Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Leukemia, Acute Myeloid
OMIM
AI-10-49 restores RUNX1 transcriptional activity, displays favorable pharmacokinetics, and delays leukemia progression in mice. Treatment of primary inv(16) AML patient blasts with AI-10-49 triggers selective cell death. ... Fong et al. (2015) used primary mouse hematopoietic stem and progenitor cells immortalized with the fusion protein MLL-AF9 (see 159555) to generate several single-cell clones that demonstrated resistance, in vitro and in vivo, to the prototypical bromodomain and extra terminal protein (BET) inhibitor I-BET. Resistance to I-BET conferred cross-resistance to chemically distinct BET inhibitors such as JQ1, as well as resistance to genetic knockdown of BET proteins. ... Fong et al. (2015) demonstrated that resistance to BET inhibitors, in human and mouse leukemia cells, is in part a consequence of increased Wnt/beta-catenin (see 116806) signaling, and negative regulation of this pathway results in restoration of sensitivity to I-BET in vitro and in vivo. ... In addition, overall survival at 5 years was 100% for 10 VLA4-negative patients and 44.4% for 15 VLA4-positive patients.
-
Nut Midline Carcinoma
Wikipedia
One of the most helpful and characteristic findings is the focal abrupt squamous differentiation, where stratification and gradual differentiation are absent, resembling a Hassall corpuscle of the thymus. [6] The defining feature of NMCs is rearrangement of the NUT gene. [4] Most common is a translocation involving the BRD4 gene and NUT gene (t(15;19)(q13;p13.1)). [5] [7] Treatment [ edit ] BET inhibitors are used for treatment [ citation needed ] Prognosis [ edit ] NUT midline carcinoma is very resistant to standard chemotherapy treatments. ... Specific molecular targeted therapies ( BET inhibitors and histone deacetylase inhibitors (HDACi)) may help to yield growth arrest of the neoplastic cells. [6] Overall, there is a mean survival of 6–9 months. [8] [9] See also [ edit ] BET inhibitor Mediastinum References [ edit ] ^ RESERVED, INSERM US14-- ALL RIGHTS. ... External links [ edit ] Classification D ICD - 10 : C80.9 External resources Orphanet : 443167BRD4, NUTM1, MYC, AFP, CDK9, SMARCA4, DNER, NSD3, HDAC9, CIC, SF3B1, ZNF532, CD274, SYP, MIR145, MIR99A, MIR3140, TP63, SMARCB1, SOX2, CCNT1, ROS1, BRD2, PIK3CG, PIK3CD, PIK3CB, PIK3CA, EZH2, EP300, EGFR, CDKN2A, CDK4, H3P10

-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Inclusion Body Myositis
OMIM
Similar aggregates were identified in 10 to 15% of the nonvacuolated normal-appearing fibers. ... The authors discussed the abnormalities of APP processing, the role of abnormal intracellular protein folding, oxidative stress, and the potential role of cholesterol in the pathogenic cascade of IBM. ... To elucidate the possible role of beta-APP mismetabolism in the pathogenesis of IBM, Sugarman et al. (2002) selectively targeted beta-APP overexpression to skeletal muscle in transgenic mice, using the muscle creatine kinase promoter. They reported that older (more than 10 months) transgenic mice exhibited intracellular immunoreactivity to beta-APP and its proteolytic derivatives in skeletal muscle. In this transgenic model, selective overexpression of beta-APP led to the development of a subset of other histopathologic and clinical features characteristic of IBM, including centric nuclei, inflammation, and deficiencies in motor performance.GNE, NT5C1A, APP, TARDBP, HLA-DRB1, SQSTM1, APOE, KHDRBS1, NUP62, DCTN4, GTF2H1, SDC1, CDR3, GSN, MAPT, TRBV20OR9-2, HLA-C, PLAAT4, FYCO1, MSTN, TNFRSF12A, NFAT5, CCR2, UBB, MALAT1, VCP, RBM45, AOC3, DCD, UCN2, DNAJB6, OPTN, KLRG1, MAP1LC3A, LILRB1, KDELR1, ICOSLG, SYNM, ROBO3, DDX58, CHMP1B, PABPC1, TIMP1, RRM2B, TWNK, FOXP3, KRT20, TTR, ACTB, THBS1, CST3, HLA-DQA1, HK1, H1-0, NR3C1, EPHB2, EMD, DES, CD47, TGFB1, CD38, CD36, CD34, MS4A1, CAPN3, BCL2, AOC2, HLA-DRB3, HMGB1, IFN1@, IFNG, TRIM21, AGER, MOK, PTPRC, PSME1, PSMB10, MAPK1, POLG, PMP22, MMP9, MMP1, MLF1, LMNA, IL6, IL1B, LOC102723996
-
Early-Onset Alzheimer's Disease
Wikipedia
This loss of brain volume affects ones ability to live and function properly, ultimately being fatal. [5] Beta-amyloid is a small piece of a larger protein called the amyloid precursor protein (APP). Once APP is activated, it is cut into smaller sections of other proteins. ... This type accounts for 30-70% of EOFAD. [4] This protein has been identified as part of the enzymatic complex that cleaves amyloid beta peptide from APP (see below). The gene contains 14 exons , and the coding portion is estimated at 60 kb, as reported by Rogaev (1997) [10] and Del-Favero (1999). [11] The protein the gene codes for (PS1) is an integral membrane protein. ... Alpha-secretase cleavage of APP, which precludes the production of Aβ, is the most common processing event for APP. 21 allelic mutations have been discovered in the APP gene. ... "A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid". ... The International Journal of Ageing and Later Life . 10 (2): 9–29. doi : 10.3384/ijal.1652-8670.16302 .
-
Chemical Eye Injury
Wikipedia
"Towards evidence based emergency medicine: best BETs from the Manchester Royal Infirmary. BET 4: use of litmus paper in chemical eye injury".
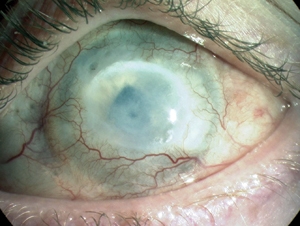
-
Party And Play
Wikipedia
For this reason, it is considered "a public health priority." [3] Contents 1 Terminology 2 Participants and drugs 3 Risks 4 Statistics 5 History and cultural significance 6 Criticism 7 See also 8 References 9 Further reading 10 External links Terminology [ edit ] The practice is nicknamed "party 'n' play" ("PNP" or "PnP") by some participants. ... Ketamine is used in chemsex encounters to "improve the experience of receptive anal intercourse or fisting". [10] A study of sauna participants in Barcelona, Spain, in 2016, found that the most commonly used drugs in chemsex are "GHB/GBL, cocaine, ecstasy, silver bars ( MDMA ), poppers and Viagra". [11] A 2014 study on chemsex in London, UK, indicated that the drugs associated with chemsex include mephedrone, GHB/GBL, crystal meth, ketamine, and cocaine. [10] Internet posts by men seeking PNP experiences often resort to slang to identify what drug they are partying with. [12] [13] These drugs tend to inhibit penile erection , [8] [9] a phenomenon known by the slang term crystal penis or tweaker dick . ... A key self-confidence issue for study participants was "body image", a concern that was heightened by the focus on social networking apps on appearance, because on these apps, there is a focus on idealized male bodies that are "toned and muscular". Men were also anxious about their sexual performance, and as such, taking drugs can reduce these anxieties and enable them to enjoy sex more. [10] [24] Criticism [ edit ] It has been observed that reliable data and relevant research are generally lacking and this situation is generating a climate of moral panic . ... David Stuart , retrieved 2019-09-01 ^ "Gay men need clear information about 'chemsex', not messages about morality" . The Guardian . 2015-11-10. ISSN 0261-3077 . Retrieved 2017-07-09 .

