Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Lichen Spinulosus
Wikipedia
Clinicopathologic review of thirty-five cases". J Am Acad Dermatol . 22 (2 Pt 1): 261–4. doi : 10.1016/0190-9622(90)70035-G . PMID 2179296 . External links [ edit ] Derm Net NZ Emedicine Thehinhso v t e Diseases of the skin and appendages by morphology Growths Epidermal Wart Callus Seborrheic keratosis Acrochordon Molluscum contagiosum Actinic keratosis Squamous-cell carcinoma Basal-cell carcinoma Merkel-cell carcinoma Nevus sebaceous Trichoepithelioma Pigmented Freckles Lentigo Melasma Nevus Melanoma Dermal and subcutaneous Epidermal inclusion cyst Hemangioma Dermatofibroma (benign fibrous histiocytoma) Keloid Lipoma Neurofibroma Xanthoma Kaposi's sarcoma Infantile digital fibromatosis Granular cell tumor Leiomyoma Lymphangioma circumscriptum Myxoid cyst Rashes With epidermal involvement Eczematous Contact dermatitis Atopic dermatitis Seborrheic dermatitis Stasis dermatitis Lichen simplex chronicus Darier's disease Glucagonoma syndrome Langerhans cell histiocytosis Lichen sclerosus Pemphigus foliaceus Wiskott–Aldrich syndrome Zinc deficiency Scaling Psoriasis Tinea ( Corporis Cruris Pedis Manuum Faciei ) Pityriasis rosea Secondary syphilis Mycosis fungoides Systemic lupus erythematosus Pityriasis rubra pilaris Parapsoriasis Ichthyosis Blistering Herpes simplex Herpes zoster Varicella Bullous impetigo Acute contact dermatitis Pemphigus vulgaris Bullous pemphigoid Dermatitis herpetiformis Porphyria cutanea tarda Epidermolysis bullosa simplex Papular Scabies Insect bite reactions Lichen planus Miliaria Keratosis pilaris Lichen spinulosus Transient acantholytic dermatosis Lichen nitidus Pityriasis lichenoides et varioliformis acuta Pustular Acne vulgaris Acne rosacea Folliculitis Impetigo Candidiasis Gonococcemia Dermatophyte Coccidioidomycosis Subcorneal pustular dermatosis Hypopigmented Tinea versicolor Vitiligo Pityriasis alba Postinflammatory hyperpigmentation Tuberous sclerosis Idiopathic guttate hypomelanosis Leprosy Hypopigmented mycosis fungoides Without epidermal involvement Red Blanchable Erythema Generalized Drug eruptions Viral exanthems Toxic erythema Systemic lupus erythematosus Localized Cellulitis Abscess Boil Erythema nodosum Carcinoid syndrome Fixed drug eruption Specialized Urticaria Erythema ( Multiforme Migrans Gyratum repens Annulare centrifugum Ab igne ) Nonblanchable Purpura Macular Thrombocytopenic purpura Actinic/solar purpura Papular Disseminated intravascular coagulation Vasculitis Indurated Scleroderma / morphea Granuloma annulare Lichen sclerosis et atrophicus Necrobiosis lipoidica Miscellaneous disorders Ulcers Hair Telogen effluvium Androgenic alopecia Alopecia areata Systemic lupus erythematosus Tinea capitis Loose anagen syndrome Lichen planopilaris Folliculitis decalvans Acne keloidalis nuchae Nail Onychomycosis Psoriasis Paronychia Ingrown nail Mucous membrane Aphthous stomatitis Oral candidiasis Lichen planus Leukoplakia Pemphigus vulgaris Mucous membrane pemphigoid Cicatricial pemphigoid Herpesvirus Coxsackievirus Syphilis Systemic histoplasmosis Squamous-cell carcinoma This condition of the skin appendages article is a stub .
