Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Necrotising Hepatopancreatitis
Wikipedia
"Asia Diagnostic Guide to Aquatic Animal Diseases" (PDF) . FAO Fisheries Technical Paper 402/2, NACA/FAO 2001. p. 207, "Chapter 4". ... Asia Diagnostic Guide to Aquatic Animal Diseases (PDF) . FAO Fisheries Technical Paper 402/2, NACA/FAO 2001. ISBN 92-5-104620-4 . ^ "Access online: Manual of Diagnostic Tests for Aquatic Animals - OIE - World Organisation for Animal Health" . www.oie.int .
-
Prion Pruritus
Wikipedia
Prion pruritus is the intense itching during the prodromal period of the Creutzfeldt–Jakob disease . [1] : 402 See also [ edit ] Pruritus References [ edit ] ^ Freedberg, et al. (2003).
-
Oculomucocutaneous Syndrome
Wikipedia
British Medical Journal . 1 (6110): 402–4. doi : 10.1136/bmj.1.6110.402 .
-
Hypospadias 4, X-Linked, Susceptibility To
OMIM
Mapping Van der Zanden et al. (2011) performed a genomewide association study using pooled DNA from 436 individuals with hypospadias and 494 controls of European descent and selected the highest ranking single-nucleotide polymorphisms (SNPs) for individual genotyping in the discovery sample, an additional Dutch sample of 133 cases and their parents, and a Swedish series of 266 cases and 402 controls. Individual genotyping of 2 SNPs (rs1934179 and rs7063116) in DGKK (300837), encoding diacylglycerol kinase-kappa, produced compelling evidence for association with hypospadias in the discovery sample (allele-specific OR = 2.5, p = 2.5 x 10(-11) and OR = 2.3, p = 2.9 x 10(-9), respectively) and in the Dutch (OR = 3.9, p = 2.4 x 10(-5) and OR = 3.8, p = 3.4 x 10(-5)), and Swedish (OR = 2.5, p = 2.6 x 10(-8) and OR = 2.2, p = 2.7 x 10(-6)) replication samples.
-
Iridoplegia
Wikipedia
New York: McGraw-Hill Medical. 2010. p. 402. ISBN 9780071741033 . OCLC 477051832 .
-
Aortoesophageal Fistula
Wikipedia
Clinical Journal of Gastroenterology . 10 (5): 393–402. doi : 10.1007/s12328-017-0762-z .
-
Smoking As A Quantitative Trait Locus 1
OMIM
Mapping In a genomewide scan using 385 microsatellite markers in 1,261 individuals representing 402 nuclear families of African American origin, Li et al. (2006) found a region near marker D10S1432 on chromosome 10q22 that showed significant linkage to indexed smoking quantity, with a maximum lod score of 4.17 at 92 cM.
-
Glycoproteinosis
Wikipedia
Carbohydrates: the essential molecules of life . Elsevier. pp. 402–. ISBN 978-0-240-52118-3 . Retrieved 3 November 2010 . ^ Bonten EJ, Wang D, Toy JN, et al.NEU1, MCOLN1, GNPTAB, GNPTG, GLB1, CTSA, NEURL1, NAGPA, EGFL7, OGA, CHIT1, DHCR7, IDS, HPRT1, FUCA2, ESA4, ERBB2, DPAGT1, IDUA

-
Mucolipidosis
Wikipedia
This article needs additional citations for verification . Please help improve this article by adding citations to reliable sources . Unsourced material may be challenged and removed. Find sources: "Mucolipidosis" – news · newspapers · books · scholar · JSTOR ( December 2007 ) ( Learn how and when to remove this template message ) Mucolipidosis Other names ML Mucolipidosis has an autosomal recessive pattern of inheritance Specialty Endocrinology Mucolipidosis is a group of inherited metabolic disorders that affect the body's ability to carry out the normal turnover of various materials within cells . [1] When originally named, the mucolipidoses derived their name from the similarity in presentation to both mucopolysaccharidoses and sphingolipidoses . [2] A biochemical understanding of these conditions has changed how they are classified. Four conditions (types I, II, III, and IV) were historically labeled as mucolipidoses. However, type I ( sialidosis ) is now classified as a glycoproteinosis , [2] and type IV ( Mucolipidosis type IV ) is now classified as a gangliosidosis . [3] Contents 1 ML II and III 2 Genetics 3 Diagnosis 4 Treatment 5 See also 6 References 7 External links ML II and III [ edit ] For details, see I-cell disease (type II) and Pseudo-Hurler polydystrophy (type III) The other two types are closely related. Mucolipidosis types II and III (ML II and ML III) result from a deficiency of the enzyme N-acetylglucosamine-1-phosphotransferase , which phosphorylates target carbohydrate residues on N-linked glycoproteins .
-
Mucolipidosis
Wikipedia
- Fundic Gland Polyp Wikipedia
-
Menarche, Age At, Quantitative Trait Locus 1
OMIM
Guo et al. (2006) performed a large-scale genomewide linkage scan in 2,461 Caucasian women from 402 pedigrees to identify quantitative trait loci underlying variations in age at menarche.
-
Murcs Association
Wikipedia
-
Murcs Association
GARD
MURCS association stands for (MU)llerian, (R)enal, (C)ervicothoracic (S)omite abnormalities and is a developmental disorder that primarily affects the reproductive and urinary systems. Most individuals with MURCS association are female, although males can also have this condition. Females with MURCS association can have an absent or abnormally shaped uterus. In rare cases, the vagina is also affected. Both males and females with MURCS association can have absent or abnormally formed reproductive tubes (usually the fallopian tubes in females and the vas deferens in males), kidney abnormalities, and short stature (adult height of less than 5 feet). Additional symptoms might include fused spinal bones in the neck and upper back and hearing loss.
-
Mayer-Rokitansky-Küster-Hauser Syndrome Type 2
Orphanet
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome type 2, a form of MRKH syndrome (see this term), is characterized by congenital aplasia of the uterus and upper 2/3 of the vagina that is associated with at least one other malformation such as renal, vertebral, or, less commonly, auditory and cardiac defects. The acronym MURCS (MÜllerian duct aplasia, Renal dysplasia, Cervical Somite anomalies) is also used. Epidemiology MRKH syndrome has an estimated worldwide incidence of 1/4500 live female births. The prevalence of MRKH syndrome type 2 is unknown. Clinical description MRKH syndrome type 2 is most often diagnosed in adolescence as the first symptom is most commonly a primary amenorrhea in young women presenting with otherwise normal development of secondary sexual characteristics and normal external genitalia. Patients lack the uterus and the upper 2/3 of the vagina. Because of this, difficulties with sexual intercourse have been reported.
-
Murcs Association
GARD
-
Villonodular Synovitis
Wikipedia
"Pigmented villonodular bursitis/diffuse giant cell tumor of the pes anserine bursa: a report of two cases and review of literature". Knee . 14 (5): 402–7. doi : 10.1016/j.knee.2007.06.004 .
-
Longevity 2
OMIM
They reviewed this cohort and compared the 213 patients who had survived to age 95 or more, whom they considered the longevity cases, with the 402 participants who had died before the age of 81, considered the average-lived controls.
-
Genetic Predisposition
Wikipedia
XCVII, "The Inheritance of Neuroticism: An Experimental Study", H. J. Eysenck and D. B. Prell, p. 402. ^ http://www.genome.gov/24519851 ^ The results of this survey are discussed here (January 20, 1998). ^ A summary of U.S.A. executive orders and proposed legislation is compiled by the National Center for Genome Resources. ^ The Intentional Stance (MIT Press; Reprint edition 1989) ( ISBN 0-262-54053-3 ) External links [ edit ] Genetic discrimination fact sheet from the National Human Genome Research Institute .
-
Congenital Heart Defects, Multiple Types, 2
OMIM
Molecular Genetics Thienpont et al. (2010) analyzed the TAB2 gene in 402 patients with cardiac outflow tract defects and identified heterozygosity for missense mutations in 2 patients with multiple types of congenital heart defects (605101.0001 and 605101.0002); neither mutation was found in 658 ethnically matched control chromosomes.
-
Osteolathyrism
Wikipedia
The Journal of Nutrition . 54 (3): 397–402. doi : 10.1093/jn/54.3.397 . PMID 13212476 . ^ a b c d Haque, Abdul; Hossain, Muffazal; Lambien, Fernand; Bell, E.
-
Fetal Resorption
Wikipedia
Journal of Reproduction and Fertility . 90 (2): 395–402. doi : 10.1530/jrf.0.0900395 . PMID 2250238 – via PubMed. ^ Hayakawa, S.; Fujikawa, T.; Fukuoka, H.; Chisima, F.; Karasaki-Suzuki, M.; Ohkoshi, E.; Ohi, H.; Kiyoshi Fujii, T.; Tochigi, M.; Satoh, K.; Shimizu, T.; Nishinarita, S.; Nemoto, N.; Sakurai, I. (1 July 2000).

-
Vanishing Twin
Wikipedia
For the band, see Vanishing Twin . This article has multiple issues. Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article includes a list of general references , but it remains largely unverified because it lacks sufficient corresponding inline citations . Please help to improve this article by introducing more precise citations. ( May 2018 ) ( Learn how and when to remove this template message ) This article needs to be updated . Please update this article to reflect recent events or newly available information. ( May 2017 ) ( Learn how and when to remove this template message ) Vanishing twin A fetus papyraceus shown with its umbilical cord next to the placenta of its dichorionic diamniotic twin Specialty Obstetrics and gynaecology A vanishing twin , also known as twin resorption , is a fetus in a multigestation pregnancy that dies in utero and is then partially or completely reabsorbed. [1] [2] In some instances, the dead twin is compressed into a flattened, parchment-like state known as fetus papyraceus. [3] Vanishing twins occur in up to one of every eight multifetus pregnancies and may not even be known in most cases. [4] "High resorption rates, which cannot be explained on the basis of the expected abortion rate,suggest intense fetal competition for space, nutrition, or other factors during early gestation, with frequent loss or resorption of the other twin(s)." [5] In pregnancies achieved by in vitro fertilization , "it frequently happens that more than one amniotic sac can be seen in early pregnancy, whereas a few weeks later there is only one to be seen and the other has 'vanished'." [6] Contents 1 See also 2 References 3 Further reading 4 External links See also [ edit ] Chimera (genetics) Mosaicism Parasitic twin Fetal resorption References [ edit ] ^ Landy, H.J.; Weiner, S.; Corson, S.L.; Batzer, F.R. (1986). "The "vanishing twin": ultrasonographic assessment of fetal disappearance in the first trimester". Am J Obstet Gynecol . 155 (1): 14–19. doi : 10.1016/0002-9378(86)90068-2 .
-
Vanishing Twin
Wikipedia
-
Hypertrophic Osteoarthropathy
Wikipedia
Description and review of the literature". Rheumatol. Int . 27 (4): 399–402. doi : 10.1007/s00296-006-0224-2 .

-
D-Glycerate Dehydrogenase Deficiency
Wikipedia
The Journal of Biological Chemistry . 274 (1): 397–402. doi : 10.1074/jbc.274.1.397 . PMID 9867856 . ^ de Koning TJ, Duran M, Dorland L, Gooskens R, Van Schaftingen E, Jaeken J, Blau N, Berger R, Poll-The BT (Aug 1998).