-
Neuroendocrine Tumor
Wikipedia
H&E stain Specialty Endocrine oncology Neuroendocrine tumors ( NETs ) are neoplasms that arise from cells of the endocrine ( hormonal ) and nervous systems . ... G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit.MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Alzheimer Disease
OMIM
Cerebral hypometabolism and impaired episodic memory were observed 10 years before expected symptom onset. ... Rovelet-Lecrux et al. (2006) estimated that in their whole cohort of 65 ADEOAD families, the frequency of the APP locus duplication was roughly 8% (5 of 65), which corresponds to half of the contribution of APP missense mutations to ADEOAD. ... Revesz et al. (2003) reviewed the pathology and genetics of APP-related CAA and discussed the different neuropathologic consequences of different APP mutations. ... To determine whether decreased neprilysin (MME; 120520) levels contribute to the accumulation of amyloid deposits in AD or normal aging, Russo et al. (2005) analyzed MME mRNA and protein levels in cerebral cortex from 10 cognitively normal elderly individuals with amyloid plaques (NA), 10 individuals with AD, and 10 controls who were free of amyloid plaques. ... By neuropathologic examination, Wilkins et al. (2006) found no difference in the presence or degree of neurofibrillary tangles, senile plaques, Lewy bodies, or amyloid angiopathy between 10 African American and 10 white individuals with AD.TOMM40, TREM2, ABCA7, APP, APOE, PSEN2, PSEN1, MAPT, SORL1, PRNP, CASP3, BACE1, GSK3B, NCSTN, IDE, IL1B, HFE, A2M, ACE, DHCR24, BIN1, ESR1, ADAM10, ADAMTS1, PGRMC1, VEGFA, ARC, CYP46A1, SLC30A4, VSNL1, PICALM, HMOX1, HLA-DRB5, IGF1R, IGF1, INPP5D, IGF2, MPO, NPY, NOS3, PLAU, PLCG2, PPARG, RELN, MTHFR, PYY, NECTIN2, SLC2A4, IGF2R, SOD2, MAOB, TF, LEP, TFAM, INSR, INS, TNF, TPI1, EPHA1, F2, ENO1, CR1, CASS4, ATP5F1A, CLU, CHRNB2, CHRNA7, MIR766, CD33, IQCK, EIF2S1, MIR505, APOC1, CALM1, MIR100, MIR146A, BDNF, BCL2, MIR375, MIR296, BCHE, MIR708, TPP1, SLC30A6, SNAR-I, DPYSL2, ACHE, CD2AP, GAPDHS, PCDH11X, CYP2D6, MIR4467, CRH, MIR3622B, BAX, AMFR, ABI3, CST3, MS4A4A, WWOX, BRCA2, FANCD2, TFF1, TAS2R64P, CTNNB1, SUCLA2, SNCA, CTSD, RNR2, NEFL, TAS2R62P, SOD1, ITPR3, ITPR2, ITPR1, FLAD1, PSENEN, TP53, CDK5R1, EIF2AK3, UBQLN1, ALG3, PIK3CG, PIK3CA, PIK3CD, SERPINA3, PIK3CB, DOCK3, APLP1, OGDH, CREB1, NOTCH1, CASP6, NGF, CCND1, FOS, DLX4, DLG4, DDIT3, RABGEF1, PEBP1, PYCARD, DAPK2, KCNIP3, CTSB, CSF2, CRMP1, CTSG, EHMT2, ENO2, ERBB4, TMED10, TERF2IP, PTK2B, FCN2, PTGES3, FGF2, ACKR1, DNM1L, SDC3, G6PD, GCHFR, ITM2B, CREBBP, MAP3K8, TRPM7, ADI1, MTCO2P12, UPK3B, ACTB, AKT1, AKT2, ANXA1, APBB1, DNLZ, STS, MIR34A, BRCA1, MIR137, C5AR1, DDR1, CAMK4, TMED10P1, MPEG1, C9orf72, ESCO1, CDCA5, PRRT2, MAP1LC3B, CAT, EHMT1, CNR2, SPPL2B, RAB9A, NRXN3, GFAP, SYNJ1, SERPINB5, CD99, MME, MNAT1, CCL2, RRAS, RPS27, RPS21, RAP1A, PYCR1, COX2, PTS, PTGS2, MTHFD1, MMUT, NCAM1, NFIA, NFIB, MAPK8, MAPK3, PRKCB, PRKCA, PPBP, MED1, NFIC, PPARA, NFIX, PKD1, NOTCH3, NRGN, MEOX2, MEF2A, SPRR2A, TTC3, GRIN2A, DENR, GRIN2B, RAB7A, LRP8, HPRT1, HSP90AA1, VIM, IDUA, UTRN, SUMO1, UBE2I, TTK, TPT1, SULT1E1, IL1A, IL6, IL12A, TSPAN6, TIE1, TGFB1, TG, KNG1, LAMC2, LGALS3, TERT, TERC, STIM1, H3P17
-
Abeta Amyloidosis, Dutch Type
Orphanet
Etiology HCHWA-D is due to a mutation in the APP gene on chromosome 21q21.2, encoding the beta-amyloid precursor protein. ... Genetic testing reveals a mutation in the APP gene. Differential diagnosis Differential diagnoses include other conditions that could cause intracerebral hemorrhage such as coagulopathies, vasculitis (see these terms), CNS neoplasms, cerebral vascular malformations, ischemic stroke and antecedent trauma. ... HCHWA-D is often fatal, either acutely at first presentation or, on average, 10 years after symptom onset and after multiple strokes.
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Pustular Psoriasis
Wikipedia
This skin eruption is often accompanied by a fever , muscle aches , nausea , and an elevated white blood cell count . [1] Annular pustular psoriasis (APP), a rare form of GPP, is the most common type seen during childhood. [6] APP tends to occur in women more frequently than in men, and is usually less severe than other forms of generalized pustular psoriasis such as impetigo herpetiformis. [6] This form of psoriasis is characterized by ring-shaped plaques with pustules around the edges and yellow crusting. [6] APP most often affects the torso, neck, arms, and legs. [6] Diagnosis [ edit ] Classification [ edit ] Pustular psoriasis is classified into two major forms: localized and generalized pustular psoriasis . [1] Within these two categories there are several variants: Classification of Localized and Generalized Pustular Psoriasis Localized pustular psoriasis Palmoplantar pustulosis (acute and chronic) Acrodermatitis continua (of Hallopeau) Generalized pustular psoriasis (von Zumbusch) acute generalized pustular psoriasis Acute generalized pustular psoriasis of pregnancy ( impetigo herpetiformis ) Infantile and juvenile Subacute circinate and annular Management [ edit ] injection of methotrexate This section is empty. ... "Acrodermatitis continua". Dermatology Online Journal . 10 (3): 9. ISSN 1087-2108 . PMID 15748579 . ^ Oumeish, Oumeish Youssef; Parish, Jennifer L. (2006). ... External links [ edit ] Classification D ICD - 10 : L40.1 ICD - 9-CM : 696.1 This cutaneous condition article is a stub .IL36RN, IL1B, CARD14, IL1A, IL1RL2, IL17A, PI3, TNF, IL22, AP1S3, TLR3, TNFAIP3, MOG, TNIP1, LDHA, IL23A, NOD2, IL1F10

