI4, MIR629, LINC01672, HSPD1, PAWR, HSPG2, PARN, ITGAV, C20orf181, MME, EPS8, ERBB4, FGF10, ITGA3, GFRA2, PRDX1, SNHG16, MIR451A, PAEP, AKR1B1, MMP11, ALDOA, P2RX7, P2RX5, ALDH1B1, GRK2, MIR296, OLR1, MNAT1, MAP3K10, ITGB2, NTS, MIR204, EPHB4, PFN1, MIR483, KCNJ11, KCNH2, MIR509-1, PER1, PDPK1, MIR497, HOXB7, MIR193B, MIR885, MIR373, HSPA4, ITK, HOXD13, GLI3, PDK1, FGFR4, MICOS10-NBL1, PDCD1, ANGPT1, MITF, TMX2-CTNND1, HSPA2, ANG, IFIT3, ROR2, PHB, TMEM238L, FBN2, MIR1231, IL6ST, IL17A, TNFRSF9, MIR135B, GAPDH, MIR1246, IL9, MTRR, FAT1, MIR148B, FAP, MIR424, MIR375, IL12A, CISD2, NEDD9, IL15, NEDD4, NCL, NBN, NBL1, MUTYH, IL13RA2, MIR374A, MTAP, IL2RA, ADAM8, NPY, NTRK2, NTF3, IRF2, FDPS, MIR1290, FCN2, FBL, GCHFR, PNP, IGF2R, ILK, MSMB, ESRRB, NGFR, NOS2, MIR1291, MSN, MIR1181, FCGR3B, MIR99B, AFP, IL18, NME1, MIR1271, FCGR3A, IKBKB, MIR744, MAP3K5, PRRX1, ELAVL1, POTEF, PPIA, GGT2, APOA2, FOXL1, EIF4EBP1, LASP1, RPSA, CCR2, EIF5A, CXCR3, FOXC1, GGTLC4P, MIR144, BIRC5, PODXL, HFE, UPK3B, HIC1, SNHG14, CDKN2B-AS1, PMS1, SMAD7, MIR15A, MR1, MIR139, LDHB, LTB, GSN, RELN, GRB7, PRSS2, AQP3, EEF1A2, MUC5B, CXCL1, GSK3A, FADS1, MIR601, MIR10A, PRKCB, MIR130B, MIR122, ANXA8L1, PRKAR1A, MIR126, GGTLC3, LRP2, GGTLC5P, ANXA8, EIF4A1, FOLR1, LTA, MAGEA3, CEACAM6, KCNN4, MCC, HNF4G, PCAT1, MCAM, GLO1, HNRNPK, KCNQ1, ENG, MIR196A1, MIR192, MIR185, PLAUR, SLC25A3, PLAT, MAP1A, LINC-ROR, MIR455, FKBP4, MIR181C, MIR183, MIR486-1, FOXA1, BMP8B, PIMREG, MTPAP, ZNF654, RHOF, NLRP2, CCL28, LTB4R2, MRGBP, PRPF40A, MIR874, ERO1B, MIR940, EIF5A2, STYK1, MIR646, MIR92B, IL17RB, LXN, MAML3, FERMT1, SMARCAD1, RIPK4, MIR652, TRPV6, ETNK1, TREM1, TERF2IP, KCMF1, SPHK2, BMP3, CHD7, ANLN, GPR137, MIR374B, MIR935, RETN, STK31, BNIP3L, GACAT2, ADPGK-AS1, OGDHL, CEP55, SNHG9, SARS2, CNDP2, OCIAD1, AGAP2-AS1, INTS11, MIR675, NKRF, ZGLP1, MSTO1, SLC35F6, LINC00462, MIR891B, MIR365A, BOK, PSENEN, ANO1, PIWIL2, KDM3A, PRR11, EXOC2, MIR876, CDKAL1, SULF2, SOX6, NSUN5, MIR1225, SLC38A7, BMP2, DIP, PARP14, KDM7A-DT, UGT1A1, MIR760, UGT1A3, PBRM1, ASPN, LOC729970, BPHL, CC2D1A, RADIL, ST7L, ALKBH5, HHAT, SLC25A36, SYCE1L, DST, MYDGF, MED29, KPNA7, PCBP3, GLS2, ERVK-11, MYB-AS1, LSM1, BANCR, LINC01111, HCAR1, GHET1, CA2, AGO2, SERPING1, LOC102723407, GATD3B, DKK4, SIK1B, CDR1-AS, CAD, CALCR, ANKRD1, SPINK4, MIR5100, UBE2S, RABGEF1, ATAD2, HIPK2, MACROD1, NOB1, ERVK-22, MIR2682, MIR4656, LAMTOR2, REM1, DLL1, SPRY4-IT1, SLCO3A1, SLCO4A1, HTRA2, DESI1, PSC, C3, DNAI1, USP21, SNORA74A, LETMD1, LOC112694756, MOB4, ZEBTR, PRPF40B, LNCRNA-ATB, EML2, CADM1, RAB38, SH3BP4, TMEFF2, ACADS, H3P13, H3P8, ABCA4, FAM215A, DDX58, SERPINA3, USP49, CASP7, IRAIN, LOC110806263, EHF, PERCC1, FGF21, PTPN22, CERNA3, KCNK15-AS1, LINC00976, CAPN1, NUTF2P3, CHD5, SIRT1-AS, ACO1, NECTIN3, CAST, CLIC4, MYRIP, TMEM158, C1GALT1C1, LINC00339, DUOX1, ADAM10, PRKAG2, CRLF3, ZMYND10, MIR1301, MIR1180, MIR634, TLR8, BTC, KLF9, C1QBP, CLDN18, CKLF, ZEB2-AS1, HSPA14, LEF1, MIR663B, MIR4306, ACVR2A, YTHDF2, SIRT6, RAB14, NT5DC3, GPR87, LOXL1-AS1, MBD3, IL17D, LINC00994, BCL11A, ADA2, MAP3K20, CDK12, CMPK1, CAB39, RAB23, SF3B6, MIR1297, PIAS4, MIR1243, MIR1266, MIR3686, A4GNT, INSIG2, IL20, DUOX2, ERVW-1, ACTA2, VSX1, KLRC4-KLRK1, ACP3, UBQLN1, BUB1B, ERVK-2, STRN4, NPC1L1, MIR3064, TSPO, BRD7, MYLIP, ERVK-12, PYCARD, ACVR1, IL21R, IRAK4, PXN-AS1, ANGPTL4, MIR642B, MIR3923, APIP, GMNN, MIR3679, ISOC1, LINC01133, BTF3, ASCC1, SMIM31, TRAT1, LINC00958, F11R, TAS2R10, LINC01006, ARHGEF4, SLC25A37, DIO3OS, EMSY, PROKR2, CD109, ADAT2, GPR151, SLCO6A1, EMB, REG3G, ANPEP, MIR208A, ANGPT2, FATE1, MIR211, TMIGD2, SPNS2, MSI2, ARR3, JDP2, SPPL3, H4-16, CCDC26, MUC17, GATA5, ANXA5, LINC00052, MIR198, MIR199A1, PTGR2, PRIMA1, FBXL14, MIR199A2, MIR19A, MIR19B1, SYT9, MIR20A, CACUL1, ANXA11, ANXA6, TMEM37, BRI3BP, SLC32A1, CYP2R1, TMEM45B, PRAP1, MAL2, MCM3AP-AS1, ALDH1A3, IL17F, MIR28, EGLN2, AGTR2, PPP1R14A, MIR302A, ART1, FOXQ1, ASAH1, DMKN, ASNS, MIR30C1, MARVELD3, LRP5L, MIR30C2, NLRP3, MIR155HG, ALPG, MIR224, MIR215, TWIST2, MRGPRX4, MRGPRX3, DCD, ANIB1, TM4SF18, CMTM5, CTHRC1, MIR219A1, NOSTRIN, MIR22, UHRF2, ALOX15, FBXO32, ALK, CSMD2, MIR197, SIK1, APAF1, MIR127, MCIDAS, MIR129-2, RSPO2, KLK3, ANO9, APRT, CHSY3, TRIM59, APOC1, ARG1, LYPD5, LINC00346, LINC01559, OR10A4, BCL9L, APOA