-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Neuroendocrine Tumor
Wikipedia
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... According to the WHO and UNICEF, deaths attributable to malaria in 2015 were reduced by 60% [77] from a 2000 estimate of 985,000, largely due to the widespread use of insecticide-treated nets and artemisinin-based combination therapies. [74] In 2012, there were 207 million cases of malaria.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
-
Premature Thelarche
Wikipedia
CPP can be differentiated from PT through biochemical testing, ultrasounds and ongoing observation . [3] There is no treatment for PT but regular observation is important to ensure it doesn’t progress to CPP. ... In some cases development may be unilateral : one breast develops. [ citation needed ] Patterns of PT [ edit ] There are four patterns of PT development. ... FSH plays a key role in development, growth and puberty, thus it is suspected to play a role in PT. Gondotropin-releasing hormone (GnRH) stimulation testing in some patients with PT has shown a dominant response from FSH. This response is linked to active mutations in the FSH receptor and Gs-a subunit in PT. Genetic investigation indicated these mutations only account for few cases of premature PT. [2] [7] PT may also be caused by transient partial activation of the HPG axis . ... These estrogenic properties may cause an imbalance in endocrine signalling pathways , leading to PT in regular users of these products. [1] Fennel tea has been studied as an endocrine disrupter linked to PT.
-
Post-Traumatic Seizure
Wikipedia
Post-traumatic seizures ( PTS ) are seizures that result from traumatic brain injury (TBI), brain damage caused by physical trauma . PTS may be a risk factor for post-traumatic epilepsy (PTE), but a person who has a seizure or seizures due to traumatic brain injury does not necessarily have PTE, which is a form of epilepsy , a chronic condition in which seizures occur repeatedly. ... The risk that a person will suffer PTS becomes progressively lower as time passes after the injury. ... Often, MRI is performed in any patient with PTS, but the less sensitive but more easily accessed CT scan may also be used. [17] Prevention [ edit ] Shortly after TBI, people are given anticonvulsant medication, because seizures that occur early after trauma can increase brain damage through hypoxia , [3] excessive release of excitatory neurotransmitters , increased metabolic demands, and increased pressure within the intracranial space. [2] Medications used to prevent seizures include valproate , phenytoin , and phenobarbital . [18] It is recommended that treatment with anti-seizure medication be initiated as soon as possible after TBI. [8] Prevention of early seizures differs from that of late seizures, because the aim of the former is to prevent damage caused by the seizures, whereas the aim of the latter is to prevent epileptogenesis. [3] Strong evidence from clinical trials suggests that antiepileptic drugs given within a day of injury prevent seizures within the first week of injury, but not after. [4] For example, a 2003 review of medical literature found phenytoin to be preventative of early, but probably not late PTS. [7] In children, anticonvulsants may be ineffective for both early and late seizures. [4] For unknown reasons, prophylactic use of antiepileptic drugs over a long period is associated with an increased risk for seizures. [1] For these reasons, antiepileptic drugs are widely recommended for a short time after head trauma to prevent immediate and early, but not late, seizures. [1] [19] No treatment is widely accepted to prevent the development of epilepsy. [3] However, medications may be given to repress more seizures if late seizures do occur. [18] Treatment [ edit ] Seizures that result from TBI are often difficult to treat. [13] Antiepileptic drugs that may be given intravenously shortly after injury include phenytoin, sodium valproate , carbamazepine , and phenobarbital. [2] Antiepileptic drugs do not prevent all seizures in all people, [5] but phenytoin and sodium valproate usually stop seizures that are in progress. [2] Prognosis [ edit ] PTS is associated with a generally good prognosis. [14] It is unknown exactly how long after a TBI a person is at higher risk for seizures than the rest of the population, but estimates have suggested lengths of 10 to over 15 years. [5] For most people with TBI, seizures do not occur after three months, and only 20–25% of people who suffer TBI have PTS more than two years after the injury. [9] However, moderate and severe TBI still confer a high risk for PTS for up to five years after the injury. [4] Studies have reported that 25–40% of PTS patients go into remission ; later studies conducted after the development of more effective seizure medications reported higher overall remission rates. [5] In one quarter of people with seizures from a head trauma, medication controls them well. [1] However, a subset of patients have seizures despite aggressive antiepileptic drug therapy. [5] The likelihood that PTS will go into remission is lower for people who have frequent seizures in the first year after injury. [5] Risk of developing PTE [ edit ] It is not known