-
Alzheimer Disease
MedlinePlus
Affected individuals usually survive 8 to 10 years after the appearance of symptoms, but the course of the disease can range from 1 to 25 years. ... The early-onset form is much less common than the late-onset form, accounting for less than 10 percent of all cases of Alzheimer disease. ... Researchers have found that this form of the disorder can result from mutations in the APP , PSEN1 , or PSEN2 genes. When any of these genes is altered, large amounts of a toxic protein fragment called amyloid beta peptide are produced in the brain. ... As a result, people with Down syndrome have three copies of many genes in each cell, including the APP gene, instead of the usual two copies. ... Learn more about the genes associated with Alzheimer disease APOE APP PSEN1 PSEN2 Inheritance Pattern Early-onset familial Alzheimer disease is inherited in an autosomal dominant pattern , which means one copy of an altered gene in each cell is sufficient to cause the disorder.APP, ACE, TREM2, ADAM10, APOE, PSEN1, GSK3B, HFE, MAPT, PLAU, NPY, BCL2, CASP3, BDNF, IDE, INSR, IL1B, LEP, BACE1, IGF2, IGF1R, ATP5F1A, INS, BAX, CR1, A2M, ABCA7, TOMM40, CD2AP, BIN1, EPHA1, CLU, PICALM, NOS3, PSEN2, APOC1, MPO, SORL1, VSNL1, INPP5D, NECTIN2, MS4A4A, PCDH11X, CASS4, BCHE, MIR146A, CYP46A1, DHCR24, CHRNA7, NCSTN, VEGFA, DPYSL2, PRNP, ESR1, PPARG, RELN, HMOX1, ACHE, CST3, MAOB, TNF, MTHFR, IGF1, CD33, TFAM, IL6, CYP2D6, CRH, SOD2, UNC5C, PLCG2, TF, ABI3, WWOX, SLC30A6, CHRNB2, ARC, PGRMC1, F2, CALM1, EIF2S1, HLA-DRB5, ENO1, TPI1, IGF2R, SLC30A4, MIR296, SLC2A4, MIR100, IQCK, MIR375, AMFR, SNAR-I, ADAMTS1, MAPK14, PIN1, PYY, PTGS2, S100B, PPARGC1A, NOS2, NGFR, NGF, NFE2L2, SOD1, SYP, CDK5, NGB, MIR505, GAPDHS, MME, MAP2, CTNNB1, TPP1, LRP1, IRS1, CHAT, GAPDH, MIR4467, MIR3622B, AGER, MIR766, MIR708, CAV1, NTRK2, PTGS1, APLP2, ADAM17, MFN2, DNM1, HSF1, GSR, IL33, CCR5, HSPD1, HSPB1, CIB1, CASP8, IKBKB, SERPINF1, ATP7A, MT2A, ADAM9, INS-IGF2, BCL2L2, CASP9, GAB2, PTK2B, PLCB1, ABCA1, GRN, CASP12, SQSTM1, FERMT2, HLA-DRB1, NFIC, CSF1R, APOB, MARK4, HSPG2, MS4A6A, CELF1, VCP, SYNJ1, ZCWPW1, MS4A4E, APH1B, APOC2, F13A1, EXOC3L2, PLXNA4, ADAMTS4, AKAP9, MADD, DST, PILRA, FRMD4A, LAMP1, SLC24A4, GLIS3, SPON1, CADPS2, IL34, COL18A1, TRIP4, SPI1, TGFB2, BCL3, MTHFD1L, AICDA, IL6R, DCHS2, MEGF10, SLC16A7, EPHX2, NDUFAF6, DSG2, OSBPL6, CELF2, UBE2L3, SPPL2A, MAPK7, CDH13, LAMA1, SGK1, SUCLG2, LUZP2, PTPRG, ST6GAL1, AP2A2, RBFOX1, SORCS3, TSPOAP1-AS1, TLN2, ZAP70, ALDH1A2, TCF7L2, FMN2, OTOF, EXOC4, HSD17B10, DNM1L, ALOX5, GULOP, HTR2A, AHCYL1, SDR42E2, HTR6, GTF2H1, GRM5, IAPP, AHSA1, STAG3, ALB, FARP1, TSHZ1, HRES1, AGFG2, DCAF7, SIGMAR1, BCKDK, RTN3, TPPP, HSPA4, HCLS1, HSP90AA1, G3BP1, PGAM5P1, BACE1-AS, KHDRBS1, ALDH2, CFH, HCRT, GPC6, ABCA8, GLP1R, NR3C1, TNRC6A, IL19, PARVB, DDX25, BZW2, FCN2, FGF10, GOLIM4, LINC00476, HPGDS, FANCD2, PDE7B, SIGLEC7, TSPAN16, TRPC4AP, POLDIP2, CNTNAP2, RNF19A, BACE2, UBQLN1, ITSN2, GRIN2B, ZGLP1, BCAS3, EPDR1, TMED9, ENO2, CYCS, ANXA1, EPHB2, EPO, RAPGEF6, APH1A, LARS1, EYA4, SNX9, ESR2, WAC, SLC8A1-AS1, DCTN4, RMDN1, QPCT, MTOR, SGK3, FBXL7, SZT2, GIP, ACSL6, WWC1, CLEC16A, KAZN, LINC01672, PRRC2C, COLGALT2, PLD3, HECW1, ZNF292, MYO16, DKK1, SIRT2, ACOT7, PSIP1, CLASRP, LINC00271, SIRT3, SIRT1, GFAP, NUP62, FYN, CBLC, INHCAP, TRIM51CP, GABPA, GABRA2, GABRG3, DAPK2, SMUG1, GAP43, GATA1, GCG, GCHFR, TARDBP, NCS1, GDNF, HDAC6, RAB3D, MVP, OGG1, LINC02268, LINC02325, SOAT1, ACTG2, ACTG1, SNCG, SNCA, SNCB, SNAP25, NOS1, ACTB, MEF2C-AS1, SLC6A4, NPC1, SLC6A3, GEMIN7-AS1, SLC1A2, LINC01508, LINC01725, NEFL, COX2, TNFRSF1B, STAG3L5P, TLR4, TLR2, MSH2, THY1, MT3, TH, SST, STAG3L5P-PVRIG2P-PILRB, TGFB1, TFF1, RNR2, LINC02653, LINC01712, TCF3, NRGN, CX3CL1, ELMO1, CCL2, PIK3CG, ABCA2, PLA2G1B, EIF2AK2, SERPINA3, PLG, MAPK8, MAPK1, PRKCB, PRKCA, PRKAB1, PRKAA2, PRKAA1, PMS2P1, POLD1, PTPA, PON1, PIK3CD, PIK3CB, PIK3CA, MOK, UPK3B, SORT1, ROS1, SERPINE1, REST, REN, RELB, RAC1, LINC01965, PVR, PVALB, PTPRA, LINC00972, ABCB1, LINC02008, MTCO2P12, TP53, OVCH1-AS1, MOBP, MNAT1, SLC4A8, AGT, INSIG1, AZIN1-AS1, EIF3E, FHL5, IRF2, GSTO1, ITM2B, GRAP2, LIPG, ADIPOQ, KL, MSC, LRAT, AP4M1, CCRL2, IL18, IL17A, IL12A, ST18, IFNG, ARL17B, AKT1, AIF1, KRBOX1, IGFALS, HDAC9, PHF14, IL10, IL1A, ALOX12-AS1, MICAL2, IL2RB, IL4, CLOCK, CXCL8, KCNN2, MPZL1, HERC2, VDR, TFEB, MAOA, COX10-AS1, ZNF232, YWHAZ, VLDLR, PARP1, UTRN, BCAM, UCHL