Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Neuroendocrine Tumor
Wikipedia
G Mitotic count (per 10 HPF ) Ki-67 index (%) GX Grade cannot be assessed G1 < 2 < 3% G2 2 to 20 3–20% G3 > 20 > 20% If mitotic count and Ki-67 are discordant, the figure which gives the highest grade is used. G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit. ... Berlin: Springer. pp. 195–233. ISBN 978-3-540-43462-7 . ^ a b c d Pommier R. 2003. ... Journal of Endocrinological Investigation . 31 (3): 277–86. doi : 10.1007/bf03345602 .MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Malaria
Wikipedia
The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity . ... According to the WHO and UNICEF, deaths attributable to malaria in 2015 were reduced by 60% [77] from a 2000 estimate of 985,000, largely due to the widespread use of insecticide-treated nets and artemisinin-based combination therapies. [74] In 2012, there were 207 million cases of malaria.ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
-
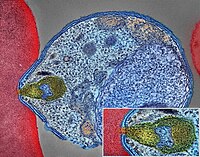
Pancreatic Neuroendocrine Tumor
Wikipedia
PanNETs are a type of neuroendocrine tumor , representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign , while some are malignant . ... "Update on pancreatic neuroendocrine tumors" . Gland Surgery . 3 (4): 258–75. doi : 10.3978/j.issn.2227-684X.2014.06.03 . ... "Therapeutic Implications of Molecular Subtyping for Pancreatic Cancer" . Oncology (Williston Park) . 31 (3): 159–66, 168. PMID 28299752 . ^ National Cancer Institute. Pancreatic Neuroendocrine Tumors (Islet Cell Tumors) Treatment (PDQ®) Incidence and Mortality [3] ^ Öberg K, Knigge U, Kwekkeboom D, Perren A (October 2012). ... Humana Press, Cham. pp. 127–140. doi : 10.1007/978-3-319-46038-3_6 . ISBN 9783319460369 . ^ a b c d e Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al.MEN1, ATRX, DAXX, ELK3, TP53, EPHB1, SLC6A2, CEACAM5, CEACAM7, UQCRFS1, DHDDS, CHPT1, RALBP1, CIB1, SEMA4D, RIPK1, CXCR4, VEGFA, TTR, GNA12, TSC2, TFE3, CDKN1B, PSG2, POMC, MYCN, CEACAM3, GRN, MUC16

-
Azotemia, Familial
OMIM
Hsu et al. (1978) described a family in which 6 persons in 3 generations had elevated serum urea with normal creatine levels, renal biopsy and all measures of renal function except urea clearance. ... Furthermore, urea is reabsorbed actively by the tubule; this process is apparently brought into play particularly in states of low protein intake. Net reabsorption might be due to exaggerated active reabsorption or to deficient secretion.
-
Mosquito Bites
Mayo Clinic
Avoid and exclude mosquitoes Limit exposure to mosquitoes by: Repairing any tears in the screens on windows, doors and camping gear Using mosquito netting over strollers and cribs Using mosquito netting when sleeping outdoors Selecting self-care products that don't have scents Use insect repellent Use insect repellent when mosquitoes are active. ... Don't use icaridin on infants younger than 6 months Check the labels of products with oil of lemon eucalyptus — some aren't suitable for children under 3 years old. Don't use para-menthane-diol on children under 3 years old Don't let young children get insect repellent on their hands, as they might get it in their mouths. ... Some sporting goods stores sell clothing pretreated with permethrin. Don't wash bed nets or set them in sunlight, as this breaks down permethrin.