-
Hyperoxaluria, Primary, Type Ii
OMIM
A number sign (#) is used with this entry because of evidence that type II primary hyperoxaluria (HP2) is caused by homozygous or compound heterozygous mutation in the glyoxylate reductase/hydroxypyruvate reductase gene (GRHPR; 604296) on chromosome 9p13. For a discussion of genetic heterogeneity of primary hyperoxaluria, see 259900. Clinical Features Seargeant et al. (1991) reported 8 HP2 patients who belonged to 3 Saulteaux-Ojibway Canadian Indian families living in 2 isolated communities in northwestern Ontario. All had increased urinary oxalic acid and L-glyceric acid. Four patients presented with symptoms resulting from calcium oxalate nephrolithiasis, including dysuria, hematuria, and urinary tract infections in infancy or early childhood; 3 did not have recurrences. The other 4 affected patients were free of symptoms, suggesting that HP2 may be a much milder disease with a better long-term prognosis for renal function than HP1 (259900).
-
Primary Hyperoxaluria Type 2
GeneReviews
Summary Clinical characteristics. Primary hyperoxaluria type 2 (PH2), caused by deficiency of the enzyme glyoxylate reductase/hydroxypyruvate reductase (GR/HPR), is characterized by recurrent nephrolithiasis (deposition of calcium oxalate in the renal pelvis/urinary tract), nephrocalcinosis (deposition of calcium oxalate in the renal parenchyma), and end-stage renal disease (ESRD). After ESRD, oxalosis (widespread tissue deposition of calcium oxalate) usually develops. Symptom onset is typically in childhood. Diagnosis/testing. The diagnosis of PH2 is established in a proband by identification of biallelic pathogenic variants in GRHPR by molecular genetic testing. If no pathogenic variants or only one pathogenic variant is identified by molecular genetic testing, identification of reduced glyoxylate reductase enzyme activity on liver biopsy can establish the diagnosis of PH2. Management. Treatment of manifestations: Reduction of urinary calcium oxalate supersaturation through adequate daily fluid intake and treatment with inhibitors of calcium oxalate crystallization (orthophosphate, potassium citrate, and magnesium); temporary intensive dialysis for ESRD, followed by transplantation.
-
Primary Hyperoxaluria Type 2
GARD
Primary hyperoxaluria type 2 is a rare condition characterized by the overproduction of a substance called oxalate (also called oxalic acid). In the kidneys, the excess oxalate combines with calcium to form calcium oxalate, a hard compound that is the main component of kidney stones. Deposits of calcium oxalate can lead to kidney damage, kidney failure, and injury to other organs. Primary hyperoxaluria type 2 is caused by the shortage (deficiency) of an enzyme called glyoxylate reductase/hydroxypyruvate reductase (GRHPR) that normally prevents the buildup of oxalate. This enzyme shortage is caused by mutations in the GRHPR gene. Primary hyperoxaluria type 2 is inherited in an autosomal recessive pattern.
-
Hyperoxaluria, Primary, Type Ii
OMIM
-
Retinitis Pigmentosa 70
OMIM
Sequencing the PRPF4 gene in another 225 Chinese RP probands revealed an 18-bp deletion (607795.0002) in a sporadic RP patient. In 402 probands with adRP, including 191 from North America who were negative for mutation in 90% of known adRP genes, 115 from Spain who were negative for known RP mutations, and 96 from France who were negative for mutation in the 10 most frequently mutated RP genes or hotspots, Benaglio et al. (2014) analyzed the PRPF4 gene but did not find any clearly pathogenic mutations.C8orf37, PDE6B, PDE6A, CRX, RPGR, RPE65, PDE6G, LRAT, ABCA4, EYS, MERTK, IMPDH1, ROM1, RHO, USH2A, CRB1, CNGB1, RPGRIP1, GUCY2D, RP2, NRL, RBP3, CLRN1, RDH12, SAG, SPATA7, CNGA1, ARL6, AIPL1, REEP6, RGR, DHX38, GUCA1B, OFD1, IDH3A, IDH3B, PRPF8, RP1, FAM161A, TULP1, SNRNP200, PRPF31, CERKL, NR2E3, CA4, MAK, PRCD, PRPF3, RLBP1, PROM1, PCARE, CHM, ARL2BP, TOPORS, BBS1, CYP4V2, BEST1, DHDDS, KLHL7, IMPG2, HGSNAT, IFT140, PRPF6, BBS2, TTC8, AHI1, SCAPER, CLN3, IFT172, CDHR1, KIZ, FLVCR1, TTPA, ARL3, PRPF4, AGBL5, RP9, SLC7A14, FSCN2, POMGNT1, ZNF513, ZNF408, RBP4, ABHD12, UNC119, NEK2, AHR, TUB, SEMA4A, ATF6, IFT88, FOXI2, UBAP1L, CCZ1B, CROCC, PDAP1, FAM71A, KIAA1549, IRX5, ARHGEF18, C1QTNF5, PRTFDC1, SLC37A3, NAALADL1, CRB2, NGF, CEP250, CWC27, CCDC66, GRIN2B, PRPH2, AGTPBP1, SLC6A6, AIFM1, FGFR2, KL, MT2A, PTEN, MYO7A, CEP290, GUCA1A, RDH5, CDH23, IQCB1, MSTO1, CACNA1A, BBS4, ATXN7, USH1C, CFAP410, ATP6, PANK2, MKKS, BBS9, SLC24A1, PEX1, RP1L1, HADHA, PNPLA6, SDCCAG8, BBS12, NDUFAF5, RRM2B, PDHA1, NDUFS8, NDUFV2, LZTFL1, FOXRED1, POMT2, RCBTB1, TST, FKRP, BBS7, CNGB3, NCAPG2, NPHP1, MKS1, NDUFB11, SURF1, SDHB, PEX2, WDPCP, PRPH, NDUFV1, NDUFA13, SCN1A, SCO1, SDHA, SDHD, PHYH, TACO1, LIPT1, SLC19A1, NDUFA12, PEX5, NGLY1, ALMS1, LARGE1, NDUFS4, PRDX1, NDUFS3, POLR3A, MFSD8, ZDHHC24, RNASEH1, CTNS, ARL13B, TRIM32, NIPAL1, ECHS1, FASTKD2, ERCC3, POMT1, ERCC6, ERG, IFT27, NDUFAF6, BBS5, NDUFS2, CLRN1-AS1, COX20, CYGB, COX15, COX10, COX8A, COX7B, CDH23-AS1, ACOX1, C8orf37-AS1, MMACHC, JAG1, AMACR, AIRE, PET100, SDHAF1, ZFYVE26, PHF3, ATP1A2, BCS1L, NDUFS7, CAV1, TTLL5, VSX2, ERCC8, COX6B1, PRRT2, MTFMT, GSS, COL18A1, GMPPB, HADHB, ND1, ND2, ND3, ND4, ND5, ND6, TRNK, TRNL1, TRNN, TRNS1, TRNV, TRNW, TRIM37, BBS10, NDUFA2, NDUFA4, NDUFA9, NDUFA10, NDUFS1, SLC19A3, WFS1, BBIP1, COA8, HCCS, NDUFAF2, HADH, TMEM14B, COX14, PLXNA2, LSM2, TRNT1, CLU, MFRP, PCDH15, DHX16, PTPRC, SLU7, KLK3, SIGMAR1, NXNL1, CLTA, CNTF, NT5C2, RPE, PROS1, PLAG1, NPHP4, TIMP3, PSAT1, NPEPPS, MYP2, CXCR6, RIMS1, RRH, SLC19A2, EXOSC2, ADIPOR1, LPAR2, USP9X, COG4, VCP, EDN1, LCA5, PDC, ATN1, OTX2, OTC, CNOT3, MVK, LPCAT1, MMP9, EDNRA, RCC1, EPO, INS, FANCF, HSPA4, HK1, GNAT1, HIF1A, OPN4, GSN, SERPINF1, GPR42, NSMCE3, GRK1, ALDH3A2, SFRP2, ACKR3, ADRA1A, ARL2, ATXN2, CC2D2A, ADRA2B, WDR19, SOD3, PLIN2, SSTR4, BRS3, STC1, ACTB, NRG4, TWIST2, GRK7, POC5, ARMS2, MFT2, SAMD11, SAMD7, NOC2L, JAKMIP1, MIR204, POLDIP2, GUCY2EP, LIN28B, NPHP3, ARSI, RD3, CENPV, PITPNM3, INVS, CENPK, TENT5A, CYCS, DCUN1D1, TWNK, SLC2A4RG, TBX20, RDH11, PNPLA2, KIDINS220, NGB, MPP4, NYX, TNMD, ENFL2, TUT1, DNER, FTO, ELOVL6, ALG12, RNF19A, COQ8B, SETD2, KLF15, PDZD7, HKDC1, C5AR2, DNAJC17, ADGRV1, CEP78, GNPTG, AAVS1, THBS2, WHRN, MMP2, HMOX1, HK2, HGF, GRM5, GRM1, GRB10, GLO1, GK, GJB2, GDNF, GDF2, G6PD, FRZB, FN1, FGF5, FGF2, FBN2, HSP90AA1, IDH2, IFNG, INSR, MEIS2, MDH1, MAP1A, SMAD4, LTB, LAMP1, ISG20, ING1, IGF1, IMPA1, CXCL8, IL6, IL2RA, IL1B, IL1A, CCN1, FASN, ETV5, ERN1, ALDH7A1, CACNA1F, C5AR1, C3, C1QBP, BSG, BMP4, BCL2, ARRB2, CANX, ARR3, ABCC6, APOE, APOB, AMFR, ADH7, ABO, CACNA1S, CCT, ERBB2, CYBB, EGF, E2F1, DUSP6, DNMT3A, CFD, ACE, CYLD, CTNNB1, CD44, MAPK14, CRYAB, CRK, CP, CORD1, COL2A1, CD74, RAB8A, MSR1, SH3BP4, CYTB, SYNJ1, FGF18, HSD17B6, DGKE, BRAP, SMC1A, USP11, AIMP2, PAX8, ZNF132, ZFP36, USH1E, TSC1, TP53, TNF, TMPRSS2, TSPAN7, SMC3, ARHGEF2, CRLF1, AKT3, TMED3, SIRT1, ARC, MAPRE3, ARPP21, AHSA1, PPIH, HEPH, SNAP29, PLEKHM1, PHYHIP, HDAC4, KNTC1, EIF2AK3, GRAP2, EFTUD2, TIMP2, TIMP1, DYNLT3, PKNOX1, HTRA1, PRNP, MAPK1, PRKCG, POLG, PMM2, PLAU, PIM1, RAC1, PGF, CFP, PDGFRB, NPTX2, NFE2L2, NAGLU, MYC, ALDH18A1, RASGRF1, ELOVL4, SFTPD, STATH, SOS1, SOD1, SNRPB, SNCA, SLC2A1, SGSH, SFRP5, RBP1, SEC14L1, CX3CL1, CCL2, S100A6, RPS6KB1, RPS6, RCVRN, H3P22
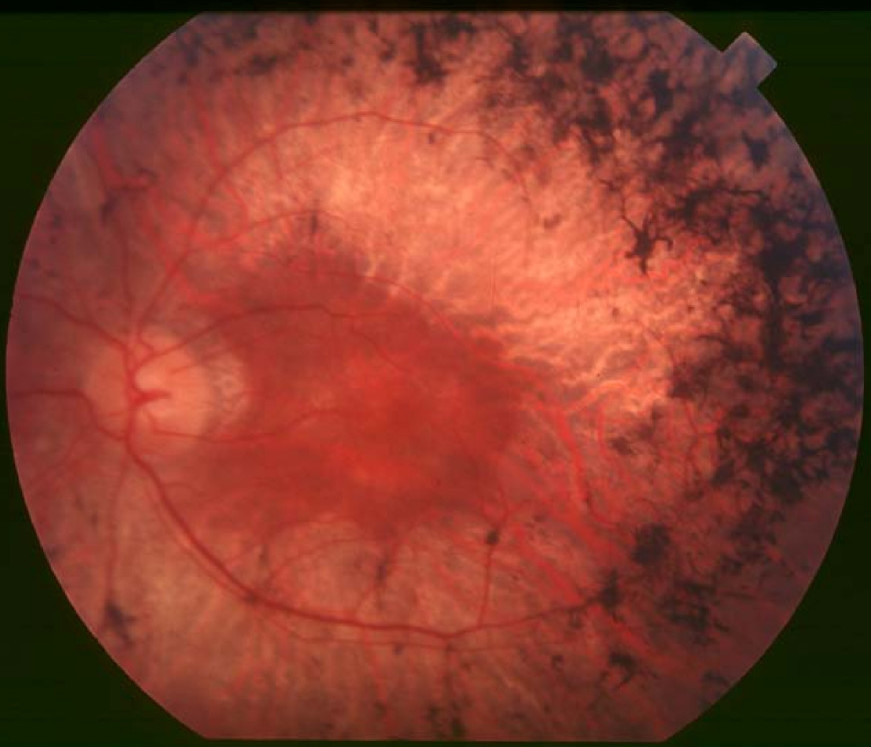
-
Retinitis Pigmentosa 48
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-48 is caused by heterozygous mutation in the gene encoding guanylate cyclase activator 1B (GUCA1B; 602275) on chromosome 6p21. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features In 7 members of 3 families in which mutation in the GUCA1B gene led to retinitis pigmentosa (RP48), Sato et al. (2005) found that the mutation was associated with RP with or without macular involvement in 5 members, macular degeneration in 1 member, and asymptomatic normal phenotype in 1 member. Sato et al. (2005) concluded that this mutation causes autosomal dominant retinal dystrophy with variable phenotypic expression and incomplete penetrance. Molecular Genetics Sato et al. (2005) screened 96 unrelated Japanese patients with retinitis pigmentosa for mutations in the GUCA1B gene and identified heterozygosity for a G157R mutation (602275.0001) in 3.