1, UBB, TYROBP, AFF1, TTR, TRPM1, MMP9, AIMP2, LTBP2, KNG1, CRADD, CACNA1G, LAMC2, RPSA, CDK5R1, SUCLA2, LCN2, LDLR, BECN1, LPL, PDE5A, ABCB11, APOC4-APOC2, KHSRP, DENR, AGPS, LPA, CDKAL1, PPARA, MEIKIN, COL4A4, CRK, CEACAM22P, SCIMP, CREB1, CR1L, ZNF862, SH2D4B, CP, PNPLA7, SIMC1, GGACT, COMT, COL12A1, FAM181A, BRCA2, PPP1R37, GPR141, TENM3-AS1, CNR2, L3MBTL4, UBXN11, ACKR2, TMEM132C, CASTOR3, CLPTM1, STRADA, FNIP1, CDCA5, NLRP3, APOA1, CRMP1, KAT8, CSMD1, CLMN, PINK1, CYP8B1, AHNAK, MIR132, MIR107, CUX1, POTEM, PPP1R3B, FAS, SAP30L, ANKRD55, CTSD, CTSB, GEMIN7, EHMT1, LINC01184, CTNNA2, LINC01185, TMC5, THSD4, CCDC134, SP6, LINC01567, PDCD1LG2, SETD7, APOD, BHMG1, CSF2, HYI, BLOC1S3, TSPO, CHRNA4, CHRNA2, TAS2R62P, C3orf67, C9orf72, CCDC83, CCDC89, KDM1B, CD14, TGM6, ATXN7L1, RSPO4, ADGRF2, STH, TAS2R64P, CALHM1, RUNX1T1, PPP1R42, ALPK2, PCSK9, CAT, ANKRD31, CASP6, NKAIN3, TRIQK, CALB1, STEAP1B, CASP1, EPHA1-AS1, CAPN1, APOC4, FAM181A-AS1, NKPD1, SPRED2, CD36, SCARB1, PLPP4, MED12L, ARAP2, CHN2, CHI3L1, ACTBL2, C10orf71, MCIDAS, LRRK2, AKR1C4, ANO4, AGBL1, CEACAM20, ZNF813, RMDN3, CETP, CDR1, LINC00343, TCAM1P, APOC1P1, IGSF23, RMDN2, CDK1, SLC25A48, NKAIN2, FSIP1, CD68, BMPER, C3, CD40, CYP19A1, CRP, NIT2, ANO3, DLG4, ARHGAP20, RCAN1, WDR41, NDUFA12, STK32B, EDEM2, DSCAML1, RNF165, SH3RF1, DYRK1A, MIR29A, SYBU, AQP4, APBB1, DLX5, DBN1, PALM2AKAP2, CEACAM19, DPP4, IL6-AS1, ARVCF, CDC42SE2, DMXL1, TULP4, DAPK1, PMS2CL, POTEKP, MIR34A, VAT1L, OLR1, HDAC2, LRP8, GSN, CCL11, S100A9, COL25A1, POTEF, KLK6, BLMH, HSD17B7, P2RX7, COX8A, ABCB6, PRRT2, IL2, SORCS1, NR1I2, MAPK3, ITGAM, CASR, ATP7B, VDAC1, EGR1, PDE4A, RAB5A, SUMO1, NRG1, OXER1, NTRK1, TFCP2, ANK1, CSNK1D, DLST, APLP1, BLVRA, NFIB, IL1RN, HTT, ACAT1, PLA2G4A, NFIX, NLRP1, GPRC6A, HMGCR, PPID, LPAR3, FZD4, REG1A, MRGPRX1, LRP2, DBH, PSENEN, VPS35, ESCO1, HSD11B1, VN1R17P, SOX2, AGTR1, XBP1, MIR155, MRGPRX4, MRGPRX3, GAL, GPR151, IL13, PAEP, OGDH, GPR166P, STAT3, SET, NFIA, PLB1, AR, LGR6, DHRS11, ABCG2, C4A, KCNIP3, HSD17B13, ABCG1, TTBK1, NOTCH1, EIF2AK3, SLCO6A1, CHMP2B, RBM45, CD44, RIPK1, APBA1, GSTK1, ADNP, ICAM1, BRCA1, APCS, TNFRSF1A, NFKB1, CNTF, MMP3, KLC1, LBP, CTNNA3, SGSM3, FGF2, C4B, HIF1A, CREBBP, SERPINA1, TMEM106B, GRIA1, GRIA2, ECE1, C4B_2, GSAP, OGA, TFRC, PLA2G6, ST3GAL4, PAWR, MFAP1, KAT5, GSTM1, APRT, COX1, HP, NTF3, MIR206, FPR2, CDC42, FUS, MARK1, FGF1, PREP, C5AR1, PON2, MIR29C, CALB2, PDIK1L, SYK, S100A1, CH25H, SREBF2, COX5A, GRIN2A, VCAM1, TMED10, GSTP1, KLK8, PHF1, CXCL10, MEFV, SP1, GJA1, IGFBP3, SLC17A7, CYP3A4, FOXO3, HMGA1, SLC11A2, XPR1, MARK2, PPIF, CRHR1, SHANK3, MYC, CD40LG, CPOX, FKBP5, ANPEP, CAST, C1D, FKBP4, HSPA1A, FLT1, MIF, PLA2G2A, CX3CR1, CSF3, IFNB1, KALRN, PLTP, STXBP3, DDR1, PWAR1, PRDX2, TP63, VIM, IL23A, F2RL3, MMP14, MEF2C, TREM1, TMED10P1, NAT2, MIR342, SAMD9, RAB7A, PGR-AS1, TRPM2, EGFR, ADRB2, CLDN5, ETS2, SYT1, TIMP1, NME8, ELANE, F2R, CD59, EPHA4, CBS, MSMB, APLN, MMP2, MYCL, CALML5, SYN1, XRCC1, TGM2, EEF2, PLA2G7, ELAVL2, EDN1, TMEM97, HMGB1, MIR455, HTRA1, BPIFA2, SLC52A2, NQO1, TUBA1B, FOS, CRYAB, SLC2A1, GPR3, LGMN, SLC2A3, RIDA, FN1, ABCA4, HSPA1B, PECAM1, PTEN, HSPA8, HLA-A, PTPN1, HAMP, TXNIP, GRM2, P4HB, LIN28A, PSPH, GSTT1, CCN2, DECR1, CPLX1, BCYRN1, NES, POU5F1P4, MIR21, GRK5, POU5F1, NTSR1, MIR212, PRKN, LINC02210-CRHR1, HSPA5, DLD, DAB1, HTRA2, POU5F1P3, MIR137, DNAH8, MAPK10, GH1, SERPING1, ADAMTS2, EEF2K, GSTO2, ROCK2, NEDD9, SPTBN1, NTN1, CEBPD, GDF2, CEBPB, PWAR4, SYNM, IGFBP2, GLUL, ABCC9, ATM, PSPN, PTPRC, MIR29B1, RANBP9, NDRG2, CNR1, RTN4R, PTBP1, AQP1, PDK1, MIR106B, PDE4D, ARNTL, PRDX1, ADM, RENBP, POMC, PTPN4, MS, PDGFRB, P2RY2, MIR142, PDE9A, SSTR4, KLK3, BCL2A1, C2, SRPK2, NFATC2, ADCYAP1, LRRTM3, PLD1, NUBP1, MIR424, ATF4, BSG, MIR29B2, PPY, BMP4, TBP, SLC18A3, POLB, NOTCH3, SLC18A2, NPTX2, MTR, SI, MIR222, SH3GL2, ND2, NR4A2, SELENOP, CXCL12, CCL5, ATXN1, CALM3, ITGAX, IFNA13, DISC1, OPTN, HTR1F, HTR4, WNT3A, COL11A2, C20orf181, IFNA1, KEAP1, HDAC4, KLK4, SEMA6A, LRRC4, CRTC1, IL9, DAO, ALOX15, AGTR2, IDO1, SLC25A27, ABCG4, CD55, APOA4, MCOLN1, REM1, ATCAY, EBPL, HSPB2, HSPA9, PLK2, GAD1, NANOG, DCX, COASY, UBE2K, DDIT3, TREML2, APBB2, MAP1LC3B, SRRM2, GZMB, FXN, HNRNPA1, HPSE, RAB10, CIP2A, FOLH1, STIM2, DIO2, MMP24, CEBPZ, GBA, CDR2, ITPR3, CDKN2A, MELTF, SLC30A3, ADRA2B, FTO, FNDC5, GGA3, XPNPEP1, VGF, NR1H2, UGCG, MFGE8, MGAT3, CXCR4, TLR9, APOC3, GPT, ELK3, NEAT1, ADORA2A, MMEL1, TRPC6, EIF4E, CAMK2A, MS4A6E, SRR, HSPA14, IRS2, MGAM, C1orf52, HDAC3, PABPC4, ACKR3, LGALS3, FAM20C, WNK1, DRD4, CYP2C9, MBTPS1, DRD1, LRP6, GRK2, CYP2B6, OGT, LIPA, AD11, GORASP1, PTGDS, SPEN, MIR200B, NPTXR, DNMBP, MIR200A, NCOA6, MIR181C, RBP4, RELA, OPN1LW, EFHD2, MIR188, TPH1, HNRNPA1P10, CTNNBL1, SLC40A1, PNO1, CHCHD2, SDF4, RETN, GOLM1, PPP3R1, PYCARD, PAG1, CCR2, DDIT4, RCBTB1, SBNO1, PPARD, CD274, PCBP4, ACE2, PROS1, PRND, PPP1R15A, CIZ1, MIR26B, TPSG1, GGA1, CFAP97, MAP2K1, AATF, SHANK2, PRL, RBMS3, LOC107987479, MAP2K2, PAXIP1, CHCHD10, SBNO2, PTGES, SPHK1, LPAR2, PPIG, NRXN3, MED23, SPP1, MAPK8IP1, BAG3, APBA3, TAP2, PRDX6, CARTPT, SNAP91, SV2A, MALAT1, MAK16, SYVN1, GDF11, TAC1, TAT, SLC6A2, TRPV1, CISD3, TPT1, TSC2, TM7SF2, TXN, UBE2I, TLE1, TIMP2, XK, CNTN2, TGFBR2, YY1, GOLGA6A, ANP32A, TAM, TERT, DCP1B, TNFSF10, PPP1R1B, GPHN, RGS2, ADAM30, SAA1, METAP2, IMMT, SDS, ADAP1, RYR3, ECHDC3, RYR2, RXRA, SCD, RPS6KB1, CIT, RPS6, CTXN3, LMTK2, NLGN1, ROCK1, RGS4, ATXN2, SRL, STUB1, SHBG, LILRB2, AKR1A1, OLFM1, SKIL, SLC9A6, CREB3, PITRM1, SCGN, CXCR6, OCM, RIN3, SGCA, SFPQ, NCKAP1, CPLX2, TP73, EDAR, CCL3, NPS, BEST1, HPS1, CXCL1, GLO1, LIF, CDH1, LHCGR, CDK4, PCNA, PCK1, L1CAM, GPR42, GPX1, GRB2, ANGPT1, ANGPT2, CETN1, MYD88, GLB1, AKT2, LMNA, DNMT3B, TSC22D3, CD38, PER1, CD69, DRD3, LOX, CD74, LMNB1, DNM2, GAS6, AVP, GC, DMRT1, SARDH, GRIN1, ANXA5, BACH1, P2RY1, INPPL1, CSF1, CS, NEFM, NTS, APEX1, STS, HTC2, NEUROD1, NPTX1, NPPA, ATF2, HTR2C, ARRB2, IGFBP7, HMGCS2, CLK1, CKB, APBA2, CYP17A1, GSTM3, CHGA, CYBB, JUN, CTSS, ORM1, ITPR1, CHRM1, CHRM2, OPRK1, OPRD1, CTRL, HK1, ADRB1, LAMP2, CASP2, EDNRA, PLD2, CAPN2, F2RL1, ACO1, ERBB4, FAT1, FGFR3, CALM2, BRS3, CALCA, CAD, EGF, CASP4, FCGR3B, FDPS, LYZ, FCGR3A, FLNA, MECP2, FABP3, ADRA1A, PLXNA2, MBP, FAAH, ENPEP, F12, BAG1, MEOX2, HOMER1, ITGB2, DBA2, ITGAL, ITGB1, AIM2, AZIN2, CD80, ITIH4, CD46, VIP, CHRNA3, ATG5, TREML1, MCL1, GPRASP2, VWF, APOA5, TMEM119, KLF4, SOCS6, WNT1, XBP1P1, FOXQ1, C3AR1, OPN4, USF1, C1QA, CXCR2, VPS26A, MOGAT3, CCR3, IL6ST, IL5, IL1RAP, LMF2, IL1R1, CREB3L1, UNG, MCU, CLSTN3, SNPH, IL9R, NPEPPS, NAPSA, USF2, MEF2A, ING1, CGB8, FTMT, CHM, VAV1, IMPA1, C1R, ILK, ADAMTS3, IL16, MAP3K5, IL15, GDF15, CGB5, CA2, KCNB1, TSPOAP1, SMAD2, DPPA2, SGO1, LIG3, IFNL3, TNK1, CP20, APCDD1, HSD17B6, CDKN1A, TTBK2, CDKN1B, SLC2A14, CFLAR, STMN1, LIPC, TAB3, CHIT1, BRAP, SPARCL1, MLKL, PTCRA, CD47, LGR5, CD8A, CCT, NR4A3, USP9X, MSRB3, CDR3, CCK, LNPEP, CASP7, CHRFAM7A, CAMP, PER3, YES1, CGA, MARK3, CGB3, SYNGR1, CALCR, IL1RL1, ARHGEF2, PER2, SLC33A1, TPH2, CHEK1, RAB7B, NOG, MBL2, CFL2, HSPB6, AHSA2P, SLC30A1, CD200R1, SOCS3, KDR, KIF5A, HAP1, CALR, CES1, TRPA1, HSPB3, WASF1, SLC32A1, ARHGEF7, CAMK4, COL3A1, H3P40, SCRN1, PTCD1, SPHK2, ATN1, PPIL2, POU2F1, FOSB, GCA, FLT4, SH2B1, APPL1, DNMT1, FLG, FOXO1, DUSP1, DUSP6, HHAT, HSPB8, E2F1, PLXNA3, DOCK2, PNPLA2, ADI1, GMFB, GPI, GPC1, KIF21B, NMNAT2, TRIB3, ALS2, DLG2, DLG3, GLS, PDSS2, ASTN2, MCF2L, GLI2, KIDINS220, CBLIF, SYNE1, DMD, GGT1, FKBP1A, SIT1, SV2C, GDE1, FOXP3, ASCC1, TMED7, FIS1, PRLH, CRYL1, ADIPOR1, LSR, F11, MBL3P, SIRT6, TRMO, NRN1, LCMT1, PRRX2, ERN1, BIN2, UBR5, HEBP1, GEMIN4, PDCD4, TBK1, SLC25A38, FGF14, PCSK1N, TRPM7, DLL1, FLVCR1, AHI1, SETD2, ELK1, IL22, NCAPH2, ELN, PADI1, BPTF, NRBF2, FABP5, EP300, PLA2G3, GRHL3, CXCR3, NAV3, SIRPB1, FLOT1, MET, KCNMB2, CRISPLD2, ARHGAP24, HNMT, SNX27, NPL, BHLHB9, TRIM13, HMOX2, KLF2, CSNK1E, LPAL2, CPQ, PPP1R2C, RAPGEF3, TET1, CRYZ, SORBS3, CTBP1, BCL2L11, COL17A1, NCAPD2, COX10, UBASH3B, NR1I3, ACOT8, PTPN5, PPP1R9B, TOM1, MINDY4, CPE, HTR7, NR1H3, HTR1B, HTR1A, LRPPRC, PDIA6, RHBDD1, NAA25, HLA-C, TPX2, BCAN, ADAMTS13, MOAP1, TNMD, GRIA3, NEUROD6, CHEK2, PADI2, HRH3, PHB2, SIL1, MGLL, FFAR1, GADD45A, MMRN1, DEFA1, IL21, NLRC4, GPR17, AZI2, HHIP, CTF1, HCRTR2, HLA-B, CTNND2, HHEX, HGF, CTSG, HDAC1, CTCF, DHX40, PTGES3, STIP1, CTSZ, CXADR, CARD14, CYP1A2, PDE10A, LILRB1, EHMT2, PDIA2, UMOD, ANGPT4, MIR339, SYN2, MSD, ACP3, APEH, ST8SIA1, AZU1, PI4KA, NCAM1, MIR144, SMIM10L2B, MSI1, NAP1L1, PRSS3, PEBP1, MASP1, SIM2, TIA1, MIR15B, ATD, RPL29, ABCC1, NCAM2, MIR125A, TLR5, SERPINF2, TNFAIP1, CXADRP1, MAPK9, ZFHX3, GGTLC4P, AD10, SGCG, MIR451A, CDR1-AS, TPTEP2-CSNK1E, MIR384, ITSN1, CBSL, MPZ, NFATC4, PKM, NCL, NTF4, LOC643387, SLC1A3, APOA2, THAS, PSMB6, SERPINB6, PSMB9, RHOA, ARMCX5-GPRASP2, SMPD1, REG3A, ATP4A, MIR193B, RFC1, NOTCH4, SLPI, PGF, AEBP1, MIR214, MIR219A1, SLC19A1, MIR22, PCSK1, ALPP, AMD1, MTNR1A, TGFBR1, COX3, ATP12A, NM, ADD3, PSD, TGM1, ARR3, NPM1, PAK1, TMED7-TICAM2, PNP, MIR195, MTHFD1, ADH1B, AMPH, ND4, AMD1P2, ARG1, SULT2A1, RRAS, PDE7A, TTPA, TYK2, TXNRD1, PPP1R1A, PPP1R10, SPG7, SPAST, RAB4A, PPP2CA, OTC, PPP2R2B, MMP1, ARMS2, RAB3A, GGTLC3, RTL1, P2RX4, ST13, SPARC, ALAS1, PPP3CA, NEFH, SEL1L, TYR, RAB6A, MICB, PPIA, CCL4, BST1, TICAM2, BNIP3, MIR326, OPRL1, PON3, BMP6, OPRM1, AHSG, H3P17, SDC2, AGRN, TYRP1, PPP1CA, BMI1, TYRO3, PNMT, DEFA1B, GGT2, ORI6, SMIM10L2A, CISD2, ARSA, EIF2AK4, PRKAR1A, LRP1-AS, SRSF2, MIR98, MIRLET7B, CD200, PDCD1, RAP1A, GGTLC5P, CCND1, ANXA6, FXYD1, S100A6, NFATC3, PLK1, ABO, PTGER3, APC, S100A12, ASL, HSP90B2P, SETMAR, PRRX1, PZP, STAT1, ODC1, CFB, CDNF, ZBTB4, PARK16, SUGP1, DIO1, SORCS2, MAGEE1, ALDH1A1, MIR1306, DES, DIAPH1, LSM2, MIR1229, XPO5, HCN3, CFD, MIR664A, KIF17, WDR48, MTRNR2L12, PRX, DHFR, EPG5, SEPTIN1, FAS-AS1, RNF213, MIR320E, MIR1908, HECW2, LINC00672, NUFIP2, ABCD1, DLG1, QRFP, ZNF410, AOC2, NBEAL1, CYP11A1, OPN1MW2, AD6, P2RY12, NMNAT1, DEPTOR, TNS3, CYP2D7, FAM72A, NUCKS1, CLEC7A, CYP26A1, ARAP3, GREM2, CDKN2B-AS1, CYP2J2, UBE2