Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Neurogenic Inflammation
Wikipedia
Smith, "Preventive treatment of migraine in adults" UpToDate 2019 [25] Additional CGRP blockers are progressing through clinical trials. [26] Anticipating later botox therapy for migraine, early work by Jancsó et al. found some success in treatment using denervation or pretreatment with capsaicin to prevent uncomfortable symptoms of neurogenic inflammation. [27] A 2010 study of the treatment of migraine with CGRP blockers had shown promise for CGRP blockers. [28] In early trials, the first oral nonpeptide CGRP antagonist, MK-0974 ( Telcagepant ), was shown effective in the treatment of migraine attacks, [29] but elevated liver enzymes in two participants were found. ... "Skeletal and hormonal effects of magnesium deficiency". J Am Coll Nutr . 28 (2): 131–41. doi : 10.1080/07315724.2009.10719764 . ... PMID 18079356 . S2CID 1001915 . ^ Chiu IM, von Hehn CA, Woolf CJ (July 2012). ... PMID 22837035 . ^ Fig. 3 of article Chiu IM, von Hehn CA, Woolf CJ (2012). "Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology" .
-
Early-Onset Alzheimer's Disease
Wikipedia
This loss of brain volume affects ones ability to live and function properly, ultimately being fatal. [5] Beta-amyloid is a small piece of a larger protein called the amyloid precursor protein (APP). Once APP is activated, it is cut into smaller sections of other proteins. ... Alpha-secretase cleavage of APP, which precludes the production of Aβ, is the most common processing event for APP. 21 allelic mutations have been discovered in the APP gene. These guarantee onset of early-onset familial Alzheimer disease and all occur in the region of the APP gene that encodes the Aβ domain. ... "A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid". ... PMID 16817891 . ^ Chow VW, Mattson MP, Wong PC, Gleichmann M (March 2010). "An overview of APP processing enzymes and products" .
-
Microvasculature Remodeling
Wikipedia
What makes vessels grow with exercise training? J App Physiol 97: 1119–28, 2004. This cardiovascular system article is a stub .
-
Problematic Smartphone Use
Wikipedia
It usually starts with social disorders, which can lead to depression and stress and ultimately affect lifestyle habits such as sleeping right and eating right. [28] According to research done by Jean M. ... For 3-4 year-old children: 180 minutes physical activity, 1 hour screen time, 10–13 hours of sleep time per day. [81] Phone settings [ edit ] Many smartphone addiction activists (such as Tristan Harris) recommend turning one's phone screen to grayscale mode, which helps reduce time spent on mobile phones by making them boring to look at. [82] Other phone settings alterations for mobile phone non-use included turning on airplane mode, turning off cellular data and/or Wi-Fi, turning off the phone, removing specific apps, and factory resetting. [83] Phone apps [ edit ] German psychotherapist and online addiction expert Bert te Wildt recommends using apps such as Offtime and Menthal to help prevent mobile phone overuse. [84] In fact, there are many apps available on Android and iOS stores which help track mobile usage. ... In Android a similar feature called "digital wellbeing" has been implemented to keep track of cell phone usage. [85] These apps usually work by doing one of two things: increasing awareness by sending user usage summaries, or notifying the user when he/she has exceeded some user-defined time-limit for each app or app category. ... The researchers implement an Android app that combined these three intervention types and found that users reduced their time with the apps they feel are a poor use of time by 21% while their use of the apps they feel are a good use of time remained unchanged. [86] AppDetox allows users to define rules that limit their usage of specific apps. [87] PreventDark detects and prevents problematic usage of smartphones in the dark. [88] Using vibrations instead of notifications to limit app usage has also been found to be effective. [89] Further, researchers have found group-based interventions that rely on users sharing their limiting behaviors with others to be effective. [90] Bans on mobile phone use [ edit ] See also: Mobile phone use in schools In some places in the world the use of mobile phones was banned in classes during instructional time, for example, in France , Ontario . ... IGI Global ^ a b "Smartphone overuse may 'damage' eyes, say opticians - BBC Newsbeat" . BBC Newsbeat . 28 March 2014 . Retrieved 12 June 2018 . ^ Bianchi, Adriana; Phillips, James G. (2005).