whether PTS increase the likelihood of developing PTE. [13] Early PTS, while not necessarily epileptic in nature, are associated with a higher risk of PTE. [20] However, PTS do not indicate that development of epilepsy is certain to occur, [21] and it is difficult to isolate PTS from severity of injury as a factor in PTE development. [13] About 3% of patients with no early seizures develop late PTE; this number is 25% in those who do have early PTS, and the distinction is greater if other risk factors for developing PTE are excluded. [21] Seizures that occur immediately after an insult are commonly believed not to confer an increased risk of recurring seizures, but evidence from at least one study has suggested that both immediate and early seizures may be risk factors for late seizures. [5] Early seizures may be less of a predictor for PTE in children; while as many as a third of adults with early seizures develop PTE, the portion of children with early PTS who have late seizures is less than one fifth in children and may be as low as one tenth. [12] The incidence of late seizures is about half that in adults with comparable injuries. [12] Epidemiology [ edit ] The relative risk of PTS increases with the severity of injury. [2] As age increases, risk of early and late seizures decreases. [5] [22] Research has found that the incidence of PTS varies widely based on the population studied; it may be as low as 4.4% or as high as 53%. [5] Of all TBI patients who are hospitalized, 5 to 7% have PTS. [4] PTS occur in about 3.1% of traumatic brain injuries, but the severity of injury affects the likelihood of occurrence. [9] The most important factor in whether a person will develop early and late seizures is the extent of the damage to the brain. [2] More severe brain injury also confers a risk for developing PTS for a longer time after the event. [4] One study found that the probability that seizures will occur within 5 years of injury is in 0.5% of mild traumatic brain injuries (defined as no skull fracture and less than 30 minutes of post-traumatic amnesia , abbreviated PTA, or loss of consciousness , abbreviated LOC); 1.2% of moderate injuries (skull fracture or PTA or LOC lasting between 30 minutes and 24 hours); and 10.0% of severe injuries (cerebral contusion, intracranial hematoma , or LOC or PTA for over 24 hours). [23] Another study found that the risk of seizures 5 years after TBI is 1.5% in mild (defined as PTA or LOC for less than 30 minutes), 2.9% in moderate (LOC lasting between 30 minutes and 1 day), and 17.2% in severe TBI (cerebral contusion, subdural hematoma, or LOC for over a day; image at right). [2] [11] Immediate seizures have an incidence of 1 to 4%, that of early seizures is 4 to 25%, and that of late seizures is 9 to 42%. [2] Age influences the risk for PTS. ... The largest risks for PTS are having an altered level of consciousness for a protracted time after the injury, severe injuries with focal lesions, and fractures. [8] The single largest risk for PTS is penetrating head trauma , which carries a 35 to 50% risk of seizures within 15 years. [2] If a fragment of metal remains within the skull after injury, the risk of both early and late PTS may be increased. [5] Head trauma survivors who abused alcohol before the injury are also at higher risk for developing seizures. [4] Occurrence of seizures varies widely even among people with similar injuries. [5] It is not known whether genetics play a role in PTS risk. [11] Studies have had conflicting results with regard to the question of whether people with PTS are more likely to have family members with seizures, which would suggest a genetic role in PTS. [11] Most studies have found that epilepsy in family members does not significantly increase the risk of PTS. [5] People with the ApoE-ε4 allele may also be at higher risk for late PTS. [1] Risks for late PTS include hydrocephalus , reduced blood flow to the temporal lobes of the brain, [1] brain contusions , subdural hematomas , [5] a torn dura mater , and focal neurological deficits . [9] PTA that lasts for longer than 24 hours after the injury is a risk factor for both early and late PTS. [1] Up to 86% of people who have one late post-traumatic seizure have another within two years. [5] See also [ edit ] Complications of traumatic brain injury References [ edit ] ^ a b c d e f g h Tucker GJ (2005). "16: Seizures".
-
Hyperphenylalaninemia, Bh4-Deficient, A
OMIM
A number sign (#) is used with this entry because tetrahydrobiopterin (BH4)-deficient hyperphenylalaninemia due to PTS deficiency (HPABH4A) is caused by mutation in the gene encoding 6-pyruvoyl-tetrahydropterin synthase (PTS; 612719). ... Oppliger et al. (1997) identified 4 novel mutations in 4 Italian families with PTS deficiency. Thony and Blau (1997) reviewed the spectrum of mutations in the PTS gene resulting in tetrahydrobiopterin deficiency. ... Population Genetics Liu et al. (1998) identified 7 single-base mutations in Chinese cases of PTS-deficient hyperphenylalaninemia. In all, 38 PTS mutant alleles from 19 unrelated Chinese families were studied. ... The N52S mutation accounted for 48% of the southern Chinese PTS mutations, but only 1 (9%) of the northern Chinese PTS mutant alleles was found to be N52S.