-
Retinitis Pigmentosa 55
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-55 (RP55) is caused by homozygous mutation in the ARL6 gene (608845) on chromosome 3q11. One such family has been reported. Mutation in the ARL6 gene can also cause a form of Bardet-Biedl syndrome (BBS3; see 209900), in which retinitis pigmentosa is one of the primary features. Mapping Aldahmesh et al. (2009) used homozygosity mapping to suggest causative genes in 52 Saudi Arabian patients with nonsyndromic retinitis pigmentosa (RP), and found linkage to the BBS3 locus on chromosome 3p12-q13. Molecular Genetics In 4 Saudi Arabian sibs with nonsyndromic retinitis pigmentosa (RP) mapping to chromosome 3p12-q13, Aldahmesh et al. (2009) identified homozygosity for a missense mutation in the ARL6 gene (608845.0006). Thorough examination of the affected sibs by Safieh et al. (2010) revealed no recognizable primary or secondary features of BBS other than retinitis pigmentosa.
-
Intellectual Developmental Disorder And Retinitis Pigmentosa
OMIM
A number sign (#) is used with this entry because of evidence that intellectual developmental disorder and retinitis pigmentosa (IDDRP) is caused by homozygous or compound heterozygous mutation in the SCAPER gene (611611) on chromosome 15q24. Description Intellectual developmental disorder and retinitis pigmentosa is characterized by mild to moderate intellectual disability and typical features of RP. Patients experience reduced night vision, constriction of visual fields, and reduced visual acuity; optic disc pallor, attenuated retinal blood vessels, and bone-spicule pigmentation are seen on funduscopy. Attention-deficit/hyperactivity disorder is observed in some patients (Tatour et al., 2017). Clinical Features Tatour et al. (2017) studied 4 patients from 3 unrelated families with intellectual developmental disorder and retinitis pigmentosa.
-
Retinitis Pigmentosa 39
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-39 (RP39) is caused by homozygous or compound heterozygous mutation in the USH2A gene (608400) on chromosome 1q41. Mutations in the same gene cause Usher syndrome type IIA (276901). For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Rivolta et al. (2002) described a patient with isolated RP who had complete paternal heterodisomy for chromosome 1. Clinical findings were typical, including fundi with attenuated retinal vessels and intraretinal bone spicule pigment around the periphery. Electroretinograms (ERGs) were reduced but detectable at the age of 54 years.
-
Cone-Rod Dystrophy 2
GARD
Cone-rod dystrophy 2 (CORD2) is an inherited eye disorder that affects the rod and cone cells in the retina . These cells process light and allow people to see the accurate shape and color of objects. Initial signs and symptoms of CORD2 usually occur in early childhood or late adolescence and include decreased sharpness of vision (visual acuity) and increased sensitivity to light (photophobia). Severity of symptoms and rate of disease progression may vary; however, most individuals experience impaired color vision, blind spots, loss of peripheral vision, and night blindness by adulthood. CORD2 is caused by mutations in the CRX gene and is inherited in an autosomal dominant manner.
-
Retinitis Pigmentosa 35
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-35 (RP35) can be caused by compound heterozygous mutation in the SEMA4A gene (607292) on chromosome 1q22. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Molecular Genetics Abid et al. (2006) screened 135 Pakistani patients with retinitis pigmentosa, 25 with cone-rod dystrophy (CORD10; 610283), and 30 with congenital blindness for mutations in the SEMA4A gene. They identified compound heterozygosity for 2 substitutions (607292.0001-607292.0002) in 2 RP and 2 CORD patients and heterozygosity for another substitution (607292.0003) in 3 patients with RP and 1 patient with congenital blindness. None of the mutations were found in 100 ethnically matched controls. INHERITANCE - Autosomal dominant - Autosomal recessive HEAD & NECK Eyes - Night blindness followed by complete blindness - Bone corpuscle-like pigmentation in equatorial and peripheral areas - Attenuated blood vessels in periphery - Macula clear in early stages MISCELLANEOUS - Allelic with cone-rod dystrophy 10 ( 610283 ) MOLECULAR BASIS - Caused by mutation in the semaphorin 4A gene (SEMA4A, 607292.0001 ) ▲ Close
-
Retinitis Pigmentosa 72
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-72 (RP72) is caused by homozygous mutation in the ZNF408 gene (616454) on chromosome 11p11. Heterozygous mutation in the ZNF408 gene has been reported to cause exudative vitreoretinopathy (see EVR6, 616468). For a general phenotypic description and discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Avila-Fernandez et al. (2015) studied 3 patients from 2 unrelated Spanish families who presented with night blindness followed by visual field loss and decreased visual acuity. Two sisters, born of unaffected parents from the same small geographic area, had onset of symptoms at 30 and 40 years of age, whereas the unrelated male patient from a consanguineous family presented at 17 years of age.
-
Retinitis Pigmentosa 66
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-66 (RP66) is caused by homozygous mutation in the RBP3 gene (180290) on chromosome 10q11. One such family has been reported. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Den Hollander et al. (2009) studied a consanguineous Italian family in which 3 brothers and a sister had retinitis pigmentosa; 2 of the brothers underwent detailed clinical evaluation, which demonstrated a wide range of severity in this family. The 42-year-old brother reported loss of central vision and onset of night blindness at 32 years of age, whereas his older brother had loss of central vision at age 60 with no night deficiency even at 67 years of age. The older brother had visual acuities of 20/60 and 20/80 and normal color vision, whereas the younger had acuities of 20/200 bilaterally, with a tritan axis of confusion on the Farnsworth-D-15 panel.
-
Retinitis Pigmentosa 19
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-19 (RP19) can be caused by homozygous or compound heterozygous mutation in the ABCR gene (ABCA4; 601691) on chromosome 1p22. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features In the population of Spain, 39% of the retinitis pigmentosa pedigrees show an autosomal recessive pattern of inheritance (Ayuso et al., 1995). Martinez-Mir et al. (1997) described a consanguineous Spanish family in which 6 of 7 sibs were affected. The parents, who were related as second cousins, were unaffected. The mean age of onset was 8 years.
-
Retinitis Pigmentosa 13
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-13 (RP13) is caused by heterozygous mutation in the PRPF8 gene (607300) on chromosome 17p13. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features The family with autosomal dominant retinitis pigmentosa (adRP) studied by Greenberg et al. (1994) was of British stock. The great-grandfather came to South Africa from Suffolk, England, in the mid-1800s. The onset of night blindness was between 4 and 10 years of age. By middle age, some patients had diffuse fundal changes and extensive retinal degeneration.
-
Retinitis Pigmentosa 23
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-23 (RP23) is caused by mutation in the OFD1 gene (300170) on chromosome Xp22. One such family has been reported. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Hardcastle et al. (2000) studied a 5-generation family with an atypical form of retinitis pigmentosa (RP) in which onset of vision loss in males was unusually early, before 2 years of age, and female obligate carriers had normal fundi and waveforms. The proband presented with limited central vision and poor night vision at age 2 years. Upon examination he was able to fix and follow with both eyes; funduscopy showed abnormal grayish macular reflexes and extensive whitish-gray spots throughout the midperiphery of the posterior pole with some areas of coalescence.
-
Retinitis Pigmentosa
OMIM
A number sign (#) is used with this entry because of the extensive genetic heterogeneity of nonsyndromic retinitis pigmentosa as well as the occurrence of retinitis pigmentosa with many generalized disorders. See INHERITANCE for a list of numbered and unnumbered forms of RP. Description Retinitis pigmentosa (RP) refers to a heterogeneous group of inherited ocular diseases that result in a progressive retinal degeneration affecting 1 in 3,000 to 5,000 people (Veltel et al., 2008). Symptoms include night blindness, the development of tunnel vision, and slowly progressive decreased central vision starting at approximately 20 years of age. Upon examination, patients have decreased visual acuity, constricted visual fields, dyschromatopsia (tritanopic; see 190900), and the classic fundus appearance with dark pigmentary clumps in the midperiphery and perivenous areas ('bone spicules'), attenuated retinal vessels, cystoid macular edema, fine pigmented vitreous cells, and waxy optic disc pallor. RP is associated with posterior subcapsular cataracts in 39 to 72% of patients, high myopia, astigmatism, keratoconus, and mild hearing loss in 30% of patients (excluding patients with Usher syndrome; see 276900).
-
Retinitis Pigmentosa, Late-Adult Onset
OMIM
Retinitis pigmentosa with onset of symptoms in the fifth or sixth decade is called senile retinitis pigmentosa. Bonneau et al. (1992) reported a family with 2 affected sisters whose parents were first cousins. Symptoms began in their fifties. The family originated from an area of France where consanguinity is not frequent. Grondahl (1987) described a Norwegian family in which 3 sibs had RP diagnosed at 58, 61, and 57 years of age; the parents came from the same Norwegian island and might have been consanguineous. In a survey of clinical aspects of RP in 93 families, Kaplan et al. (1990) found that autosomal recessive RP represented 21.5% of cases or 25.8% if isolated cases with consanguineous parents were considered.