- Dowling-Degos Disease GARD
-
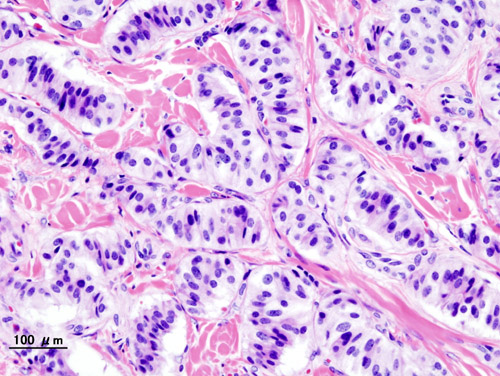
Insulinoma
GARD
Insulinoma is a type of pancreatic neuroendocrine tumor (pancreatic NET), which refers to a group of rare tumors that form in the hormone-making cells of the pancreas.MEN1, RPS15, CDKN2B, CDKN2C, IAPP, GCG, CDKN1B, CDKN1A, SST, FOXM1, GLP1R, PDX1, INS, IL1B, RIT2, PTPRN2, GAD1, EHMT1, IGF2, ZGLP1, CDKN2A, SLC30A8, SLC30A10, GCK, SSTR2, FFAR1, YY1, LEP, DPP4, INSM1, MNX1, HSPD1, GAD2, SLC2A2, CASR, RALBP1, RIPK1, PDHX, BTC, UQCRFS1, TP53, TGM2, SSTR5, CDKN1C, INSR, ABCC8, SLC6A2, SSTR4, SSTR3, WFS1, NIT1, SERPINA1, PTPRN, GIP, GCKR, CORO1A, H3P47, PRL, H3P10, ERBB2, GAST, EGR1, ELK3, CALCA, CASP3, EPHB1, G6PC, DLK1, CCN5, SQSTM1, PTTG1, GCM2, LHX2, KL, MAPK8IP1, INSL5, IRS2, ZNRD2, KHDRBS1, DCTN6, LILRB1, FASTK, CCND1, PDIA5, FAS, ATF6, KDM1A, PDZD2, BCL2, BRCA1, TNKS, PLA2G6, HNF1A, TCF19, TGFA, TGFB1, CASP8, THBD, TKT, TSPAN7, TPD52, TRP-AGG2-5, TRPC1, EIPR1, TXN, TYRP1, UCP2, VDR, CACNA1D, BRAF, STAB1, ERP44, NUP62, KCNH4, CAT, KCNH8, GPR119, STOML3, AKT1, HCAR2, GOLGA6A, TICAM2, HES3, MIR107, MIR144, MIR155, MIR204, MIR21, MIR375, INS-IGF2, ADSS2, TMED7-TICAM2, ECT, LINC02210-CRHR1, H3P23, ADM, SLC22A12, TXNDC5, TRABD, RCBTB1, FGF21, MCAT, MCTS1, TMED7, ADIPOR1, DCTN4, CDKAL1, SLC25A38, BANK1, MEG3, ZC3H12A, APOC2, SOX6, SELENOS, IGSF9, SEMA6A, HAMP, G6PC2, PDIA2, ANGPT2, SYP, STAT5A, STC1, STAT5B, KCNJ1, KCNJ6, KRT8, KRT16, KRT19, DECR1, LEPR, LGALS3, LMO2, EPCAM, SMAD2, SMAD3, SMAD4, MAPT, MC2R, MDK, RAB8A, CUX1, MET, CIITA, MLH1, EGF, EGFR, INPPL1, HK1, MTOR, FGF13, GNA12, GPD2, FBN1, GRN, GSK3B, GSR, GTF2H1, ESR2, ELK1, HLA-DQB1, HMGN2, HNF4A, EPHB2, IFI27, IGFBP1, IGFBP2, IL4, IL10, MRC1, NCAM1, NEDD4, SLC2A1, RAP1A, REG1A, CPE, CMA1, S100A8, SCT, CCL2, CXCL12, SDHD, CHGA, RAB3A, CDKN2D, SLC16A1, SNX1, CDC42, CDK1, CCND3, CCNC, CCK, STAT1, RANBP2, CR2, NF1, PIK3CG, NFE2L1, CTSB, NME1, OPA1, PAX4, PAX6, PCSK1, ENPP1, CTNNB1, PKD1, CRHR1, POLD1, MAPK1, MAPK3, MAPK8, ADCYAP1, PRSS1, PSEN2, PSMD9, PTEN, ACO2
- Sneddon Syndrome GARD
-
Clanging
Wikipedia
This is associated with the irregular thinking apparent in psychotic mental illnesses (e.g. mania and schizophrenia ). [1] Gustav Aschaffenburg found that manic individuals generated these "clang-associations" roughly 10–50 times more than non-manic individuals. [2] Aschaffenburg also found that the frequency of these associations increased for all individuals as they became more fatigued. [3] Clanging refers specifically to behavior that is situationally inappropriate. ... Livingstone. p. 32 . ^ Spitzer, Manfred (1999). The mind within the net: Models of learning, thinking, and acting .
-
Esophageal Food Bolus Obstruction
Wikipedia
] and the use of large-bore tubes inserted into the esophagus to forcefully lavage it. [17] [ unreliable medical source? ] Endoscopic [ edit ] The Roth net can be inserted through the endoscope to remove pieces of the obstructed food. ... Primary and definitive diagnosis and therapy". Surgical Endoscopy . 3 (4): 195–8. doi : 10.1007/BF02171545 . ... British Medical Journal . 1 (5957): 561–3. doi : 10.1136/bmj.1.5957.561 . PMC 1672660 . ... Diseases of the Esophagus . 15 (4): 330–3. doi : 10.1046/j.1442-2050.2002.00271.x . ... Digestive Diseases and Sciences . 25 (2): 100–3. doi : 10.1007/bf01308305 . PMID 7353455 . ^ a b Katsinelos P, Kountouras J, Paroutoglou G, Zavos C, Mimidis K, Chatzimavroudis G (2006).