-
Cerebral Amyloid Angiopathy, App-Related
OMIM
A number sign (#) is used with this entry because cerebral amyloid angiopathy (CAA) can be caused by mutation in the gene encoding the amyloid precursor protein (APP; 104760). Mutations in the APP gene can also cause autosomal dominant Alzheimer disease-1 (AD1; 104300), which shows overlapping clinical and neuropathologic features. ... Revesz et al. (2003) reviewed the pathology and genetics of APP-related CAA and discussed the different neuropathologic consequences of different APP mutations. ... In 4 affected members of an Italian family with cerebral amyloid angiopathy, Obici et al. (2005) identified a mutation in the APP gene (104760.0019). In 2 brothers from an extensive Iowa kindred with progressive dementia and cerebroarterial amyloidosis, Grabowski et al. (2001) identified a heterozygous mutation in the APP gene (N694D; 104760.0016). ... Human APP mRNA was detected in neurons and neuronal processes, but not in vessel walls. ... Herzig et al. (2006) extended their earlier studies by developing several murine models of APP-related CAA and APP-related parenchymal amyloid deposition.
-
Pleomorphic Anaplastic Neuroblastoma
Wikipedia
Pleomorphic anaplastic neuroblastoma (PAN) is a striking aspect of neuroblastoma first described by Cozzutto and Carbone in 1988. [1] Another case was thereafter reported by Cowan, et al. with cytogenetic and immunohistological analysis in a 28-year-old man. [2] The case described by Navarro, et al. showed MYCN amplification (more than 10 copies) and a 1p36 deletion as measured with FISH in 13% of cells. [3] Additionally there was a main cell population with a DNA index of 2 indicating a tetraploid DNA content and a high expression of MIBI (Ki-67), bel 2, p53, and P-glycoprotein , either correlated with rapid progression of disease. ... Arch Pathol Lab Med 113:11-12. ^ Shimada H, Ambros IM, Dehner LP, Joshi VV, Roald B (1999). ... Cancer 100(2):390-397. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B (1999).
-
Occupational Hearing Loss
Wikipedia
Within the United States of America alone, 10 of the 28 million people that have experienced hearing loss related to noise exposure. ... "Evaluation of smartphone sound measurement applications (apps) using external microphones-A follow-up study" . ... "Smartphone-based sound level measurement apps: Evaluation of compliance with international sound level meter standards". ... Retrieved May 3, 2016 . ^ a b "CDC - NIOSH Topic: Occupational Hearing Loss (OHL) Surveillance" . www.cdc.gov . Retrieved 2016-03-28 . ^ a b "Ototoxic chemicals - chemicals that result in hearing loss" . Department of Commerce Western Australia . 2014-01-08 . Retrieved 2016-03-28 . ^ Campo P, Morata TC, Hong O (April 2013).

-
Inclusion Body Myositis
OMIM
The authors discussed the abnormalities of APP processing, the role of abnormal intracellular protein folding, oxidative stress, and the potential role of cholesterol in the pathogenic cascade of IBM. ... Accumulation of the amyloid-beta peptide, which is derived from proteolysis of the larger beta-APP, seems to be an early pathologic event in both Alzheimer disease and IBM; in the latter, it occurs predominantly intracellularly within affected myofibers. To elucidate the possible role of beta-APP mismetabolism in the pathogenesis of IBM, Sugarman et al. (2002) selectively targeted beta-APP overexpression to skeletal muscle in transgenic mice, using the muscle creatine kinase promoter. They reported that older (more than 10 months) transgenic mice exhibited