-
Retinitis Pigmentosa 38
OMIM
A number sign (#) is used with this entry because this form of retinitis pigmentosa (RP38) is caused by homozygous or compound heterozygous mutation in the MER tyrosine kinase protooncogene (MERTK; 604705) on chromosome 2q13. Description Retinitis pigmentosa (RP) describes a group of disorders with progressive degeneration of rod and cone photoreceptors in a rod-cone pattern of dysfunction. RP has a prevalence of 1 in 3,500, and is genetically and phenotypically heterogeneous (summary by Mackay et al., 2010). For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Gal et al. (2000) described 3 individuals with degenerative retinal disease who carried mutation in the MERTK gene (RP38).
-
Retinitis Pigmentosa 20
OMIM
A number sign (#) is used with this entry because autosomal recessive retinitis pigmentosa-20 (RP20) is caused by homozygous or compound heterozygous mutation in the RPE65 gene (180069) on chromosome 1p31. Mutations in the RPE65 gene also cause Leber congenital amaurosis (LCA2; 204100). For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Gu et al. (1997) described a 5-generation consanguineous Indian family with 4 members with childhood-onset severe retinal dystrophy (RP20). Onset of severe visual impairment was between 3 and 7 years of age. Night blindness was a typical and early symptom in all patients.
-
Retinitis Pigmentosa 45
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-45 (RP45) is caused by homozygous mutation in the gene encoding the beta-1 subunit of the rod photoreceptor cyclic nucleotide-gated channel (CNGB1; 600724) on chromosome 16q21. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Bareil et al. (2001) identified a consanguineous French family in which typical and severe retinitis pigmentosa segregated as an autosomal recessive trait in 3 affected individuals. The proband had night blindness since early childhood. Later, she experienced loss in the peripheral visual field. At 30 years of age, the fundus showed typical bone spicule-shaped pigmentary deposits.
-
Retinitis Pigmentosa 47
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-47 (RP47) is caused by homozygous mutation in the S-antigen gene (SAG; 181031) on chromosome 2q37. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Nakazawa et al. (1998) identified 3 unrelated patients with retinitis pigmentosa (RP47) who carried the same homozygous mutation in the SAG gene (181031.0001; see MOLECULAR GENETICS). Patient 1 had a sib with Oguchi disease (258100) associated with the same mutation. Patient 2 demonstrated pigmentary retinal degeneration associated with golden-yellow reflex in the peripheral fundus.
-
Retinitis Pigmentosa 44
OMIM
A number sign (#) is used with this entry because this form of retinitis pigmentosa is caused by homozygous or heterozygous mutation in the gene encoding the RPE-retinal G protein-coupled receptor (RGR; 600342) on chromosome 10q23. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Using a single-strand conformation technique, Morimura et al. (1999) searched for mutations in the exons and flanking intron sequences of RGR in 182 unrelated patients with dominantly inherited retinitis pigmentosa, 182 with recessive retinitis pigmentosa, 383 with simplex retinitis pigmentosa (sporadic cases), 45 with Leber congenital amaurosis (see 204000), 28 with retinitis punctata albescens (see 136880), 22 with choroidal sclerosis (see 303100), and approximately 95 normal controls. They identified 2 probands with mutations in the RGR gene. One proband with autosomal recessive RP had 4 affected sibs. All affected sibs (aged 35 to 44) had visual acuity of 20/200 or worse, severely constricted visual fields, attenuated retinal vessels, diffuse depigmentation of the retinal pigment epithelium (RPE), and intraretinal pigment deposits in the periphery.
-
Retinitis Pigmentosa 57
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-57 (RP57) is caused by homozygous mutation in the PDE6G gene (180073). For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Dvir et al. (2010) studied an extended Israeli Muslim Arab pedigree segregating severe early-onset autosomal recessive retinitis pigmentosa in which affected individuals had markedly constricted visual fields, ranging from 10% to less than 5%, with tiny residual islands of central vision. Nevertheless, best-corrected visual acuity was relatively good. Both scotopic and photopic electroretinograms were severely reduced or completely extinct as early as 4 years of age. Funduscopy showed typical bone spicule-type pigment deposits spread mainly at the midperiphery, as well as optic disc pallor.
-
Retinitis Pigmentosa 4
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-4 (RP4) is caused by heterozygous, and rarely by homozygous, mutation in the RHO gene (180380) on chromosome 3q22. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Bradley et al. (1989, 1989) reported a large 5-generation Irish family segregating autosomal dominant early-onset retinitis pigmentosa. Affected status was determined by using titally extinguished electroretinogram (ERG) and/or symptoms characteristic of RP, including nyctolopia and peripheral visual field loss. In addition, all affected individuals exhibited funduscopic disturbances typical of RP: disc pallor, attenuation of retinal vessels, and classic bone-spicule pigmentary deposits in the retinal periphery.
-
Cone-Rod Dystrophy 15
OMIM
A number sign (#) is used with this entry because this form of cone-rod dystrophy (CORD15) can be caused by homozygous mutation in the CDHR1 gene (609502) on chromosome 10q23. An adult-onset form of retinitis pigmentosa (RP65) can also be caused by homozygous mutation in the CDHR1 gene. For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy, see 120970. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Cone-Rod Dystrophy 15 Ostergaard et al. (2010) studied 6 affected members of a 3-generation consanguineous pedigree from the Faroe Islands segregating autosomal recessive cone-rod dystrophy.
-
Leber Congenital Amaurosis 14
OMIM
A number sign (#) is used with this entry because of evidence that Leber congenital amaurosis-14 (LCA14) is caused by homozygous mutation in the LRAT gene (604863) on chromosome 4q32. Mutation in the LRAT gene can also cause juvenile retinitis pigmentosa and a form of early-onset severe retinal dystrophy. Description Autosomal recessive childhood-onset severe retinal dystrophy is a heterogeneous group of disorders affecting rod and cone photoreceptors simultaneously. The most severe cases are termed Leber congenital amaurosis, whereas the less aggressive forms are usually considered juvenile retinitis pigmentosa (Gu et al., 1997). For a general phenotypic description and a discussion of genetic heterogeneity of Leber congenital amaurosis, see LCA1 (204000); for retinitis pigmentosa, see 268000.
-
Retinitis Pigmentosa 74
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-74 (RP74) is caused by homozygous or compound heterozygous mutation in the BBS2 gene (606151) on chromosome 16q13. Clinical Features Shevach et al. (2015) reported a Moroccan Jewish family in which 3 sibs had nonsyndromic retinitis pigmentosa. The proband received a diagnosis of RP at age 20 years following complaints of impaired night vision and constricted visual fields. At age 32, her electroretinographic responses were undetectable. At age 49, her visual acuity was at the level of hand motions, and posterior polar cataract and posterior subcapsular opacity were evident. Fundus findings were compatible with advanced RP, including moderate pallor of the optic disc, widespread atrophy, and pigmentary changes.
-
Retinitis Pigmentosa 51
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-51 (RP51) is caused by homozygous mutation in the TTC8 gene (608132) on chromosome 14q31. Mutation in the TTC8 gene can also cause Bardet-Biedl syndrome-8 (BBS8; 615985), in which retinitis pigmentosa is one of the primary features. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Riazuddin et al. (2010) studied 4 affected and 7 unaffected members of a large consanguineous Pakistani family segregating autosomal recessive retinitis pigmentosa (RP). Medical records of the 4 affected individuals were suggestive of early-onset RP, with diagnoses made between 2 years and 4 years of age.
-
Retinitis Pigmentosa 54
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-54 (RP54) is caused by homozygous mutation in the C2ORF71 gene (PCARE; 613425) on chromosome 2p23. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features In a 4-generation consanguineous family with retinitis pigmentosa studied by Nishimura et al. (2010), the phenotype was variable in that 6 of 8 affected individuals had adult-onset RP, whereas 2 patients had a much earlier onset of disease, at less than 5 years of age, and had severe generalized dystrophy associated with nystagmus. Collin et al. (2010) described 4 Dutch sibs from a large pedigree, originally reported by Pinckers et al. (1973), segregating autosomal dominant granular corneal dystrophy, autosomal recessive retinal degeneration, and intermediately transmitted albinism. The 4 affected sibs were the only family members who manifested retinal degeneration, consisting of a photoreceptor dystrophy resembling RP or rod-cone dystrophy in most aspects.
-
Retinitis Pigmentosa, Y-Linked
OMIM
Clinical Features Zhao et al. (1995) reported a 4-generation Chinese family in which retinitis pigmentosa affected only males. All sons of affected males were affected, but all 4 daughters of affected males (and all children of these daughters) were healthy. Inheritance According to Zhao et al. (1995) the probability of autosomal dominant inheritance in the family they reported is only 1:8,192. Therefore, the authors proposed Y-linked inheritance as a possible explanation for the pattern in this family. INHERITANCE - Y-linked HEAD & NECK Eyes - Retinitis pigmentosa MISCELLANEOUS - Based on 1 4-generation Chinese family - Limited clinical information provided ▲ Close
-
Retinitis Pigmentosa 46
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-46 (RP46) is caused by homozygous mutation in the isocitrate dehydrogenase 3B gene (IDH3B; 604526) on chromosome 20p13. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Hartong et al. (2008) provided results of an ophthalmologic examination of 2 individuals with autosomal recessive IDH3B-related retinitis pigmentosa. The index case and the other individual, identified in a screen of patients with recessive or simplex retinitis pigmentosa, were evaluated at ages 47 and 38 years, respectively. Both had subnormal visual acuities, concentrically constricted visual fields, fundi typical of retinitis pigmentosa (pale optic discs, attenuated arterioles, and intraretinal pigment deposits), impaired dark adaptation, and reduced electroretinogram amplitudes indicating substantial loss of rod and cone photoreceptor function.
-
Retinitis Pigmentosa 79
OMIM
A number sign (#) is used with this entry because of evidence that autosomal dominant retinitis pigmentosa-79 (RP79) is caused by heterozygous mutation in the HK1 gene (142600) on chromosome 10q22. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Sullivan et al. (2014) studied a large 6-generation family (UTAD003) segregating autosomal dominant retinitis pigmentosa (adRP). The retinal dystrophy in the family could be traced back to an Acadian ancestor in the early 1800s, who was from the same region of south-central Louisiana where the family still lived. Affected members of the family exhibited a highly variable phenotype, with some reporting night blindness and/or patchy vision from early childhood, whereas others did not have discernible symptoms until well into the sixth or seventh decade of life.
-
Retinitis Pigmentosa 37
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-37 (RP37) is caused by heterozygous or homozygous mutation in the NR2E3 gene (604485) on chromosome 15q23. Mutation in the NR2E3 gene is also the cause of enhanced S-cone syndrome (ESCS; 268100). For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Coppieters et al. (2007) described a 4-generation Belgian family which segregated autosomal dominant retinitis pigmentosa (adRP) in 25 individuals. The phenotype corresponded to that seen in classic adRP, with progressive degeneration of rods and subsequent involvement of cones, further characterized by progressive intraretinal pigment migration, generally of the spicular type.