1, SLC25A45, MACC1, PWAR4, SBF2-AS1, DNAAF3, CCSER1, PAIP2B, LINC01121, C16orf74, HRNR, MIRLET7C, SERPINA13P, CEP85L, MIRLET7D, LINC-PINT, MIR100, MIR106A, FASLG, ENTPD8, GSTK1, TMPRSS9, MIR125A, H19, ASPM, MIR193A, MUC20, ARID2, B3GNT6, MIR182, ADAMTS18, OIT3, BHLHA15, OXER1, IFNLR1, SLFN5, EML5, CLEC14A, MIR186, SLC5A8, RHOB, FEZF1-AS1, NKAIN2, DAB2IP, MIR181A2, CENPX, MRGPRX1, MED19, MIR149, SGMS1, NPCA1, FAM133B, ARG2, MIR152, RICTOR, IL27, SEMA3D, BIRC2, GPRC6A, FOXK1, KDM1B, MIR18A, ALKBH3, MARCHF8, VWCE, LINC00473, MIR32, SMYD2, SMURF2, NIBAN2, YTHDC2, PORCN, IL25, DEPTOR, ILRUN, S100PBP, SMYD3, TMPRSS3, PNPT1, HIF3A, COP1, RHBDF1, MMS19, LINC01060, BGLAP, MIIP, SAV1, MIR520H, PINK1, MIR519D, MIR181D, BCL6, DHX40, DYNC2H1, TMEM204, MIR489, MIR146B, BCR, MIR202, PLEKHF1, MIR492, BDNF, MIR493, CFB, GGCT, STK33, ADRB3, CDK15, AFAP1, LGR6, ACE2, SLURP1, ADH4, PNPLA2, CXADRP1, CIAPIN1, ADAMTS9, PRDM1, H3P38, MIR539, SNORA25, MIR421, MIR584, KIF15, OTUD7B, MIR607, MIR608, MIR613, MIR623, CD248, NORAD, IL22RA1, ADH6, SNX6, SINHCAF, HAMP, ANKRD36B, KLHL1, MRTFA, CFAP97, PCDH10, CERNA2, CRNDE, MIB1, RPTOR, ARID1B, PPM1H, BGN, AICDA, PARP4, MIR485, MIR410, BCKDHB, VN1R17P, EBPL, GPR166P, FOXB2, XRCC6P5, ZGPAT, NT5C1A, MCHR2, HOPX, MCM8, RAB11FIP4, PROK1, RPAIN, MIR331, AP2A1, MIR340, USP44, MIR367, TRIM63, KDM2B, MIR370, MIAT, TSLP, NKD1, MIR320A, MIR34B, SNHG7, MIR93, JAG1, RAB2B, ATAD1, PARP10, MIR99A, RIOX2, KMT5C, PSRC1, CRISP1, ASTL, HINT2, MYCBPAP, ATF3, SH3D21, PPP1R2C, GRHL2, DUXAP8, JADE1, DUXAP10, NANOG, UBA5, NBDY, MFSD13A, KDM8, MIR448, MIR449A, H4C15, MIR431, MMRN2, MYH14, BCHE, MIR452, NAA25, CEP70, MIR371A, MIR425, RERE, IMMP2L, HASPIN, SUMO1P3, SESN2, VANGL1, NETO2, MIR377, MIR381, MIR384, NECTIN4, AZGP1, ZFP91, ITIH5, SLC44A4, SRCIN1, BAK1, GPX2, HS3ST3B1, LPAR3, PLOD2, EFNB2, PKN1, EGR1, PRKAR2A, PRKACG, EGR3, EIF4A2, PRELP, PPP5C, PPP1R1A, PPID, PPIB, MED1, PPARA, PON2, PON1, POLE, POLD1, PNMT, PNLIP, PMS2, PMS2P3, PMAIP1, PRKCZ, PRKDC, EFNA2, PTAFR, PTPRJ, PTPRG, PTPRA, PTPN13, QSOX1, PTN, E2F2, PTGS1, PTH, PTGER4, PSMD10, EEF1G, PYY, PRTN3, KLK10, HTRA1, KLK6, PRSS3, EDNRB, PROX1, EEF1A1, MAPK9, PLXNA1, CELA1, DYRK1A, PLEK, CDK16, PCBP1, PCBD1, PBX3, PAX6, PAX5, PAK3, PAK2, EREG, SERPINB2, ERCC4, PAFAH1B2, PCSK6, P2RY6, P2RY2, OSM, OSBP, ORM1, OPRM1, OPRD1, TNFRSF11B, ERCC5, ODC1, PCYT1A, PDE8A, PDGFA, PIGR, ELK3, PLCB3, EMD, PLA2G4A, PKLR, PKD1, PIN1, EPHA1, EPHA4, EPHB1, EPHB3, PDGFB, PGR, PFKP, PFKM, PFKFB4, PFDN5, PEG3, PECAM1, PDZK1, ENPP1, PDK4, PVR, RGL2, PSD4, SAFB, SLC1A2, DCX, SKIL, SKI, SIM2, ST3GAL4, ST6GAL1, DDB2, SFRP1, DDT, SELP, CX3CL1, XCL1, CXCL5, DLD, CCL8, CCL7, DMD, SRL, SCTR, SCN10A, SCN9A, SARS1, ACE, SLC5A1, DCTD, DAG1, SPP2, CYP2C19, SPG7, CYP3A4, SOX12, SOX15, CYP51A1, SOS1, SOD3, DACH1, SNCG, SLC6A8, DAP, SMO, SMN2, SMN1, SMARCC1, DAPK1, SLPI, DAPK3, SLC9A1, SLC8A1, SAI1, S100B, RAB27A, S100A10, RGS16, TRIM27, REST, REN, RDX, RBP4, RBP1, RBM3, KDM5A, DPEP1, RASGRF1, PLAAT4, RARB, RAP1B, RAP1A, RALA, RAG2, DRD2, HBEGF, RAD17, DVL2, RABIF, RAB27B, RNASE1, RNASE3, RNASEL, RPS8, S100A7, S100A6, DNA2, SORT1, S100A1, RXRA, DNAH5, DNASE1, DYNC1H1, RPS15A, RPS6KA3, RNF2, RPS6KA1, RPL39, RPL34, RPL29, RPL26, RPE65, RORB, RORA, SNORA73A, BRD2, ERG, NTRK3, ERN1, IARS1, GAS6, GATA1, GATA2, GATA3, IGSF1, GC, IGFBP7, IGFBP2, GCK, GCKR, GCNT1, IFNB1, IFNAR1, IFNA17, GCY, GDF2, GDF10, IFN1@, GFPT1, IDH1, ID2, IRF8, ICAM2, CXCR1, GALNT3, GAD2, ITGB4, KRT8, FRZB, FUS, KCNK2, FUT3, FUT4, GABBR1, JAK1, JAG2, EIF6, GABRA3, GAD1, ITGA5, ITGA1, ISG20, IRS1, IRF1, ITGA6, GABRP, INSR, INPP4A, IDO1, GFRA1, HYAL1, NT5E, HTR1D, HLA-DRB1, HLA-DOA, HLA-C, HLA-B, GOT2, GP2, HGFAC, GPI, HDGF, HCRTR1, HCRT, H2AX, H1-5, H1-4, GUSB, GUCY2C, GPT, GSTM3, GPX1, GRM1, GRIA3, GRB10, GRB2, GNRHR, HMGB2, HMGCR, HPD, HTR1B, HSPE1, GHR, GHRH, HSPA9, GJB1, HES1, AGFG1, GPC3, HPGD, HOXB9, GLUL, HOXB8, HOXB6, HOXB2, HOXA1, HNRNPU, HNRNPC, ONECUT1, HNF4A, GLUD1, HMMR, KRT17, KRT18, KRT19, FPR2, NEDD8, MYH9, MYD88, FANCD2, MMUT, TRIM37, PTK2B, MTR, ND2, FBLN2, MT1M, MRE11, MPST, MPO, MPL, MMP16, MMP15, GPC4, MMP13, MMP12, FER, MMP3, GPC5, NELL1, NEO1, NEU2, NQO2, YBX1, NRTN, NPY2R, NPM1, NPC1, CCN3, NOTCH2, NOS1, NOP2, NNMT, NM, NEUROD1, NHS, ETV4, NFYA, ETV5, EWSR1, EYA2, NFE2, F9, F10, NEUROG1, KMT