-
Coinfection
Wikipedia
Global prevalence or incidence of coinfection among humans is unknown, but it is thought to be commonplace, [1] sometimes more common than single infection. [2] Coinfection with helminths affects around 800 million people worldwide. [3] Coinfection is of particular human health importance because pathogen species can interact within the host. The net effect of coinfection on human health is thought to be negative. [4] Interactions can have either positive or negative effects on other parasites. ... The Journal of General Virology . 91 (Pt 6): 1373–87. doi : 10.1099/vir.0.020818-0 .GPT, IL10, TNF, IFNA1, IFNA13, IL1B, MBL2, ATAD1, IL6, TLR4, S100A8, NLRP3, IFNG, CRP, TLR2, TMPRSS13, MSMB, MPO, TCN1, MST1, LMLN, CRYL1, GLUD1, UGDH, MYD88, PTPN11, LPL, ALB, H6PD, TP53, CXCL10, UBE2B, STIL, IVNS1ABP, MMP1, TLR9, PMEL, IFNA2, IFNB1, IL2, CXCL8, IL17A, KLRC2, EXT1, SMOX, PDLIM7, ARG1, CACNA1B, BMPER, BRCA1, IL33, CCR5, LINC01194, CD14, MIR146A, CCL2, DEFB4A, AFP, LXN, DEFB4B, COPD, SOAT1, SNAP25, HACD1, SMARCB1, ST13, ST14, IL32, IL1RL1, FCGR2C, STXBP1, HSP90B1, SULT2A1, TAP1, TPO, TPI1, TLR3, UTRN, ZAP70, CXCR4, SLBP, NR4A3, BCAR3, TIMP4, DENR, TGM3, TGFB1, PRDX2, TNFSF10, PROM1, CD163, ARTN, NAT2, PPIG, TICAM1, HAMP, SRR, ERVK-6, MRPL41, RNF34, DNAJC5, UBASH3B, TSLP, HPS4, TRIM69, NLRP6, GDF15, IFNL3, VN1R2, VN1R4, VN1R5, TREML1, MIR20B, KLKP1, CCR2, TRAP, AD12, PGPEP1, RHOF, IER3IP1, HEBP1, TP53I3, APOBEC3B, STAM2, TRIM22, TACC3, SORBS1, C1QL1, METAP2, NT5C2, BRD4, SEC14L2, SUMF2, PARS2, IL17C, PRSS50, RMC1, OBP2A, ERVW-1, IL22, MBL3P, NTM, CAP1, PPARG, CXCL12, F12, DECR1, DHCR7, DHFR, DPP4, E2F1, EGR1, ELANE, ELAVL1, EPHA3, EPRS1, F8, FCGR3A, CXCL11, FCGR3B, FGF2, FGF9, FLNA, GAPDH, CBLIF, GJA1, GLB1, GRN, CXCL1, HDLBP, DAPK1, CYP26A1, CYP3A4, CYP2B6, ACTB, AGRP, AHR, AKT1, AMBP, ANXA6, AZU1, BCL2, OPN1SW, CFB, BMP1, CAV2, CD33, CD38, ENTPD1, CEACAM5, CEACAM3, CEACAM7, CHAT, CHML, COX8A, CRH, CTAA1, CFH, HLA-B, HLA-DQB1, OAS1, PCNA, PF4, SLC25A3, SERPINA1, SERPINB6, SERPINB9, PIK3CA, PIK3CB, PIK3CD, PIK3CG, ABCA1, PRCP, MAP2K1, EIF2AK2, PSEN1, PSG2, PSMB8, PTPN2, RAD51, RAG1, RTN2, SCD, CCL5, SERPINE1, NM, HLA-G, NFKB1, HMOX1, FOXA2, HSPA4, IFNA4, IFNAR2, IL1A, IL4, IL15, IL18, IRF3, ITGA2B, ITPA, KLKB1, KIF22, LNPEP, LTF, LYZ, MBP, CXCL9, MMP9, MUC2, NDUFS3, NFE2L2, ERVK-20
-
Post-Thrombotic Syndrome
Wikipedia
With PTS, these symptoms typically are worse after walking or standing for long periods of time and improve with resting or elevating the leg. [1] PTS lowers a person's quality of life after DVT, specifically with regards to physical and psychological symptoms and limitations in daily activities. [2] [3] [4] Cause [ edit ] Despite ongoing research, the cause of PTS is not entirely clear. ... They may be useful to treat edemas. [7] Upper extremities [ edit ] Patients with upper-extremity DVT may develop upper-extremity PTS, but the incidence is lower than that for lower-extremity PTS (15-25%). [22] [23] No treatment or prevention methods are established, but patients with upper-extremity PTS may wear a compression sleeve for persistent symptoms. [20] Epidemiology [ edit ] PTS can affect 23-60% of patients in the two years following DVT of the leg. Of those, 10% may go on to develop severe PTS, involving venous ulcers. [24] Socioeconomic [ edit ] Treatment of PTS adds significantly to the cost of treating DVT. The annual health care cost of PTS in the United States has been estimated at $200 million, with costs over $3800 per patient in the first year alone, and increasing with disease severity. [24] [25] PTS also causes lost work productivity: people with severe PTS and venous ulcers lose up to 2 work days per year. [26] Research directions [ edit ] The field of PTS still holds many unanswered questions that are important targets for more research. Those include Fully defining the pathophysiology of PTS, including the role of inflammation and residual thrombus after completion of an appropriate duration of anticoagulant therapy Developing a PTS risk prediction model Role of thrombolytic ("clot-busting") drugs in PTS prevention Defining the true efficacy of elastic compression stockings for PTS prevention (and if effective, elucidating the minimum compression strength necessary and the optimal timing and duration of compression therapy) Whether PTS prevention methods are necessary for patients with asymptomatic or distal DVT Additional treatment options for PTS with demonstrated safety and efficacy (compression and pharmacologic therapies) [ citation needed ] References [ edit ] ^ a b c d Kahn SR (November 2009).

-
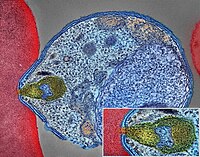
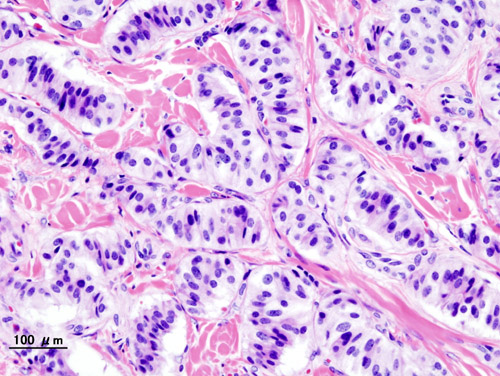
Pancreatic Neuroendocrine Tumor
Wikipedia
PanNETs are a type of neuroendocrine tumor , representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign , while some are malignant . ... However, morphological imaging alone is not sufficient for a definite diagnosis [14] [16] On biopsy , immunohistochemistry is generally positive for chromogranin and synaptophysin . [17] Genetic testing thereof typically shows altered MEN1 and DAXX / ATRX . [17] Staging [ edit ] The 2010 WHO classification of tumors of the digestive system grades all the neuroendocrine tumors into three categories, based on their degree of cellular differentiation (from well-differentiated "NET G1" through to poorly-differentiated "NET G3"). ... Combinations of several medicines have been used, such as doxorubicin with streptozocin and fluorouracil (5-FU) [12] and capecitabine with temozolomide. [ citation needed ] Although marginally effective in well-differentiated PETs, cisplatin with etoposide has some activity in poorly differentiated neuroendocrine cancers (PDNECs), [12] particularly if the PDNEC has an extremely high Ki-67 score of over 50%. [8] : 30 Several targeted therapy agents have been approved in PanNETs by the FDA based on improved progression-free survival (PFS): everolimus (Afinitor) is labeled for treatment of progressive neuroendocrine tumors of pancreatic origin in patients with unresectable, locally advanced or metastatic disease. [20] [21] The safety and effectiveness of everolimus in carcinoid tumors have not been established. [20] [21] sunitinib (Sutent) is labeled for treatment of progressive, well-differentiated pancreatic neuroendocrine tumors in patients with unresectable locally advanced or metastatic disease. [22] [23] Sutent also has approval from the European Commission for the treatment of 'unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors with disease progression in adults'. [24] A phase III