-
Retinitis Pigmentosa
Wikipedia
Gradual retinal degeneration leading to progressive sight loss Retinitis pigmentosa Back of the eye of a person with retinitis pigmentosa, mid stage. Note pigment deposits in the mid periphery along with retinal atrophy . While the macula is preserved there is some loss of pigmentation around it. Specialty Ophthalmology Symptoms Trouble seeing at night , decrease peripheral vision [1] Usual onset Childhood [1] Causes Genetic [1] Diagnostic method Eye examination [1] Treatment Low vision aids , portable lighting, orientation and mobility training [1] Medication Vitamin A palmitate [1] Frequency 1 in 4,000 people [1] Retinitis pigmentosa ( RP ) is a genetic disorder of the eyes that causes loss of vision . [1] Symptoms include trouble seeing at night and decreased peripheral vision (side vision). [1] As peripheral vision worsens, people may experience " tunnel vision ". [1] Complete blindness is common. [2] Onset of symptoms is generally gradual and often in childhood. [1] [2] Retinitis pigmentosa is generally inherited from a person's parents . [1] Mutations in one of more than 50 genes are involved. [1] The underlying mechanism involves the progressive loss of rod photoreceptor cells in the back of the eye . [1] This is generally followed by loss of cone photoreceptor cells . [1] Diagnosis is by an examination of the retina finding dark pigment deposits. [1] Other supportive testing may include an electroretinogram , visual field testing , or genetic testing . [1] There is currently no cure for retinitis pigmentosa. [2] Efforts to manage the problem may include the use of low vision aids , portable lighting, or orientation and mobility training. [1] Vitamin A palmitate supplements may be useful to slow worsening. [1] A visual prosthesis may be an option in certain people with severe disease. [1] It is estimated to affect 1 in 4,000 people. [1] Contents 1 Signs and symptoms 2 Causes 2.1 Genetics 3 Pathophysiology 4 Diagnosis 5 Treatment 6 Prognosis 7 Epidemiology 8 Research 9 Notable cases 10 See also 11 References 12 External links Signs and symptoms [ edit ] Example of tunnel vision (bottom) The initial retinal degenerative symptoms of retinitis pigmentosa are characterized by decreased night vision ( nyctalopia ) and the loss of the mid-peripheral visual field. [3] The rod photoreceptor cells, which are responsible for low-light vision and are orientated in the retinal periphery, are the retinal processes affected first during non-syndromic forms of this disease. [4] Visual decline progresses relatively quickly to the far peripheral field, eventually extending into the central visual field as tunnel vision increases. Visual acuity and color vision can become compromised due to accompanying abnormalities in the cone photoreceptor cells, which are responsible for color vision, visual acuity, and sight in the central visual field. [4] The progression of disease symptoms occurs in a symmetrical manner, with both the left and right eyes experiencing symptoms at a similar rate. [5] A variety of indirect symptoms characterize retinitis pigmentosa along with the direct effects of the initial rod photoreceptor degeneration and later cone photoreceptor decline.
-
Leber Congenital Amaurosis 4
OMIM
A number sign (#) is used with this entry because Leber congenital amaurosis-4 (LCA4) is caused by homozygous or compound heterozygous mutation in the gene encoding arylhydrocarbon-interacting protein-like-1 (AIPL1; 604392) on chromosome 17p13. Heterozygous mutation in the AIPL1 gene can cause juvenile retinitis pigmentosa and a form of cone-rod dystrophy. Description Autosomal recessive childhood-onset severe retinal dystrophy is a heterogeneous group of disorders affecting rod and cone photoreceptors simultaneously. The most severe cases are termed Leber congenital amaurosis (LCA), whereas the less aggressive forms are usually considered juvenile retinitis pigmentosa (Gu et al., 1997). Various intermediate phenotypes between LCA and retinitis pigmentosa are known and are sometimes described as 'early-onset severe rod-cone dystrophy' or 'early-onset retinal degeneration' (Booij et al., 2005).
-
Retinitis Pigmentosa 61
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-61 (RP61) is caused by homozygous mutation in the CLRN1 gene (606397) on chromosome 3q25. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Khan et al. (2011) described 2 consanguineous Pakistani families segregating autosomal recessive retinitis pigmentosa. Analysis of the fundus of affected individuals of both families indicated attenuation of the retinal vessels, waxy appearance of the optic disc, and bone spicule pigmentation in the periphery of the retina. ERG analysis revealed that the scotopic responses, which are indicative of the activity of the rod photoreceptors, were more severely diminished than the photopic responses.
-
Retinitis Pigmentosa 12
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-12 (RP12) is caused by homozygous or compound heterozygous mutations in the CRB1 gene (604210) on chromosome 1q31. Homozygous or compound heterozygous mutation in CRB1 can also cause a more severe retinal dystrophy, Leber congenital amaurosis (LCA8; see 604210). For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Heckenlively (1982) described 5 patients with retinitis pigmentosa of probable autosomal recessive inheritance who showed relative preservation of retinal pigment epithelium adjacent to and under retinal arterioles and hypermetropia (RP patients tend to be myopic). Affected sibs and parental consanguinity were noted. Bleeker-Wagemakers et al. (1992) reported a consanguineous Dutch family of 90 members from a genetically isolated population in the north of the Netherlands.
-
Retinitis Pigmentosa 26
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-26 (RP26) is caused by homozygous or compound heterozygous mutation in the CERKL gene (608381), which encodes a ceramide kinase, on chromosome 2q31. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Mapping Bayes et al. (1998) performed linkage analysis in a large nuclear Spanish family with 5 sibs affected by autosomal recessive retinitis pigmentosa (arRP). After excluding several genomic regions containing loci for retinal dystrophies, a genomewide search for linkage was undertaken. Positive lod scores were obtained with markers on chromosome 2q31-q33, defining an interval of about 7 cM for this novel ARRP locus, designated RP26, between D2S148 and D2S161.
-
Retinitis Pigmentosa 62
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-62 (RP62) is caused by homozygous mutation in the MAK gene (154235) on chromosome 6p24. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Ozgul et al. (2011) studied 8 patients with retinitis pigmentosa who were found to have mutations in the MAK gene. The age at diagnosis in 3 patients was in the third decade of life, whereas the other 5 patients were diagnosed in the fourth through sixth decades of life. Visual acuity was relatively preserved, with 6 of the 8 patients having 20/40 acuity in at least 1 eye.
-
Retinitis Pigmentosa 49
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-49 (RP49) is caused by homozygous or compound heterozygous mutation in the alpha-1 cyclic nucleotide-gated channel gene (CNGA1; 123825) on chromosome 4p12. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Molecular Genetics In a screen of 267 patients with autosomal dominant or autosomal recessive retinitis pigmentosa, Dryja et al. (1995) identified 4 probands with autosomal recessive RP carrying mutations in the CNCG1 (CNGA1) gene. Five mutant sequences cosegregated with the disease among 4 unrelated families with autosomal recessive RP. In 3 of the families, the affected individuals were either homozygous for a mutation or were compound heterozygotes.
-
Retinitis Pigmentosa
GARD
Retinitis pigmentosa (RP) is a group of inherited eye diseases that affect the light-sensitive part of the eye (retina). RP causes cells in the retina to die, causing progressive vision loss. The first sign of RP usually is night blindness . As the condition progresses, affected individuals develop tunnel vision (loss of peripheral vision), and eventually loss of central vision. RP may be caused by mutations in any of at least 50 genes. Inheritance can be autosomal dominant, autosomal recessive, or X-linked. Treatment options to slow the progression of vision loss include light avoidance, use of low-vision aids, and vitamin A supplementation.
-
Retinitis Pigmentosa 9
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-9 (RP9) is caused by heterozygous mutation in the RP9 gene (607331) on chromosome 7p14. One such patient has been reported. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Description Autosomal dominant retinitis pigmentosa (ADRP) is characterized by a typical fundus appearance, narrowed retinal vessels, and changes in the electrophysiological responses of the eye. Early signs are night blindness and constriction of the visual fields with a variable ages of onset (summary by Jay et al., 1992). Clinical Features Jay et al. (1992) described a 9-generation family (family N) with autosomal dominant retinitis pigmentosa.
-
Retinitis Pigmentosa 1
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-1 (RP1) is caused by heterozygous, homozygous, or compound heterozygous mutation in the ORP1 gene (RP1; 603937) on chromosome 8q12. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Blanton et al. (1991) described a large extended American family (UCLA-RP01) segregating autosomal dominant retinitis pigmentosa with relatively late onset of night blindness, usually by the third decade of life, and with slow progression. Characteristic clinical findings included diffuse retinal pigmentation, progressive decrease in recordable ERGs, and concentric visual field loss. Funduscopic findings included retinal atrophy, bone-spicule-like pigment deposits, and vascular attenuation.
-
Retinitis Pigmentosa 58
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-58 (RP58) is caused by homozygous mutation in the ZNF513 gene (613598) on chromosome 2p23. One such family has been reported. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Naz et al. (2010) studied a consanguineous Pakistani family segregating autosomal recessive retinitis pigmentosa (RP). All affected individuals had progressive night blindness since the age of 8 to 10 years. Vision in affected individuals was limited to light perception or hand movement with no peripheral vision.
-
Retinitis Pigmentosa 50
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-50 (RP50) is caused by heterozygous mutation in the BEST1 gene (607854) on chromosome 11q12. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Mapping In a nonconsanguineous family of European descent segregating autosomal dominant retinitis pigmentosa, Davidson et al. (2009) performed linkage analysis and obtained a maximum lod score of 2.9 on chromosome 11; haplotype analysis revealed a 64.9-Mb critical region between markers rs536715 and rs17304039, containing the BEST1 gene. Molecular Genetics In the proband of a nonconsanguineous family of European descent segregating autosomal dominant retinitis pigmentosa (adRP) mapping to chromosome 11, Davidson et al. (2009) sequenced the candidate gene BEST1 and identified heterozygosity for a missense mutation (I205T; 607854.0022) that was subsequently detected in 9 other affected family members and was absent from 5 unaffected family members. Analysis of BEST1 in a panel of 95 adRP patients who were negative for mutation in 10 known RP genes and in 12 patients with concentric RP revealed heterozygosity for a D228N missense mutation (607854.0023) in affected members of 1 adRP family and 1 concentric RP family, and heterozygosity for a Y227C mutation (607854.0024) in another concentric RP family.
-
Retinitis Pigmentosa 67
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-67 (RP67) is caused by homozygous mutation in the NEK2 gene (604043) on chromosome 1q32. One such family has been reported. Description Retinitis pigmentosa (RP) is the name given to a group of hereditary retinal conditions in which degeneration of rod photoreceptors, responsible for vision under dark conditions, is more pronounced than that of cone photoreceptors, which mediate daylight vision. Individuals with RP typically experience night blindness at first, followed by progressive and unstoppable visual impairment in daytime conditions as well. Their visual fields become reduced gradually and sight is lost from the midperiphery to the periphery, then from the midperiphery to the center, resulting eventually in complete or near-complete blindness if left untreated. Most patients show intraretinal pigment in a bone-spicule configuration around the fundus periphery as well as retinal arteriolar attenuation, elevated final dark-adapted thresholds, and reduced and delayed electroretinograms.
-
Retinitis Pigmentosa 77
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-77 (RP77) is caused by homozygous or compound heterozygous mutation in the REEP6 gene (609346) on chromosome 19p13. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Arno et al. (2016) examined the probands from 5 unrelated families with autosomal recessive retinitis pigmentosa. All probands presented with nyctalopia, with onset varying from early childhood to 20 years of age. There was a gradual decline in vision, characterized by reduced peripheral visual fields followed by reduced visual acuity.