2A, MAP3K9, MKI67, FLNA, LMO2, LMNB1, LLGL1, LIPE, LIMS1, LIMK2, LIMK1, LIG4, LIG3, LIFR, FLNB, LPA, LGALS3BP, FOLH1, LFNG, LEPR, FOLR2, LBR, LAMC2, LAMP1, LAMB3, LAG3, LOX, LRP1, MICB, VEGFD, MFAP1, FH, MAP3K4, MAP3K3, MEF2D, MEF2A, MECP2, ME2, FHL2, MDH1, MATK, LRP6, MAT1A, MAP2, SMAD6, SMAD3, SMAD1, FOXD1, TACSTD2, FLI1, BCAM, LTA4H, SPOCK1, CYP2B7P, SREBF2, MVP, GDF11, TRIM13, RBM5, RBM6, TRIM28, G3BP1, ARFRP1, FRY, LRPPRC, RAD50, PPIF, ACTR2, ARPC4, EDIL3, IL18BP, ABCB6, GNE, HDAC6, AKT3, HNRNPDL, NUP153, NR1H4, NR1I3, TRIB1, CNKSR1, SF3B4, CD74, DDX17, CD63, SEMA3C, CARM1, CAP1, MERTK, TACC3, GPNMB, BASP1, RACK1, DLC1, CDK2AP2, IRF9, TUBA1B, MICU1, KLF2, WARS2, TLR6, TNIP1, RTN3, SF3A1, STUB1, MED12, USP15, MSC, GPX4, LITAF, MAGED1, ROCK2, ATG5, RASAL2, MAP4K4, AIM2, GSTO1, QKI, NTN1, RECQL5, CD101, KIF3B, SLC9A3R1, SLC9A3R2, CD163, SOCS6, SNORD22, ADGRG1, BCL7B, PIWIL1, MAPKAPK2, AIMP1, SCAMP1, BAG5, CDH13, SDC3, DLEC1, KIF14, NUAK1, TLK1, CDC25C, MTSS1, SETD1A, USP34, CDH5, DCAF1, MDC1, PTGES, SOCS5, TTLL4, WTAP, PRDX6, APOBEC3B, SPAG6, CDH11, CHD1L, CXCL14, EI24, NEBL, CD59, ARPC1A, HTATIP2, KLRK1, ARHGEF15, MLXIP, FASTKD2, NLRP1, VASH1, CCNA2, ITGA11, CBX3, TUSC2, PDAP1, PHB2, GPR182, SLCO2B1, KLF12, FILIP1L, SLC6A14, GALNT5, CCNB1, WIF1, MAP4K5, RPP14, ADAMTS8, KIFAP3, SIRT2, STK38L, NCAPH, ZNF281, PES1, ICMT, CELA3B, GPR161, TPSD1, CAV2, RUNX1, SIRT4, DICER1, NCSTN, CBR1, SATB2, SIN3B, BICD2, MPRIP, CBL, KDM6B, PEG10, KDM4C, PDS5B, TNIK, RER1, TRIM31, TOPBP1, KHDRBS1, PLK2, KDM5B, RBBP9, PLK4, NFAT5, POLQ, USP39, CCT8, DLL3, TNFSF13B, SPINT2, WASF3, SCGB1D2, IGF2BP2, CD34, IGF2BP1, ENTPD1, PDLIM5, SLCO1B1, ENTPD2, SMC2, OLFM4, SEPTIN9, FRS2, UBE2C, STIP1, OGFR, RIPK3, DSTN, RAB31, CCND2, CCND3, KCNQ1OT1, COPS6, FERMT2, CKAP4, CCNG1, CD28, EHMT2, PAPOLA, GADD45G, BRD8, MALT1, PPARGC1A, MRPS30, NMU, ME3, ADAM28, GCNT3, CDKN1C, SRPK2, TNFAIP1, COMP, COMT, SLC31A1, CPE, NR1H2, UCK2, UGT1A, UGP2, UCHL3, UCHL1, UBE2N, CPN1, TULP3, TUFT1, TTR, TRH, HSP90B1, TPO, TPD52L2, CREBBP, CREM, TOP1, TNFRSF1B, BEST1, TRPV1, VTN, XRCC2, NELFE, DEK, SCLC1, PAX8, BTG2, ZNF154, ZBTB16, CNBP, ZFX, YES1, COL5A2, WARS1, COL6A1, XPA, XK, COL6A2, COL6A3, WNT7B, WNT2, WNT1, COL17A1, WAS, TNFRSF1A, CRKL, PTTG1, CLDN5, CTPS1, HNF1B, CTRL, TBX3, TCEA3, TCEA2, TCEA1, CTSB, TAGLN, ADAM17, CTSD, SULT1A1, CTSE, CTSH, STIM1, CX3CR1, STAT2, CXADR, ST13, CYP2A6, CYP2A13, SSR2, TRIM21, CST1, PPP1R11, TDG, CS, TMSB4X, TMPRSS2, TM7SF2, TLR3, TLR2, TLE1, CRY1, THBS2, THBD, THAS, TGIF1, PRDX2, CSF1R, TGFB1I1, CSF3, CSN2, TFF2, TFCP2, TF, CSNK2A2, TERC, VCAN, TFEB, COL1A1, CNC2, ABCC2, SPHK1, ARHGEF7, IER3, ASAP2, DLEU2, CCN6, NRP2, INPP4B, CHGA, RAB11A, CHKA, ADAM15, CHRM3, CHRNA7, TNK1, BECN1, EIF3F, IRS2, DYNLL1, CLCA1, NUMB, PTCH2, RUVBL1, FUBP1, NAE1, MBD2, ATP6V0D1, MTA2, AURKB, CDKN2D, ARHGEF2, SCAF11, TMSB10, EBAG9, EXO1, ATG12, CCNE2, CDO1, WASF1, DIRAS3, CLDN8, CDX1, BRSK2, CLIC3, MAP3K14, F2RL3, CEBPB, P4HA2, CDK5R2, NOP14, AKR7A2, MADD, UBL4A, H4C11, H4C12, H4C6, H4C4, H4C1, FZD6, FZD4, CDC7, H4C9, ARID1A, GATD3A, H4C8, SLC7A5, ANP32A, GPR68, YEATS4, KMT2D, MFAP5, RASSF7, ADAM12, SLC25A16, CUBN, H4C3, H4C2, LMO4, TTF2, HAT1, PARG, PIK3R3, PPFIA4, CLTC, GEMIN2, SEMA7A, OGT, SORBS2, KLF11, DOC2A, H4C5, RAD54L, STK24, TAGLN2, SPARCL1, PIP4K2B, PIP5K1B, PIP5K1A, NME5, H4C14, H4C13, H3P40
-
Mosquito-Borne Disease
Wikipedia
Depending on the mosquito vector, and the affected community, a variety of prevention methods may be deployed at one time. Insecticidal nets and indoor residual spraying [ edit ] The use of insecticide treated mosquito nets (ITNs) are at the forefront of preventing mosquito bites that cause malaria. ... Because the Anopheles gambiae feeds indoors (endophagic) and rests indoors after feeding (endophilic), insecticide treated nets (ITNs) interrupt the mosquito's feeding pattern. The ITNs continue to offer protection, even after there are holes in the nets, because of their excito-repellency properties which reduce the number of mosquitoes that enter the home. ... "The Impact of Pyrethroid Resistance on the Efficacy of Insecticide-Treated Bed Nets against African Anopheline Mosquitoes: Systematic Review and Meta-Analysis" . PLOS Medicine . 11 (3): e1001619. doi : 10.1371/journal.pmed.1001619 .