Z, MIR1246, TSPY3, MIR632, CTSK, GTDC1, CTSL, MIR650, MIR660, SNORD118, TNFAIP8L2, LYNX1, MUL1, PAGR1, CYLD, MAPKAP1, APOF, PDCL3, CYP2C19, NOC3L, CYP27A1, DPEP2, MFT2, PROK2, HPSE2, AD14, AKR1C2, ALPI, TRPV4, NTN4, PRM3, PDF, JAM2, ALOX5AP, TSPY10, DEFB4A, DEFB4B, ALOX12, PTBP2, DCN, NECAB3, FKBPL, NEUROG2, DGKQ, SLC25A4, MIR873, MIR301B, CENPK, DAXX, GFRA4, GOLPH3, ERVK-6, MTUS1, DBI, MIR937, ANG, ACE3P, SOD2-OT1, ANK3, DRD2, ZNF608, NAT10, DYM, LOC102724334, TRIT1, EIF4G2, TET2, EIF5, SERPINB1, ELAVL4, PDP1, ACO2, THRA1/BTR, UGT1A1, CCHCR1, CPVL, SMOX, TOLLIP, TERF2IP, SNTG1, EMP1, LOC102723407, EIF4EBP1, MIR6845, EIF2S3, ADCY2, NUDT11, EGR2, MSTO1, ADARB1, SLC6A15, ADA, TAPBPL, TESC, MIR6840, ACVRL1, FOCAD, EIF4A1, CASZ1, QRICH1, PGPEP1, EIF4A2, NDE1, ASIC2, CTTN, ACADVL, GSKIP, LNCRNA-ATB, ATP6V1H, H3P7, TDP2, ERBB2, CINP, ZCCHC17, H3P13, DTL, GPRC5B, ERCC1, DCDC2, NAT8B, GULP1, H3P23, ERG, H3P28, H3P11, PPIL1, STIN2-VNTR, NANS, EPHA8, H2BS1, POLE3, ACACA, SLC29A1, FXYD6, LRP1B, CST12P, SIRT1-AS, INPP5K, MSRB1, ARID4B, EPOR, ABL1, AAVS1, NR2F6, ERVK-32, LOC110366354, MNS16A, EFNA5, SLC47A1, ALAD, EEF1A1, SNHG19, MICA, DNTT, SOX21-AS1, DOCK3, DPYSL3, MIR626, XAB2, MFF, DUSP22, ARNTL2, SPPL2B, MCCC1, TMX2-CTNND1, ANKS1B, DPYSL5, FXYD6-FXYD2, BARHL1, DSC1, TWSG1, TLE5, DNASE1, DNA2, OCLN, NLN, AMIGO1, AHR, PLEKHG5, SLC24A3, SPC25, TTC7A, PELI1, JAG1, TMEM159, RTN4, APMAP, CD177, CAMK1D, PLAAT1, NR0B1, TIGAR, P2RX5-TAX1BP3, PARD3, GKN1, ADH6, INAVA, CDK5RAP2, OGDHL, LINC01080, ATF7IP, IPO9, VAC14, DVL1, PPP4R3A, OPN1MW3, EBM, OTUB1, SOX6, SLC30A10, SMPD3, MEG3, PLIN2, FBXW7, TDP1, ADORA1, DSC3, ACSS2, BTNL2, KIAA1217, ZNF253, CFC1, MIR4668, DSG1, APOM, MYO5C, MIR4487, NOTCH2NLC, USE1, SELENOS, GDNF-AS1, DSPP, ADCY10, ADRA2A, ZNF415, LINC-ROR, NARS2, CSF2RB, MIR616, MIR20A, CDC25C, PROM2, ATP6V1E1, IL23R, GLIS1, PM20D1, PHF13, CDH2, ZNF569, MIR191, CDK9, MIR192, PRIMA1, CDKN2D, MIR196A1, OR2AG1, LAYN, PIWIL4, MIR19B1, GPBAR1, CDC25B, GDF7, ZDHHC15, MIR139, CD63, SGMS2, MIR140, TMPRSS6, RHBDL3, AVPR2, MIR15A, CBLL2, MIR186, PRUNE2, AMOTL1, CD81, MIR18A, SLC2A12, CDA, MIR181A2, ATP5PO, SESN3, ATP6V1B2, UBR1, ATP5PF, PPME1, MIR224, LYZL4, KCNH8, MTERF4, MIR23A, CPO, ACMSD, MIR23B, BHLHE23, MIR25, CFL1, OSCAR, SPNS2, SEZ6, SLC38A10, MSI2, CFTR, MIR27A, MIR223, ALDH7A1, MIR221, SELENOM, ATP5MC2, CACUL1, HECTD2, SREK1, CTCFL, CBLN4, CDSN, ATP5MC1, DEFB104A, OCIAD2, MAGEC3, MIR210, CEACAM5, CECR, PPARGC1B, ATP5F1B, IL31RA, GNPDA2, SCARB2, NSMCE1, SOCS4, UBE2L1, BNC1, CACNA1C, SLC25A20, SERPINA13P, BLM, SREK1IP1, MIF-AS1, C20orf203, SYPL2, ZNF763, CCL4L1, BID, BGN, ZSCAN1, ZADH2, SMIM20, MILR1, PGP, GOLGA6L2, TMEM189-UBE2V1, TMEM189, IL31, C4BPA, BTK, AMIGO2, HCN1, NHLRC2, ATP9B, SBSN, BMP1, OSTN, C5, CFAP410, BARHL2, NANOS3, C9, STING1, GADL1, ARMH1, VPS51, HCAR2, CAPG, LINC00639, TMEM201, LIN28B, CD5L, MIR127, CD19, KIF6, MS4A1, MS4A3, HYLS1, STOX1, FOLH1B, OR8J1, TRIML2, CD28, KHDRBS2, GAPT, CENPV, KLHDC8B, MIR134, CD86, PIKFYVE, SLC29A4, CCNC, KRIT1, PTF1A, DAOA-AS1, BDKRB2, HCA1, BRD3OS, ASPM, BCS1L, SGMS1, BCR, MIRLET7D, MIR122, RUNX1, ANKK1, BCL6, PHYHD1, BAK1, MIR10A, EBF3, CCKAR, MIR28, FRMD6, MIR613, ADAMTS10, TMEM175, XIAP, MAF1, BIRC3, SLA2, ANTXR1, ASCC2, CRHBP, EVA1A, QRFPR, MAGT1, NCALD, LOC646506, ROPN1L, L3MBTL2, GMNC, SCFV, CSE1L, RNF146, PHF6, HOOK3, BRSK1, MBOAT4, COX15, MIR497, MFSD2A, MIR501, ACCS, ARG2, FAM126A, CPB1, CPN1, ECSCR, MAP1LC3A, MIR484, AQP9, CPS1, SNORD35B, CPT1A, ABLIM2, FASLG, NETO1, DOCK8, RNFT2, C1QBP, TM2D3, ASRGL1, PTGES2, PANK2, MIR590, SCD5, CSNK2A1, VCAN, ZC3H14, CSPG4, CTBS, MIR592, MIR598, CAMKMT, CTNS, SLTM, PTCD2, CTNND1, MIR603, NUBPL, WDR26, SPHKAP, GSTT2B, TMEM163, NCF1, LBH, SPAG11A, SFTPA1, CSF3R, ZNF436, CSN2, NDFIP1, MIR545, SLC44A4, SLC19A3, FAM72B, AIRE, IQCJ, CSNK1G2, DNAJC5, CSNK1G3, LINGO1, ATG4C, ATP2B4, MIR346, SERPINC1, CISH, ASPA, CLC, UCN3, TMEM54, ASS1P1, CLCN3, NACC1, ASIP, STX1B, IFT43, MIR133B, MIR151A, CLK2, TP53INP1, MIR330, MIR335, MIR338, MIR93, SLC26A7, OMA1, CHGB, PLD4, TDRD9, CHD1, MIR299, ATIC, ATHS, H4-16, LRIG3, EXOSC6, CHRM3, MIR30B, MIR30E, AGAP2, MIR31, MIR34C, MIR9-1, GRIN3B, GRIN3A, ASAH1, MIR369, H2BC12, KPRP, LRSAM1, MIR429, H4C15, SHF, GADD45GIP1, COL11A1, ZNF628, MIR431, HNP1, NAV2, MIR409, SLC31A1, RPPH1, SNORD14E, SNORD14D, SNORD14C, SNORD14B, COX6B1, MIR485, CNTN1, DNM1P33, CNTFR, CHRDL1, CLN3, MIR361, MYOCD, PRDM6, DNER, SPECC1, MIR377, CLN5, EXOC3L4, CNP, MIR425, ARRB1, ZNF804A, BDNF-AS, NLRP12, CCR6, ABCC2, DEFB104B, LRRC15, POU3F4, HOOK1, TERC, NAT1, MCM2, EZR, MDH1, VEGFC, MDH2, MDM4, MEF2D, UVRAG, UROD, UQCRC1, UGT1A, SLC35A2, UCP2, UBTF, UBP1, MID1, UBE3A, UBE2V1, CXCL9, ATXN3, UBE2D2, UBE2A, UBC, MAP3K10, MC1R, WARS1, WAS, ZMYM2, MANF, SCG2, FZD5, SMAD7, MAG, MAP3K12, MAP1A, MAP1B, ZNF236, ZNF224, ZNF217, RNF112, WEE1, MZF1, ZIC1, MARS1, MAS1, MAT1A, MAT2A, MAZ, XIST, WT1, WNT2B, WNT5A, UBA52, KMT2A, MLLT3, THBS1, TLR3, TLE3, MSH3, TKT, TIMP4, TIMP3, MSR1, MSRA, THRA, THOP1, THBS4, CYTB, MRE11, NUDT1, TGFBI, TGFB3, ND1, TFF3, TFDP1, MTNR1B, MTRR, TRNL1, MUC1, TERF2, TSPAN7, MRC1, TWIST1, TRAF2, TUBA4A, NR3C2, TTN, TSPY1, TSHR, TSG101, TSC1, MMP7, MMP8, TRPC1, TRH, NR2C2, TNFAIP6, MMP13, TPM1, MOG, MOV10, TP53BP2, TP53BP1, MPG, TNR, TNNI3, MPI, MPST, SLBP, REEP5, DEK, SNX3, TNFRSF6B, RAB11A, LDHA, LEPR, LGALS4, LGALS9, GPAA1, RNMT, GBF1, ADAM19, URI1, TRADD, LCK, B3GALT4, LIFR, LIG1, SOCS1, NUMB, LIMS1, AOC3, PDE8B, USO1, TNFSF11, STK16, LCT, CES2, KMO, KLRC1, BRSK2, KCNQ1, KIR2DL2, NOL3, ATP6V0E1, SELENBP1, USP13, KLKB1, CDK5R2, RAB29, MBD2, KIF11, TMEM11, ENDOU, EIF2S2, TAX1BP1, NAE1, KRT14, KRT18, LAMC1, LBR, PROM1, LCAT, SOCS2, PRKRA, DEGS1, DDX39B, PABPN1, EOMES, BAP1, LTF, H4C9, COLQ, DYSF, CHAF1B, LYN, NRIP1, COIL, SLC7A5, AD5, H4C1, FGF23, ADAM12, BRD3, PSCA, ARHGEF5, TFPI2, FZD3, GHS, M6PR, MARCKS, SMAD1, LTC4S, H4C4, BHLHE40, IRS4, MAPKAPK5, LMO4, LOXL1, CST7, DDO, DGKZ, GAS7, PIK3R3, PKP4, PPFIA1, LRPAP1, SORBS2, H4C6, CUL4A, GNPAT, LTB, H4C14, H4C13, H4C5, H4C2, H4C8, H4C3, H4C11, H4C12, TERF1, MUC4, KCNMA1, TDO2, PCP4, CDK18, RBM3, RBL2, RBBP6, RB1, RASGRF1, RASA1, RARRES2, RAN, RAF1, RAD52, PCYT1A, RAD23B, RAC2, PDB1, RAB27B, RAB27A, PDC, PDE2A, PURA, PDGFB, ENPP2, PDYN, PTPN13, PC, PAX6, PARN, RPL15, P2RX1, S100A8, P2RX3, P2RX5, P2RY4, RREB1, RPS23, RPS21, RPS6KB2, P2RY6, RPS3A, RPL13, RET, RPA1, PAFAH1B2, RORA, ROM1, SNORD15A, BRD2, RNASE1, RHO, RHD, PAK3, TRIM27, PTPN11, PENK, PTN, PMM2, PLAUR, PLCL1, PLEK, PRKCE, PRKCD, PLP1, PRKAR1B, PRKACB, PRKACA, PLXNB1, PML, PRG2, PLAT, PMP22, PRB1, PPT1, PPP2R5E, POLG, PPP2R1A, PPP1CB, PPL, PPIC, PPIB, POR, MAP2K3, PLAG1, PFDN5, PSMD2, PFKFB3, PTGER2, PTGER1, PGD, PTGDR, PTCH1, PGR, PHB, PSMD9, PSMD7, PSMD3, PSMB2, PITX2, SERPINE2, SERPINI1, KLK10, PIK3C3, PIK3R1, KLK7, PIK3R2, PRS, PROS2P, PIN4, PROC, S100A10, OXT, OXA1L, SQLE, STAR, ST14, ST2, NDUFA6, SSTR3, SSTR2, NDUFA9, NDUFB8, SRM, SRF, NEDD4, SEPTIN2, STC1, SP4, NEU1, SOX5, SOX3, SOS2, SOS1, SOD3, NFE2L1, NFKB2, SNRPG, SNRNP70, NDUFA5, STIM1, NME1, MYH9, TRBV20OR9-2, TCP1, TCN2, MMUT, MUTYH, TCF4, ELOC, TBX2, MX1, MYH6, TARBP2, MAP3K7, STK11, MYO6, TACR2, NACA, VAMP2, VAMP1, SURF1, ABCC8, SUOX, NDP, STXBP1, STX1A, NINJ2, NME2, SAA2, OMP, SFTPC, TRA2B, SRSF6, SRSF5, SRSF3, SRSF1, SFRP1, OCA2, MAP2K4, ODF1, SELE, OPA1, OAS3, CXCL11, CCL21, CCL20, CCL19, CCL8, CCL1, SCP2, SCN1A, ORM2, OSM, TSPAN31, OAT, SGSH, SUMO2, SLC8A3, SMPD2, SLN, SLIT3, SLC22A5, SLC22A2, NQO2, SLC18A1, SLC16A1, SLC12A3, SLC11A1, SLC10A2, SLC8A1, NUP98, SLC6A12, NPHP1, SLC6A1, SLC5A2, NRCAM, NRDC, SLC1A1, NRF1, PMEL, YBX1, NT5E, NAT8, SEMA5A, ESRRA, RAB31, MACF1, HEY2, BRD4, TRAM1, CBX5, ANGPTL2, OPN1MW, GRIP1, KCNH4, MSTN, GFER, GFRA1, SIRT5, COTL1, GFRA3, GHR, ZNF629, UBR4, GHSR, WASHC4, GLI1, NUP160, CLUH, GLI3, SCFD1, KCTD2, CLCF1, SEC14L2, NR5A1, SLC24A2, PRPF6, TFIP11, MAFF, EID1, RAB38, FSHR, TMEFF2, PLA2G15, SLC7A11, FTH1, SNHG1, TSPAN15, GAST, ACKR1, G6PD, GAB1, NTSR2, GABBR1, GAD2, GALNS, DDAH1, PADI4, GART, NBEAL2, GMPR, PLEKHM2, WDHD1, GPR39, CARD8, ARHGEF15, AAK1, SYNPO, GPX4, ECD, PARK7, KLF8, TREX1, WIF1, WDR45, ATF6, CORO1A, TBC1D8, SLC7A9, GRIA4, FAF1, GRIK4, RER1, GRM1, STMN2, RAPGEF4, ADRM1, MSRB2, RAB3GAP1, GNA12, ZNF423, ATG4B, UBXN4, RCOR1, SEPTIN8, STAB1, GNAI1, MAPK8IP3, GRAMD4, GNB3, NFASC, KIF1B, GOLGA2, GPER1, SETX, GOLGA4, PDZD2, SAMD4A, KDM1A, GPM6A, RAB21, GPR6, P2RX2, GPR20, SNW1, FRK, FBXO7, BRI3, SNX12, SOCS7, CD209, FABP6, TBX21, NOP53, FABP7, SLC2A8, UBQLN2, A1CF, PSAT1, FANCG, HOOK2, DELEC1, FASN, SNX8, NPC1L1, BLNK, MS4A2, FCGR1A, FCGR2A, MYLIP, SCG3, DROSHA, TMEM230, NOX4, PCA3, SPCS1, VRK3, ETFA, SLC25A37, TLR8, SLC22A17, ECSIT, CD320, EZH2, DNAJC27, CLEC1B, NT5C3A, MZB1, F2RL2, HP1BP3, F3, F9, IRAK4, SAR1B, UTP11, F13B, SH3GLB1, SIDT2, SHANK1, TRAT1, F11R, RGCC, TMEM176B, NOCT, FBXO2, NPTN, TPK1, VCX, GREM1, FKBP1AP2, FKBP1AP3, AGO1, SEZ6L2, FKBP1AP4, FGF21, CLDN17, NOC2L, SND1, FOXM1, EPC2, ATRNL1, LRP10, FLNB, FMR1, POU2F3, TXN2, FBXL2, FOLR1, FOLR2, FKBP1AP1, B3GAT1, IGHV1-68, TNFRSF21, FEB1, FES, FGF9, RABGEF1, PRPF19, FGF13, FGFR1, FGFR4, PDLIM3, RND1, KLHL20, COQ2, CACYBP, HCAR1, FHL2, VPS4A, IL37, GLS2, NAAA, CYTH4, DKK2, DKK3, BBC3, SDCBP2, GRM3, IL24, SPAG9, CXCL2, PCLAF, IGFBP1, ACAP1, IGFBP5, SART3, KDM4A, IGHG3, SDC3, SH3PXD2A, RGS6, SNCAIP, TCL1B, IL4R, IL7, BCAR1, CCL4L2, CXCR1, BAG2, IL12B, BAG5, TMEM59, TBPL1, GAL3ST1, AKAP5, STX8, PIEZO1, BMS1, PTDSS1, IDH1, SH2D3C, GNE, SH2B3, IRF8, SRA1, TANK, KCNE3, HNRNPDL, CCS, NUP153, MED12, HS3ST1, TOMM20, RBM8A, IDH2, CFI, SV2B, TECPR2, IFI27, TOMM70, KIAA0319, IFIT3, IFNAR1, IGBP1, INSRR, PCYT1B, IL27RA, CCNE2, NOLC1, TIAF1, JUND, ZMYM3, KCNC4, KCNJ13, HGS, SYNGR3, ATG12, CBFA2T2, RABEP1, P2RX6, RAB11B, SLC16A3, SLC16A4, ATP6V0D1, RGN, USP10, USP2, USP14, DNAJA3, CLDN1, CLDN8, ARTN, JUNB, GPR50, PICK1, ITPKB, IRAK1, HOMER2, IREB2, IRF3, IRF6, IRF7, ITGAV, CYP7B1, ITGB3, NRXN1, SLC22A8, ITPR2, AIMP1, JAG2, ITGBL1, LHX2, SLIT2, TAOK2, CD163, JAK2, GPR37L1, PIWIL1, MAPKAPK2, PDLIM7, IARS1, TNC, IL18BP, CCT2, HCK, NRG3, USP39, DCTN6, CD226, HDC, HDLBP, CAMKK2, HIP1, TXNRD2, NPC2, SLC35A1, HBG2, PRDX4, ZNRD2, HYOU1, HLA-DQA1, SEMA4D, HLA-DQB1, NXF1, HLA-DRA, ATP5PD, COG5, GPNMB, CHL1, HAS3, FAM3C, GYPA, GSK3A, COPS5, GSM1, GSTM2, EBNA1BP2, PRSS21, PRDX3, GSTZ1, GTS, GUSB, C1QL1, GYPB, HAS1, GYPC, CCL27, ALDH1L1, GYPE, HAGH, WASF3, HSPH1, GJB6, HARS1, ARPP19, DHS, HLA-DRB4, HLA-G, PQBP1, MPHOSPH6, STAM2, GLYAT, HOXA@, RABEPK, HPCA, CALCOCO2, HRC, OLIG2, DDX39A, TOPORS, EIF1, HSD17B1, RAMP2, WASF2, HSD17B4, HSPA2, HSP90AB1, DNAJB1, NAMPT, BCAP31, CTDSP2, TSPAN3, ACTR2, CERT1, ABCC4, HNRNPK, HMBS, NPM3, CFDP1, PRMT5, HMGB2, YAP1, IFITM3, SPAG11B, PEMT, RACK1, SYCP2, TUBB4B, SEMA3A, WARS2, HNRNPC, FOXA1, CCL26, TLR6, FOXA2, LAMC3, LANCL1, HNF4A, TCIRG1, APBB3, APC2, HNRNPA2B1, MTCH2

-
Angioedema Induced By Ace Inhibitors, Susceptibility To
OMIM
Clinical Features Blais et al. (1999) and Adam et al. (2002) reported significantly lower plasma aminopeptidase P (APP) activities in patients with a history of AEACEI. ... Measured genotype analysis strongly suggested that the linkage signal for APP activity at this locus was accounted for predominantly by the SNP association. ... There was a significant association between the -2399A allele and decreased serum APP activity in both men and women, but the APP activity was lower in men regardless of genotype. ... This haplotype was associated with decreased plasma APP activity and decreased luciferase gene expression compared to other haplotypes of these SNPs. Cilia La Corte et al. (2011) concluded that the ATG haplotype of XPNPEP2 is functional and contributes to the development of ACEi-angioedema through a reduction in APP activity.
-
Problematic Smartphone Use
Wikipedia
For 3-4 year-old children: 180 minutes physical activity, 1 hour screen time, 10–13 hours of sleep time per day. [81] Phone settings [ edit ] Many smartphone addiction activists (such as Tristan Harris) recommend turning one's phone screen to grayscale mode, which helps reduce time spent on mobile phones by making them boring to look at. [82] Other phone settings alterations for mobile phone non-use included turning on airplane mode, turning off cellular data and/or Wi-Fi, turning off the phone, removing specific apps, and factory resetting. [83] Phone apps [ edit ] German psychotherapist and online addiction expert Bert te Wildt recommends using apps such as Offtime and Menthal to help prevent mobile phone overuse. [84] In fact, there are many apps available on Android and iOS stores which help track mobile usage. ... In Android a similar feature called "digital wellbeing" has been implemented to keep track of cell phone usage. [85] These apps usually work by doing one of two things: increasing awareness by sending user usage summaries, or notifying the user when he/she has exceeded some user-defined time-limit for each app or app category. ... PMID 27151528 . ^ "Internet Gaming" . www.psychiatry.org . Retrieved 10 May 2019 . ^ "Gaming disorder" . Gaming disorder . World Health Organization . 1 September 2018 . Retrieved 10 May 2019 . ^ "ICD-11 - Mortality and Morbidity Statistics" . icd.who.int . ... "Problematic Use of Mobile Phones in Australia…Is It Getting Worse?" . Front. Psychiatry . 10 : 105. doi : 10.3389/fpsyt.2019.00105 .

-
Alzheimer Disease 18
OMIM
Q170H and R181G mutant mice showed significant attenuation of APP processing compared to wildtype, with a decrease in APP-CTF-alpha levels and an increase in sAPP-beta levels, indicating that the mutations attenuated Adam10 alpha-secretase activity on APP. Crossing these Adam10 mutant mice with the Tg2576 AD mouse model showed that the Adam10 mutations increased amyloidogenic APP processing, as manifest by a shift from the alpha-secretase to the amyloidogenic beta-secretase pathway. ... Collectively, these findings suggested that diminished alpha-secretase activity of ADAM10 on APP resulting from mutations in the ADAM10 prodomain can cause AD-related pathology.
-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... The other species usually cause only febrile disease. [132] Severe and complicated malaria cases are medical emergencies since mortality rates are high (10% to 50%). [133] Recommended treatment for severe malaria is the intravenous use of antimalarial drugs.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
-
Compulsive Gambling
Mayo Clinic
If you have a problem with compulsive gambling, you may continually chase bets that lead to losses, use up savings and create debt. ... Tell yourself it's too risky to gamble at all. One bet typically leads to another and another. ... Recognize and then avoid situations that trigger your urge to bet. Family members of people with a compulsive gambling problem may benefit from counseling, even if the gambler is unwilling to participate in therapy.DRD3, PLTP, DRD2, DRD4, SCLY, KRT7, TAL1, SLC6A3, BDNF, CHPT1, FZD10, CIT, TRIM31, BAG3, VLDLR, MAOB, OPRM1, COMT, MAOA, LEP, HTR2A, HTR1B, DRD1, DBH, CREB1, DHDDS