-
Retinitis Pigmentosa 60
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-60 (RP60) can be caused by heterozygous mutation in the PRPF6 gene (613979) on chromosome 20q13.33. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Tanackovic et al. (2011) described the proband from a 4-generation family of European descent segregating autosomal dominant retinitis pigmentosa, who presented at 37 years of age for evaluation of decreased night vision and loss of peripheral vision. Funduscopy revealed bilateral normal discs with atrophy just temporal to the disc, clear macula, attenuated retinal arterioles, and bone spicule pigment all around the periphery. Reexamination almost 20 years later at 55 years of age demonstrated progression of disease, with decline in visual acuity to hand motions OD and 20/100 OS, as well as reductions in visual fields and full-field ERG amplitudes; funduscopy at that time showed waxy pallor of the discs bilaterally, atrophy of the retinal pigment epithelium and prominent choroidal vessels in each macula, attenuated retinal vessels, and bone spicule pigment visible in the midperiphery.
-
Retinitis Pigmentosa 56
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-56 (RP56) is caused by homozygous mutation in the gene encoding interphotoreceptor matrix proteoglycan-2 (IMPG2; 607056) on chromosome 3q12. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Bandah-Rozenfeld et al. (2010) examined 12 patients from 7 families with retinal disease and mutations in the IMPG2 gene. In general, affected individuals with IMPG2 mutations displayed typical symptoms and signs of RP, including night blindness, visual field loss, optic disc pallor, attenuated vessels, and bone-spicule-like pigmentation. In 11 of the 12 patients, lens abnormalities were observed: 7 had posterior subcapsular cataracts, 2 had mild cortical cataracts, 1 had pseudophakia, and 1 patient, who was diagnosed with mild maculopathy (see VMD5, 616152) rather than RP, had mild nuclear sclerosis.
-
Retinitis Pigmentosa
MedlinePlus
Retinitis pigmentosa is a group of related eye disorders that cause progressive vision loss. These disorders affect the retina, which is the layer of light-sensitive tissue at the back of the eye . In people with retinitis pigmentosa, vision loss occurs as the light-sensing cells of the retina gradually deteriorate. The first sign of retinitis pigmentosa is usually a loss of night vision, which becomes apparent in childhood. Problems with night vision can make it difficult to navigate in low light.
-
Retinitis Pigmentosa 76
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-76 (RP76) is caused by homozygous mutation in the POMGNT1 gene (606822) on chromosome 1p34. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Xu et al. (2016) studied an Italian sister and brother with retinitis pigmentosa. The 78-year-old sister, who became aware of nyctalopia and vision problems at age 12 years, showed visual acuity of 20/80 in the right eye and light-perception only in the left eye, constricted visual fields, and peripapillary atrophy; funduscopy revealed bone spicule pigmentation and attenuated retinal vessels. Her 69-year-old brother, who was aware of vision problems at age 10 years, had visual acuities of 20/100 and very restricted visual fields bilaterally, with bone spicules, narrow retinal vessels, peripapillary atrophy, and a tigroid appearance of the fundus.
-
Retinitis Pigmentosa 73
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-73 (RP73) is caused by homozygous mutation in the HGSNAT gene (610453) on chromosome 8p11. Mutation in the HGSNAT gene also causes mucopolysaccharidosis type IIIC (252930), the features of which include late-onset RP. Clinical Features Haer-Wigman et al. (2015) studied 3 affected individuals from 2 Ashkenazi Jewish Israeli families and 3 sibs from a Dutch family with nonsyndromic retinitis pigmentosa. All had night blindness and/or visual field loss as initial symptoms, but in the Israeli families, the 3 patients noted onset in childhood or adolescence and were diagnosed with RP in the fourth decade of life, whereas in the Dutch family, onset of symptoms did not occur until the fifth or sixth decade of life. The proband from the first Ashkenazi Jewish family was diagnosed at age 34 years, at which time full-field electroretinography (ERG) responses were undectectable.
-
Retinopathy, Pericentral Pigmentary, Dominant
OMIM
Grondahl (1987) diagnosed autosomal dominant pericentral retinal dystrophy in 4 families from northern Norway. Three of these families had a pigmentary retinal degeneration of night blindness starting in the teens and leading to blindness in the sixth and seventh decades of life, after the development of bony spicules, attenuation of retinal blood vessels, and retinal atrophy. Thus, the changes were quite different from those in the sibs reported by Traboulsi et al. (1988); see 268060. Eyes - Pericentral retinal dystrophy - Pigmentary retinal degeneration - Night blindness - Late blindness - Bony spicule pigmentary retinopathy - Attenuation of retinal blood vessels - Retinal atrophy Inheritance - Autosomal dominant ▲ Close
-
Retinitis Pigmentosa 71
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-71 (RP71) is caused by homozygous or compound heterozygous mutation in the IFT172 gene (607386) on chromosome 2p23. Mutation in the IFT172 gene also results in short-rib thoracic dysplasia-10 with or without polydactyly (SRTD10; 615630). For a general phenotypic description and discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Bujakowska et al. (2015) reported an unrelated woman and man with retinitis pigmentosa. The 33-year-old French woman developed night blindness in the second decade of life; examination showed RP as well as a lamellar macular hole on the left and optic nerve drusen on the right.
-
Retinitis Pigmentosa 69
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-69 (RP69) is caused by homozygous or compound heterozygous mutation in the KIZ gene (615757) on chromosome 20p11. Description Retinitis pigmentosa (RP), also designated rod-cone dystrophy, is characterized by initial night blindness due to rod dysfunction, with subsequent progressive constriction of visual fields, abnormal color vision, and eventual loss of central vision due to cone photoreceptor involvement (summary by El Shamieh et al., 2014). For a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features El Shamieh et al. (2014) studied 3 unrelated patients with rod-cone dystrophy (retinitis pigmentosa) and mutations in the KIZ gene (see MOLECULAR GENETICS). All were diagnosed in their late teens on the basis of night blindness followed by changes in midperipheral visual fields and undetectable responses on full-field electroretinography by approximately 35 years of age.
-
Retinitis Pigmentosa 82 With Or Without Situs Inversus
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-82 with or without situs inversus (RP82) is caused by homozygous mutation in the ARL2BP gene (615407) on chromosome 16q13. Clinical Features Davidson et al. (2013) studied a consanguineous family of Arab Muslim origin in which 3 sibs were diagnosed with retinitis pigmentosa (RP) in their 20s, after complaints of night vision and visual field impairment. At 36 years of age, the oldest sib could detect hand motion with her right eye and had bare light perception with her left eye, with no significant refractive errors. Mild posterior subcapsular cataracts were present, and funduscopy revealed typical signs of RP, including optic disc pallor, mild to moderate bone-spicule-like pigmentation in the midperiphery, and attenuated retinal blood vessels. All 3 sibs displayed a marked component of macular atrophy, with smaller and larger atrophic patches, as well as epiretinal membranes with wrinkling of the retina.
-
Retinitis Pigmentosa 29
OMIM
For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Hameed et al. (2001) reported a 6-generation, consanguineous Pakistani family with autosomal recessive retinitis pigmentosa (arRP). All affected individuals had pigmentary retinopathy associated with symptoms of night blindness and the loss of peripheral visual fields by the age of 20 years, loss of central vision between the ages of 25 and 30 years, and complete blindness between the ages of 40 and 50 years. Mapping By linkage analysis in a Pakistani family with arRP, Hameed et al. (2001) mapped the disease locus to chromosome 4q32-q34, with a maximum lod score of 3.76 for marker D4S415, with no recombination. Further analyses gave a probable disease interval of 4.6 cM between markers D4S3035 and D4S2417.
-
Retinitis Pigmentosa 32
OMIM
For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Zhang et al. (2005) identified a severe form of retinitis pigmentosa (RP) in a large consanguineous Pakistani family, in which all affected individuals had night blindness in early childhood, early complete loss of useful vision, and typical RP fundus changes plus macular degeneration. Mapping After exclusion of known RP loci in a consanguineous Pakistani family, Zhang et al. (2005) performed a genomewide scan which showed linkage to markers in a 10.5-cM region of chromosome 1p21.2-p13.3, between D1S2896 and D1S457. The highest lod score, 6.54 at theta = 0.0, was obtained at D1S485. Zhang et al. (2005) noted that this locus, designated RP32, lies approximately 4.9 cM from the ABCA4 gene (601691), which was excluded from the linked region.
-
Retinitis Pigmentosa 7
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-7 (RP7) is caused by heterozygous mutation in the RDS gene (PRPH2; 179605) on chromosome 6p21. Homozygous mutation in the PRPH2 gene causes retinal dystrophy of earlier onset, diagnosed clinically as Leber congenital amaurosis (LCA18). One patient was reported to have juvenile retinitis pigmentosa. Heterozygous mutation in the PRPH2 gene has also been reported to cause various retinal disorders with overlapping or related phenotypes, e.g., retinitis punctata albescens (see 136880), central areolar choroidal dystrophy (CACD2; 613105), vitelliform macular dystrophy-3 (VMD3; 608161), and patterned macular dystrophy (169150). A digenic form of retinitis pigmentosa resulting from a mutation in the RDS gene (179605.0004) and a null mutation of the ROM1 gene (see 180721.0001) has been reported. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000; for Leber congenital amaurosis, see 204000.
-
Retinitis Pigmentosa 2
OMIM
A number sign (#) is used with this entry because X-linked retinitis pigmentosa-2 (RP2) is caused by mutation in the RP2 gene (300757) on chromosome Xp11. Description Retinitis pigmentosa is characterized by constriction of the visual fields, night blindness, and fundus changes, including 'bone corpuscle' lumps of pigment. RP unassociated with other abnormalities is inherited most frequently (84%) as an autosomal recessive, next as an autosomal dominant (10%), and least frequently (6%) as an X-linked recessive in the white U.S. population (Boughman et al., 1980). For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features The X-linked form of retinitis pigmentosa is also called choroidoretinal degeneration, or pigmentary retinopathy.
-
Retinitis Pigmentosa 43
OMIM
A number sign (#) is used with this entry because this form of retinitis pigmentosa (RP43) is caused by homozygous or compound heterozygous mutation in the PDE6A gene (180071), encoding the alpha subunit of cGMP phosphodiesterase, on chromosome 5q31-q33. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Huang et al. (1995) studied 2 unrelated families segregating autosomal recessive retinitis pigmentosa (RP). Affected individuals reported night blindness since early childhood. Visual field testing revealed marked peripheral field loss. Fundus examination showed abnormalities typical of RP including waxy pallor of the optic discs, attenuated retinal vessels, Corton et al. (2010) described a 5-generation consanguineous Spanish pedigree segregating autosomal recessive RP, in which 3 sibs and their nephew exhibited common symptoms such as bilateral vision impairment and night blindness from early childhood, abolished electroretinogram (ERG) responses, and fundus features typically associated with RP, including optic disc pallor, attenuated vessels, and intraretinal clumping of pigment.
-
Retinitis Pigmentosa 33
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-33 (RP33) is caused by heterozygous mutation in the SNRNP200 gene (601664) on chromosome 2q11. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Zhao et al. (2006) reported a Chinese family in which 13 members spanning 4 generations developed retinitis pigmentosa in an autosomal dominant pattern of inheritance. Affected individuals developed night blindness at about 16 to 18 years of age. Subsequently, visual acuity gradually decreased with accompanying progressive loss of peripheral visual fields.