study of sunitinib treatment in well differentiated pNET that had worsened within the past 12 months (either advanced or metastatic disease) showed that sunitinib treatment improved progression-free survival (11.4 months vs. 5.5 months), overall survival , and the objective response rate (9.3% vs. 0.0%) when compared with placebo. [25] Genetics [ edit ] Pancreatic neuroendocrine tumors may arise in the context of multiple endocrine neoplasia type 1 , Von Hippel–Lindau disease , neurofibromatosis type 1 (NF-1) or tuberose sclerosis (TSC) [26] [27] Analysis of somatic DNA mutations in well-differentiated pancreatic neuroendocrine tumors identified four important findings: [28] [6] as expected, the genes mutated in NETs, MEN1 , ATRX , DAXX , TSC2 , PTEN and PIK3CA , [28] are different from the mutated genes previously found in pancreatic adenocarcinoma . [29] [30] one in six well-differentiated pancreatic NETs have mutations in mTOR pathway genes, such as TSC2 , PTEN and PIK3CA . [28] The sequencing discovery might allow selection of which NETs would benefit from mTOR inhibition such as with everolimus , but this awaits validation in a clinical trial . mutations affecting a new cancer pathway involving ATRX and DAXX genes were found in about 40% of pancreatic NETs. [28] The proteins encoded by ATRX and DAXX participate in chromatin remodeling of telomeres ; [31] these mutations are associated with a telomerase -independent maintenance mechanism termed ALT (alternative lengthening of telomeres) that results in abnormally long telomeric ends of chromosomes . [31] ATRX / DAXX and MEN1 mutations were associated with a better prognosis . [28] References [ edit ] ^ Burns WR, Edil BH (March 2012).MEN1, ATRX, DAXX, ELK3, TP53, EPHB1, SLC6A2, CEACAM5, CEACAM7, UQCRFS1, DHDDS, CHPT1, RALBP1, CIB1, SEMA4D, RIPK1, CXCR4, VEGFA, TTR, GNA12, TSC2, TFE3, CDKN1B, PSG2, POMC, MYCN, CEACAM3, GRN, MUC16

- Dowling-Degos Disease GARD
-
Azotemia, Familial
OMIM
Furthermore, urea is reabsorbed actively by the tubule; this process is apparently brought into play particularly in states of low protein intake. Net reabsorption might be due to exaggerated active reabsorption or to deficient secretion.
-
Insulinoma
GARD
Insulinoma is a type of pancreatic neuroendocrine tumor (pancreatic NET), which refers to a group of rare tumors that form in the hormone-making cells of the pancreas.MEN1, RPS15, CDKN2B, CDKN2C, IAPP, GCG, CDKN1B, CDKN1A, SST, FOXM1, GLP1R, PDX1, INS, IL1B, RIT2, PTPRN2, GAD1, EHMT1, IGF2, ZGLP1, CDKN2A, SLC30A8, SLC30A10, GCK, SSTR2, FFAR1, YY1, LEP, DPP4, INSM1, MNX1, HSPD1, GAD2, SLC2A2, CASR, RALBP1, RIPK1, PDHX, BTC, UQCRFS1, TP53, TGM2, SSTR5, CDKN1C, INSR, ABCC8, SLC6A2, SSTR4, SSTR3, WFS1, NIT1, SERPINA1, PTPRN, GIP, GCKR, CORO1A, H3P47, PRL, H3P10, ERBB2, GAST, EGR1, ELK3, CALCA, CASP3, EPHB1, G6PC, DLK1, CCN5, SQSTM1, PTTG1, GCM2, LHX2, KL, MAPK8IP1, INSL5, IRS2, ZNRD2, KHDRBS1, DCTN6, LILRB1, FASTK, CCND1, PDIA5, FAS, ATF6, KDM1A, PDZD2, BCL2, BRCA1, TNKS, PLA2G6, HNF1A, TCF19, TGFA, TGFB1, CASP8, THBD, TKT, TSPAN7, TPD52, TRP-AGG2-5, TRPC1, EIPR1, TXN, TYRP1, UCP2, VDR, CACNA1D, BRAF, STAB1, ERP44, NUP62, KCNH4, CAT, KCNH8, GPR119, STOML3, AKT1, HCAR2, GOLGA6A, TICAM2, HES3, MIR107, MIR144, MIR155, MIR204, MIR21, MIR375, INS-IGF2, ADSS2, TMED7-TICAM2, ECT, LINC02210-CRHR1, H3P23, ADM, SLC22A12, TXNDC5, TRABD, RCBTB1, FGF21, MCAT, MCTS1, TMED7, ADIPOR1, DCTN4, CDKAL1, SLC25A38, BANK1, MEG3, ZC3H12A, APOC2, SOX6, SELENOS, IGSF9, SEMA6A, HAMP, G6PC2, PDIA2, ANGPT2, SYP, STAT5A, STC1, STAT5B, KCNJ1, KCNJ6, KRT8, KRT16, KRT19, DECR1, LEPR, LGALS3, LMO2, EPCAM, SMAD2, SMAD3, SMAD4, MAPT, MC2R, MDK, RAB8A, CUX1, MET, CIITA, MLH1, EGF, EGFR, INPPL1, HK1, MTOR, FGF13, GNA12, GPD2, FBN1, GRN, GSK3B, GSR, GTF2H1, ESR2, ELK1, HLA-DQB1, HMGN2, HNF4A, EPHB2, IFI27, IGFBP1, IGFBP2, IL4, IL10, MRC1, NCAM1, NEDD4, SLC2A1, RAP1A, REG1A, CPE, CMA1, S100A8, SCT, CCL2, CXCL12, SDHD, CHGA, RAB3A, CDKN2D, SLC16A1, SNX1, CDC42, CDK1, CCND3, CCNC, CCK, STAT1, RANBP2, CR2, NF1, PIK3CG, NFE2L1, CTSB, NME1, OPA1, PAX4, PAX6, PCSK1, ENPP1, CTNNB1, PKD1, CRHR1, POLD1, MAPK1, MAPK3, MAPK8, ADCYAP1, PRSS1, PSEN2, PSMD9, PTEN, ACO2
-
Primary Testicular Diffuse Large B-Cell Lymphoma
Wikipedia
Primary testicular diffuse large B-cell lymphoma Other names PT-DLBCL ; testicular diffuse large B-cell lymphoma; diffuse large B-cell lymphoma of the testes Specialty Hematology ; oncology Symptoms enlarging testicular mass Complications metastases , particularly to the central nervous system and uninvolved testicle Prognosis guarded Primary testicular diffuse large B-cell lymphoma (PT-DLBCL), also termed testicular diffuse large B-cell lymphoma and diffuse large B-cell lymphoma of the testes, is a variant of the Diffuse large B-cell lymphomas (DLBCL). DLBCL are a large and diverse group of B-cell malignancies with the great majority (-85%) being typed as diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS). PT-DLBCL is a variant of DLBCL, NOS that involves one or, in uncommon cases, both testicles . Other variants and subtypes of DLBCL may involve the testes by spreading to them from their primary sites of origin in other tissues. PT-DLBCL differs from these other DLBCL in that it begins in the testes and then may spread to other sites. [1] The B-cells in PT-DLBCL are malignant lymphocytes that normally function in the humoral immunity component of the adaptive immune system by secreting antibodies that, for example, bind to and neutralize invasive pathogens . [1] In ~75% of PT-DLBCL cases these malignant B-cells are termed "activated B-cells" to distinguish them from "germinal center B-cells." ... These and possibly other unidentified gene abnormalities cause the complete lose of the expression of MHC class I and MHC class II proteins in >65% of all PT-DLBCL cases. MHC class I and II proteins are required for immune cells to identify and attack them. The neoplastic B cells in PT-DLBCL also show gains and amplifications of CD274 and PDCD1LG2 , which are the genes for the pro-death ligands, PDL1 and PDL2, respectfully.
- Sneddon Syndrome GARD
-
Mosquito Bites
Mayo Clinic
Avoid and exclude mosquitoes Limit exposure to mosquitoes by: Repairing any tears in the screens on windows, doors and camping gear Using mosquito netting over strollers and cribs Using mosquito netting when sleeping outdoors Selecting self-care products that don't have scents Use insect repellent Use insect repellent when mosquitoes are active. ... Some sporting goods stores sell clothing pretreated with permethrin. Don't wash bed nets or set them in sunlight, as this breaks down permethrin.