-
Dogger Bank Itch
Wikipedia
Dogger Bank itch Specialty Dermatology Dogger Bank itch is a cutaneous condition characterized by a long-lasting dermatitis caused by exposure to the sea chervil , Alcyonidium diaphanum , a bryozoan . [1] The disease, common in fishermen who work in the North Sea , has been recognized by the Danish Workman's Compensation Act since 1939. [2] Contents 1 Pathogenesis 2 Treatment 3 Epidemiology 4 History 5 See also 6 References Pathogenesis [ edit ] The structural formula of the toxin responsible for Dogger Bank itch The rash is caused by a type of cell-mediated hypersensitivity reaction; this type of hypersensitivity normally occurs in people who become sensitized to volatile organic compounds . ... In Dogger Bank itch, sensitivity is acquired after repeated handling of the sea chervils that become entangled in fishing nets. [ citation needed ] The specific toxin responsible for the rash was determined to be the sulfur -bearing salt (2-hydroxyethyl) dimethylsulfoxonium chloride. [3] This salt is also found in some sea sponges and has potent in vitro activity against leukemia cells. [4] Treatment [ edit ] A study of two cases in 2001 suggests that the rash responds to oral ciclosporin . ... The sea chervil, abundant in the area, frequently came up with the fishing nets and had to be thrown back into the water. ... British Journal of Dermatology . 69 (3): 92–93. doi : 10.1111/j.1365-2133.1957.tb13235.x . v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal Outline
-
Mosquito-Borne Disease
Wikipedia
The prevalence of ITNs in sub-Saharan Africa has grown from 3% of households to 50% of households from 2000 to 2010 with over 254 million insecticide treated nets distributed throughout sub-Saharan Africa for use against the mosquito vectors Anopheles gambiae and Anopheles funestus which carry malaria. ... PLOS Medicine . 11 (3): e1001619. doi : 10.1371/journal.pmed.1001619 . ... PMID 28579763 . ^ Sharma, Anshika; Lal, Sunil K (February 3, 2017). "Zika Virus: Transmission, Detection, Control, and Prevention" . ... Current Opinion in Infectious Diseases . 3 (3): 434–438. doi : 10.1097/00001432-199006000-00020 . ... Transactions of the Royal Society of Tropical Medicine and Hygiene . 94 (3): 253–255. doi : 10.1016/S0035-9203(00)90310-9 .

-
West Nile Fever
Wikipedia
"West Nile virus neuroinvasive disease". Annals of Neurology . 60 (3): 286–300. doi : 10.1002/ana.20959 . ... Vector-Borne and Zoonotic Diseases . 5 (3): 252–7. doi : 10.1089/vbz.2005.5.252 . ... "Punctate exanthem of West Nile Virus infection: report of 3 cases". J. Am. Acad. Dermatol . 51 (5): 820–3. doi : 10.1016/j.jaad.2004.05.031 . ... Vector-Borne and Zoonotic Diseases (Submitted manuscript). 3 (3): 99–110. doi : 10.1089/153036603768395799 . ... Journal of Virological Methods . 51 (2–3): 201–10. doi : 10.1016/0166-0934(94)00105-P .CCR5, ERVK-32, ROBO3, MAVS, DDX58, PLAAT4, IFIT2, ERVK-6, STAT1, SPP1, OAS1, IL1B, IFNB1, RNASEL, CASP8, HLA-DRB1, PELI1, SELENBP1, ARHGEF2, LRRFIP1, NAMPT, TRAIP, RIPK3, SEC14L2, CSF1R, LAMP3, ERVW-1, FOXP3, ZMYND10, DDX56, CCR7, VCP, CDKN2A, IFIH1, DHX58, ZBP1, HAVCR2, PIK3IP1, NLRP3, TNFRSF13C, TRIM6, RBM45, CCR2, ERVK-20, ERVK-18, VAMP8, TNFRSF1A, IFNA1, TNF, IFNA13, HLA-DQA1, IL1A, HLA-C, IL10, IL17A, IL18, IRF3, IRF5, KIR2DL2, KIR3DL1, KIR3DS1, LSAMP, CD180, SMAD4, MMP9, HLA-A, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PZP, GLS, CASP1, SNCA, GEM, DDX3X, TAP1, TLR3, ATF4

-
Uric Acid Concentration, Serum, Quantitative Trait Locus 1
OMIM
The combined p value for white individuals from all 3 cohorts was 2.5 x 10(-60), and further analysis showed that the SNP was direction-consistent with the development of gout in white participants (OR of 1.74; p = 3.3 x 10(-15)). ... Evidence for both an increased rate of uric acid synthesis and an impaired net elimination of uric acid by the kidney has been advanced. ... Autosomal dominant form Lab - Increased rate of uric acid synthesis - Impaired net elimination of uric acid by the kidney - Hyperuricemia Skin - Urate tophi ▲ Close
-
Epileptic Encephalopathy, Early Infantile, 45
OMIM
Lien et al. (2016) reported a 32-month-old boy with severe developmental delay and hypotonia who developed refractory epilepsy at age 3 months. Brain imaging was normal. ... In vitro functional studies in HEK293 cells showed that the mutation altered the kinetic properties of the channel, resulting in the net loss of GABAergic inhibition. In a boy with EIEE45, Lien et al. (2016) identified a de novo heterozygous missense mutation in the GABRB1 gene (T287I; 137190.0002).