-
Retinitis Pigmentosa 3
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-3 (RP3) is caused by mutation in the RPGR gene (312610) on chromosome Xp11. Mutations in the RPGR gene can also cause X-linked cone-rod dystrophy (CORDX1; 304020) and a syndromic form of retinitis pigmentosa with deafness and sinorespiratory infections (300455). Description X-linked retinitis pigmentosa (XLRP) is a severe form of inherited retinal degeneration that primarily affects the rod photoreceptors (Demirci et al., 2002). It typically causes an early-onset night blindness and loss of peripheral vision, often causing patients to become legally blind by the age of 30 to 40 years. In RP3, affected males have a severe phenotype, and carrier females show a wide spectrum of clinical features ranging from completely asymptomatic to severe RP (Jin et al., 2007).
-
Retinitis Pigmentosa 27
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-27 (RP27) is caused by heterozygous mutation in the neural retina leucine zipper gene (NRL; 162080) on chromosome 14q11. One family with a clinical diagnosis of clumped pigment-type retinal degeneration has been reported with compound heterozygous mutation in the NPL gene. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features In a large 3-generation British family (RP251) segregating autosomal dominant retinitis pigmentosa, Bessant et al. (1999) excluded previously identified autosomal dominant loci and identified a heterozygous mutation in the NRL gene (S50T; 162080.0001) in affected members. Bessant et al. (2000) identified 3 other British RP families who carried the same mutation and determined that all 4 families were descended from a common founder.
-
Retinitis Pigmentosa 84
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-84 (RP84) is caused by homozygous mutation in the DHX38 gene (605584) on chromosome 16q22. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Ajmal et al. (2014) reported 4 affected sibs from a consanguineous Pakistani family with early-onset retinitis pigmentosa and macular coloboma. All of the sibs had developed night blindness at 4 years of age, and all were completely blind (no light perception). Funduscopic examination of the proband showed severely attenuated retinal vessels throughout the fundus, and the maculae were severely affected bilaterally, with unusually prominent and deep macular colobomas devoid of neuroretinal tissue.
-
Retinitis Pigmentosa 63
OMIM
For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Kannabiran et al. (2012) described a large Indian family in which 14 of 34 individuals studied had retinitis pigmentosa. Age at presentation ranged from 16 to 65 years, and in most cases the initial symptoms consisted of night blindness associated with blurred vision. Fundus examination revealed a range of features, including degeneration of the retinal pigment epithelium (RPE) to varying extents, arterial attenuation, disc pallor, and pigment migration. Electroretinography (ERG) showed diminished or extinguished responses.
-
Retinitis Pigmentosa 36
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-36 (RP36) is caused by homozygous mutation in the PRCD gene (610598) on chromosome 17q25. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Zangerl et al. (2006) examined a 32-year-old woman from Bangladesh who first noticed difficulty seeing at night as a child. Funduscopic examination revealed optic discs with fairly normal color but markedly attenuated arterioles. Extensive bone-spicule-like pigmentation was present in all 4 quadrants of both eyes, with highest density at the equator, and was admixed with lighter colored deposits at the level of the retinal pigment epithelium.
-
Retinitis Pigmentosa 28
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-28 (RP28) is caused by homozygous or compound heterozygous mutation in the FAM161A gene (613596) on chromosome 2p15. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Gu et al. (1999) described a consanguineous Indian family in which 4 members in 2 generations had autosomal recessive RP. Age of onset was between 5 and 15 years. The 3 oldest members, aged 39 to 47, had severe visual handicap. Langmann et al. (2010) studied 3 German patients with RP28 and reviewed the phenotype of the Indian family studied by Gu et al. (1999), stating that this type of retinal dystrophy shows no unique clinical features.
-
Leber Congenital Amaurosis 3
OMIM
A number sign (#) is used with this entry because Leber congenital amaurosis-3 (LCA3) and a form of juvenile retinitis pigmentosa are caused by homozygous or compound heterozygous mutation in the SPATA7 gene (609868) on chromosome 14q31. Description Autosomal recessive childhood-onset severe retinal dystrophy is a heterogeneous group of disorders affecting rod and cone photoreceptors simultaneously. The most severe cases are termed Leber congenital amaurosis, whereas the less aggressive forms are usually considered juvenile retinitis pigmentosa (Gu et al., 1997). Mackay et al. (2011) concluded that SPATA7 retinopathy is an infantile-onset severe cone-rod dystrophy with early extensive peripheral retinal atrophy but with variable foveal involvement. For a general phenotypic description and a discussion of genetic heterogeneity of Leber congenital amaurosis, see LCA1 (204000); for retinitis pigmentosa, see 268000.
-
Retinitis Pigmentosa 25
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-25 (RP25) is caused by homozygous or compound heterozygous mutation in the EYS gene (612424) on chromosome 6q12. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Collin et al. (2008) examined 6 affected individuals from 3 families with retinitis pigmentosa, all but 1 of whom displayed characteristic RP abnormalities including night blindness as the initial symptom, retinal bone-spicule pigmentation and attenuated retinal vessels, constriction of visual fields, and a nonrecordable ERG or ERG responses in a rod-cone pattern. Two unrelated patients had posterior subcapsular cataracts. The authors observed differences in the photoreceptor dystrophy between the families: in 1 patient from family A, the cones were more severely affected than the rods (cone-rod pattern) and kinetic visual fields were not constricted but showed bilateral central scotomas; fundus examination revealed central abnormalities at the level of the retinal pigment epithelium and moderate attenuation of retinal vessels. Her 60-year-old brother also had central fundus lesions, but his ERG showed neither rod nor cone activity.
-
Retinitis Pigmentosa 10
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-10 (RP10) is caused by heterozygous mutation in the IMPDH1 gene (146690) on chromosome 7q32. Heterozygous mutation in the IMPDH1 gene can also cause Leber congenital amaurosis-11 (LCA11; 613837). Description Retinitis pigmentosa-10 (RP10) is characterized in most patients by early onset and rapid progression of ocular symptoms, beginning with night blindness in childhood, followed by visual field constriction. Some patients experience an eventual reduction in visual acuity. Funduscopy shows typical changes of RP, including optic disc pallor, retinal vascular attenuation, and bone-spicule pattern of pigmentary deposits in the retinal midperiphery. Electroretinography demonstrates equal reduction in rod and cone responses (Jordan et al., 1993; Bowne et al., 2002; Bowne et al., 2006).
-
Retinitis Pigmentosa 30
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-30 (RP30) is caused by mutation in the retinal fascin gene (FSCN2; 607643) on chromosome 17q25. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Wada et al. (2001) studied 4 Japanese families segregating autosomal dominant retinitis pigmentosa (adRP). All 14 affected patients had had night blindness from childhood. Their visual acuity ranged from hand motion to 1.0. Fundus examination of affected members of 3 generations of 1 family disclosed the progression of retinal degeneration with increasing age.
-
Retinitis Pigmentosa 34
OMIM
For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Melamud et al. (2006) described a 3-generation family in which retinitis pigmentosa appeared to be transmitted in an X-linked manner. Four affected individuals who were examined described onset of night blindness, and some described color vision difficulties in the second decade of life, with the earliest at 13 years of age. Indirect ophthalmoscopy revealed peripheral bone spicules, waxy pallor of the optic nerve, and attenuated retinal blood vessels in all patients. All had evidence of constricted peripheral vision. Visual acuity ranged from 20/40 to 20/80+.
-
Retinitis Pigmentosa 22
OMIM
For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Mapping Finckh et al. (1998) collected DNA samples from 20 large consanguineous Indian families in which autosomal recessive retinitis pigmentosa (arRP) segregated and that were suitable for homozygosity mapping of the disease locus. After excluding linkage to all known arRP loci, a genomewide scan was initiated. In 2 families, homozygosity mapping, haplotype analysis, and linkage data mapped the disease locus (RP22) in an approximately 16-cM region between D16S287 and D16S420 on the proximal short arm of chromosome 16. The location of RP22 was given as 16p12.3-p12.1. Molecular Genetics By direct sequencing, Finckh et al. (1998) found no mutation in the CRYM gene (123740), which encodes mu crystallin and maps to the same region.
-
Retinitis Pigmentosa 24
OMIM
For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Gieser et al. (1998) described a 5-generation family with X-linked retinitis pigmentosa designated RP24. Affected males (hemizygotes) had early onset of rod photoreceptor dysfunction; cone-receptor function was normal at first, but there was progressive loss. Patients at advanced stages showed little or no detectable rod or cone function and had clinical hallmarks of typical RP. Mapping Gieser et al. (1998) demonstrated linkage of X-linked retinitis pigmentosa in a 5-generation family to Xq26-q27.
-
Retinitis Pigmentosa 78
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-78 (RP78) is caused by homozygous or compound heterozygous mutation in the ARHGEF18 gene (616432) on chromosome 19p13. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Arno et al. (2017) studied 3 unrelated individuals with simplex retinitis pigmentosa. All presented in their third to fourth decades with central visual disturbance, visual field defects, and mild nyctalopia. Examination at ages 37, 38, and 51 years, respectively, revealed visual acuities ranging from 20/30 to 20/1250, with the worst in the oldest individual (patient 2; GC3626).
-
Retinitis Pigmentosa 17
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-17 (RP17) is caused by heterozygous mutation in the gene encoding carbonic anhydrase IV (CA4; 114760) on chromosome 17q23. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Yang et al. (2005) reported 13 affected individuals in a large Caucasian family with RP17. Reduction in night and peripheral vision manifested at around age 15, followed by rod photoreceptor atrophy, bone spicule pigmentation, and concomitant cone photoreceptor dysfunction manifested by photophobia, color vision changes, and decreased central vision. ERG demonstrated reduction of both rod and cone responses. Genealogic records indicated that this family was an offshoot of the South African family reported by Bardien et al. (1995).
-
Retinitis Pigmentosa 75
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-75 (RP75) is caused by homozygous mutation in the AGBL5 gene (615900) on chromosome 2p23. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Kastner et al. (2015) studied a consanguineous Turkish family in which 2 sisters and a brother had retinitis pigmentosa with onset between 7 and 12 years of age. All 3 exhibited tunnel vision and night blindness; other features included myopia in 1 sister and mixed astigmatism in the brother. Fundus examination revealed bone spicule pigmentation, attenuation of retinal vessels, and waxy pallor of the optic disc.
-
Retinitis Pigmentosa 83
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-83 (RP83) is caused by heterozygous mutation in the ARL3 gene (604695) on chromosome 10q24. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Description Retinitis pigmentosa-83 (RP83) is characterized by onset of night blindness in the first decade of life, with decreased central vision in the second decade of life in association with retinal degeneration. The retinal dystrophy is associated with cataract, and macular edema has also been reported in some patients (Holtan et al., 2019). Clinical Features Strom et al. (2016) reported a mother and her son and daughter (pedigree RP02) with retinitis pigmentosa.
-
Retinitis Pigmentosa 80
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa 80 (RP80) is caused by homozygous or compound heterozygous mutation in the IFT140 gene (614620) on chromosome 16p13. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Xu et al. (2015) studied a 43-year-old Han Chinese man who developed night blindness in childhood and vision loss at age 36 years. Fundus images showed widespread retinal bone spicule pigmentation with a 'gold foil' macular reflex. Ocular coherence tomography (OCT) revealed loss of inner and outer segment photoreceptor cells.
-
Retinopathy, Pericentral Pigmentary, Autosomal Recessive
OMIM
Traboulsi et al. (1988) described a brother and sister, born to parents related as third cousins, who had pigmentary retinopathy in a pericentral distribution. The retinopathy was noted in infancy when the sibs were examined for strabismus. The optic discs, maculae, and retinal vessels were normal. Both sibs had moderate hyperopic astigmatism and esotropia. The fundus and visual acuity remained unchanged for 9 and 13 years in the brother and sister, respectively. Results of eye examinations in the father, mother, and older sister were normal.
-
Retinitis Pigmentosa 68
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-68 (RP68) is caused by homozygous or compound heterozygous mutation in the SLC7A14 gene (615720) on chromosome 3q26. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa (RP), see 268000. Clinical Features Jin et al. (2014) studied a Chinese man, born of first-cousin parents, who in childhood developed night blindness and a visual field defect. Visual acuity in both eyes progressively decreased to hand motion only by 35 years of age. Fundus photography and optical coherence tomography (OCT) revealed typical intraretinal bone-spicule pigmentation, large spots of retinal atrophy, and overall thinning of the outer retinal layer.
-
Cone-Rod Dystrophy 16
OMIM
A number sign (#) is used with this entry because of evidence that retinal dystrophy, including a form of cone-rod dystrophy (CORD16) and a form of retinitis pigmentosa (RP64), can be caused by homozygous or compound heterozygous mutation in the C8ORF37 gene (614477) on chromosome 8q22. Mutation in C8ORF37 can also cause Bardet-Biedl syndrome (BBS21; 617406), in which retinal dystrophy is associated with other features. For a general phenotypic description and a discussion of genetic heterogeneity of cone-rod dystrophy (CORD), see 120970; for a similar discussion regarding retinitis pigmentosa (RP), see 268000. Description Cone-rod dystrophy (CORD) and retinitis pigmentosa (RP) are clinically and genetically overlapping heterogeneous retinal dystrophies. RP is characterized initially by rod photoreceptor dysfunction, giving rise to night blindness, which is followed by progressive rod and cone photoreceptor dystrophy, resulting in midperipheral vision loss, tunnel vision, and sometimes blindness.
-
Retinitis Pigmentosa 42
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-42 (RP42) is caused by heterozygous mutation in the KLHL7 gene (611119) on chromosome 7p15. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Andreasson (1991) studied 26 affected and 6 unaffected members of 4 families from central Norway and Sweden segregating autosomal dominant retinitis pigmentosa. Although fundus examination was similar in patients from all 4 families, showing narrowed vessels and bone spicule pigment, 2 different types of RP could be distinguished by electroretinography (ERG) based on cone b-wave implicit times, which were normal in 1 family ('family 59') but prolonged in the other 3 families. ERG recordings from 10 RP patients in different age groups from a 6-generation family ('family 72') revealed that cone b-wave amplitude decreased with age but implicit time did not, suggesting that amplitude is useful for monitoring the progression of disease whereas implicit time is more suitable for distinguishing different types of disease.
-
Leber Congenital Amaurosis 13
OMIM
A number sign (#) is used with this entry because Leber congenital amaurosis-13 (LCA13) is caused by homozygous or compound heterozygous mutation in the photoreceptor-specific retinal dehydrogenase gene RDH12 (608830) on chromosome 14q24. Heterozygous or homozygous mutation in RDH12 has also been shown to cause a form of retinitis pigmentosa (RP53). For a general phenotypic description and a discussion of genetic heterogeneity of Leber congenital amaurosis, see 204000; for retinitis pigmentosa, see 268000. Clinical Features Janecke et al. (2004) reported 3 consanguineous Austrian kindreds segregating Leber congenital amaurosis. Affected individuals in these families, as well as 2 Austrian individuals with sporadic LCA and 3 non-Austrian individuals with LCA, carried mutations in the RDH12 gene (see MOLECULAR GENETICS).
-
Retinitis Pigmentosa 59
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-59 (RP59) is caused by homozygous mutation in the DHDDS gene (608172) on chromosome 1p36. Congenital disorder of glycosylation type Ibb (CDG1BB) can be caused by compound heterozygous mutation in the DHDDs gene. One such patient has been reported. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000; for congenital disorder of glycosylation, see 212065. Clinical Features Zuchner et al. (2011) studied an Ashkenazi Jewish family in which 3 of 4 sibs were diagnosed with retinitis pigmentosa (RP) in their teenage years. Early symptoms consisted of impaired night and peripheral vision. Clinical examination of the affected individuals revealed pigmentary retinal degeneration, and the diagnosis of RP was confirmed by rod and cone responses on electroretinograms (ERGs).
-
Retinitis Pigmentosa 85
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-85 (RP85) is caused by homozygous mutation in the AHR gene (600253) on chromosome 7p21. One such family has been reported. For a general phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Zhou et al. (2018) studied 2 distantly related Indian boys with retinitis pigmentosa, aged 8 years (RD-ICP-79) and 10 years (RD-ICP-78), from a large consanguineous family. Both boys reported early-onset progressive difficulty with night vision and gradually decreasing visual acuity bilaterally. Typical features of RP were observed in both patients, with fundi showing pigment deposits and a pale retina.
-
Retinitis Pigmentosa 41
OMIM
A number sign (#) is used with this entry because of evidence that autosomal recessive retinitis pigmentosa-41 (RP41) is caused by homozygous mutation in the PROM1 gene (604365) on chromosome 4p15. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Maw et al. (2000) reported a consanguineous Indian family in which 4 of 8 sibs had night blindness and loss of peripheral vision from childhood with progression to profound visual impairment and extinguished electroretinograms (ERG) by their third decade. Fundus examination revealed narrowed arteries, optic disc pallor, pigment deposits, and macular degeneration. One sister also had polydactyly on one foot. Polydactyly and retinal degeneration are manifestations of Bardet-Biedl syndrome (see 209900); however, the authors stated that none of the other manifestations of that disorder were present in this family.
-
Retinitis Pigmentosa 31
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-31 (RP31) is caused by heterozygous mutation in the gene encoding topoisomerase I-binding arginine/serine-rich protein (TOPORS; 609507) on chromosome 9p21. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Papaioannou et al. (2005) examined 14 members of a French Canadian family segregating early-onset retinitis pigmentosa in an autosomal dominant fashion. Onset of symptoms ranged from 10 to 50 years of age and differed between the generations. Visual acuities were well maintained in most patients, with 13 of 14 having better than 20/40 and 9 of 14 having 20/20 acuities at their last examination.
-
Retinitis Pigmentosa 40
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-40 (RP40) is caused by homozygous or compound heterozygous mutation in the PDE6B gene (180072), encoding the beta subunit of rod phosphodiesterase, on chromosome 4p16. Mutations in the PDE6B gene also cause congenital stationary night blindness (CSNBAD2; 163500). For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features McLaughlin et al. (1993) identified 6 patients from 3 families with autosomal recessive retinitis pigmentosa (RP) and mutations in the PDE6B gene. All affected individuals had clinical findings typical of RP. They reported absent night vision from early childhood.
-
Retinal Dystrophy And Obesity
OMIM
A number sign (#) is used with this entry because of evidence that retinal dystrophy and obesity (RDOB) is caused by homozygous mutation in the TUB gene (601197) on chromosome 11p15. One such family has been reported. Clinical Features Borman et al. (2014) studied a consanguineous Caucasian family from the United Kingdom in which 3 sibs had retinal dystrophy and obesity. The proband was an 18-year-old male who presented at age 11 with a 2-year history of deteriorating vision, at which time he had no perception of light in the left eye and decreased visual acuity (20/40) in the right eye. Examination revealed a bilateral myopic and astigmatic refractive error, with a 'blonde' fundus on the right and total retinal detachment with vitreous hemorrhage on the left. At 18 years of age, visual acuity was measured at 20/30 on the right with no light perception on the left, and bilateral myopic and astigmatic refractive errors were confirmed.
-
Retinitis Pigmentosa 6
OMIM
For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Mapping Two separate loci for X-linked recessive retinitis pigmentosa have been mapped to the short arm of the X chromosome, RP2 at Xp11.4-p11.23 (312600) and RP3 at Xp21.1 (300029). On the basis of different linkage relationships, it has been suggested that there is a third RP locus on Xp, designated RP6 and thought to be located at Xp21.3-p21.2. Ott et al. (1990) performed a multilocus linkage analysis of 62 family pedigrees with X-linked RP. In 60 to 75% of the families, location of an XLRP gene was estimated at 1 cM distal to OTC, and in 25 to 40% of the families, an XLRP locus was located halfway between DXS14 (p58-1) and DXZ1 (Xcen), with an estimated recombination fraction of 25% between the 2 XLRP loci.
-
Retinitis Pigmentosa 18
OMIM
A number sign (#) is used with this entry due to evidence that this form of retinitis pigmentosa, designated RP18, is caused by heterozygous mutation in the PRPF3 gene (607301) on chromosome 1q21. For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa, see 268000. Clinical Features Xu et al. (1996) studied a large Danish family of 7 generations in which autosomal dominant retinitis pigmentosa segregated. Clinical diagnosis was based on a history of night blindness and ocular fundus findings typical of retinitis pigmentosa and included peripheral bone spicule formation, severe constriction of retinal arterioles, and progressive visual field defects beginning as midperipheral ring scotomas. Pathologic dark adaptation did not occur until the end of the first decade.
-
Retinitis Pigmentosa 11
OMIM
A number sign (#) is used with this entry because retinitis pigmentosa-11 (RP11) is caused by heterozygous mutation in the PRPF31 gene (606419) on chromosome 19q13. Description Retinitis pigmentosa (RP) is a clinically and genetically heterogeneous group of retinal dystrophies characterized by a progressive degeneration of photoreceptors, eventually resulting in severe visual impairment. For a discussion of genetic heterogeneity of RP, see 268000. Clinical Features Moore et al. (1993) described 4 families with autosomal dominant retinitis pigmentosa. Of the 15 patients who were studied from 3 of the families (families 1, 3, and 4), 14 had early onset of night blindness, 10 before age 10 years and 4 before age 20 years, and evidence of severe disease. Patients in all families showed typical fundus features of RP. The majority also had posterior subcapsular lens opacities, macular edema, and/or macular atrophy.
-
Retinitis Pigmentosa 14
OMIM
A number sign (#) is used with this entry because of evidence that retinitis pigmentosa-14 (RP14) is caused by homozygous or compound heterozygous mutation in the TULP1 gene (602280) on chromosome 6p21. Mutation in TULP1 can also cause a form of Leber congenital amaurosis (LCA15; 613843) as well as juvenile retinitis pigmentosa (see 613843). For a phenotypic description and a discussion of genetic heterogeneity of retinitis pigmentosa and juvenile RP, see 268000. Mapping In a large pedigree from the Dominican Republic showing segregation for autosomal recessive retinitis pigmentosa, Knowles et al. (1994) demonstrated linkage to microsatellite markers on 6p. The results of 2-point and multipoint analyses suggested that the most likely location of the disease gene is near D6S291, which is located approximately 20 cM telomeric of the photoreceptor peripherin locus (PRPH2; 179605).
-
Retinitis Pigmentosa
Orphanet
Retinitis pigmentosa (RP) is an inherited retinal dystrophy leading to progressive loss of the photoreceptors and retinal pigment epithelium and resulting in blindness usually after several decades. Epidemiology Prevalence of RP is reported to be 1/3,000 to 1/5,000. No ethnic specificities have been reported although founder effects are possible. Clinical description Retinitis pigmentosa is slowly progressive but relentless. There is however broad variability in age of onset, rate of progression and secondary clinical manifestations. Affected individuals generally first develop night blindness (nyctalopia) due to loss of rod function, often in adolescence or earlier.
-
Retinitis Pigmentosa 48
OMIM