intracellular immunoreactivity to beta-APP and its proteolytic derivatives in skeletal muscle. In this transgenic model, selective overexpression of beta-APP led to the development of a subset of other histopathologic and clinical features characteristic of IBM, including centric nuclei, inflammation, and deficiencies in motor performance.GNE, NT5C1A, APP, TARDBP, HLA-DRB1, SQSTM1, APOE, KHDRBS1, NUP62, DCTN4, GTF2H1, SDC1, CDR3, GSN, MAPT, TRBV20OR9-2, HLA-C, PLAAT4, FYCO1, MSTN, TNFRSF12A, NFAT5, CCR2, UBB, MALAT1, VCP, RBM45, AOC3, DCD, UCN2, DNAJB6, OPTN, KLRG1, MAP1LC3A, LILRB1, KDELR1, ICOSLG, SYNM, ROBO3, DDX58, CHMP1B, PABPC1, TIMP1, RRM2B, TWNK, FOXP3, KRT20, TTR, ACTB, THBS1, CST3, HLA-DQA1, HK1, H1-0, NR3C1, EPHB2, EMD, DES, CD47, TGFB1, CD38, CD36, CD34, MS4A1, CAPN3, BCL2, AOC2, HLA-DRB3, HMGB1, IFN1@, IFNG, TRIM21, AGER, MOK, PTPRC, PSME1, PSMB10, MAPK1, POLG, PMP22, MMP9, MMP1, MLF1, LMNA, IL6, IL1B, LOC102723996
-
Alzheimer's Disease
Wikipedia
The hypothesis holds that an amyloid-related mechanism that prunes neuronal connections in the brain in the fast-growth phase of early life may be triggered by ageing-related processes in later life to cause the neuronal withering of Alzheimer's disease. [64] N-APP, a fragment of APP from the peptide's N-terminus , is adjacent to beta-amyloid and is cleaved from APP by one of the same enzymes. N-APP triggers the self-destruct pathway by binding to a neuronal receptor called death receptor 6 (DR6, also known as TNFRSF21 ). [64] DR6 is highly expressed in the human brain regions most affected by Alzheimer's, so it is possible that the N-APP/DR6 pathway might be hijacked in the ageing brain to cause damage. ... Osaka mutation A Japanese pedigree of familial Alzheimer's disease was found to be associated with a deletion mutation of codon 693 of APP. [65] This mutation and its association with Alzheimer's disease was first reported in 2008. [66] This mutation is known as the Osaka mutation. ... A β is a fragment from the larger amyloid precursor protein (APP). APP is a transmembrane protein that penetrates through the neuron's membrane. APP is critical to neuron growth, survival, and post-injury repair. [103] [104] In Alzheimer's disease, gamma secretase and beta secretase act together in a proteolytic process which causes APP to be divided into smaller fragments. [105] One of these fragments gives rise to fibrils of amyloid beta, which then form clumps that deposit outside neurons in dense formations known as senile plaques . [98] [106] AD is also considered a tauopathy due to abnormal aggregation of the tau protein .APP, ACE, TREM2, ADAM10, APOE, PSEN1, GSK3B, HFE, MAPT, PLAU, NPY, BCL2, CASP3, BDNF, IDE, INSR, IL1B, LEP, BACE1, IGF2, IGF1R, ATP5F1A, INS, BAX, CR1, A2M, ABCA7, TOMM40, CD2AP, BIN1, EPHA1, CLU, PICALM, NOS3, PSEN2, APOC1, MPO, SORL1, VSNL1, INPP5D, NECTIN2, MS4A4A, PCDH11X, CASS4, BCHE, MIR146A, CYP46A1, DHCR24, CHRNA7, NCSTN, VEGFA, DPYSL2, PRNP, ESR1, PPARG, RELN, HMOX1, ACHE, CST3, MAOB, TNF, MTHFR, IGF1, CD33, TFAM, IL6, CYP2D6, CRH, SOD2, UNC5C, PLCG2, TF, ABI3, WWOX, SLC30A6, CHRNB2, ARC, PGRMC1, F2, CALM1, EIF2S1, HLA-DRB5, ENO1, TPI1, IGF2R, SLC30A4, MIR296, SLC2A4, MIR100, IQCK, MIR375, AMFR, SNAR-I, ADAMTS1, MAPK14, PIN1, PYY, PTGS2, S100B, PPARGC1A, NOS2, NGFR, NGF, NFE2L2, SOD1, SYP, CDK5, NGB, MIR505, GAPDHS, MME, MAP2, CTNNB1, TPP1, LRP1, IRS1, CHAT, GAPDH, MIR4467, MIR3622B, AGER, MIR766, MIR708, CAV1, NTRK2, PTGS1, APLP2, ADAM17, MFN2, DNM1, HSF1, GSR, IL33, CCR5, HSPD1, HSPB1, CIB1, CASP8, IKBKB, SERPINF1, ATP7A, MT2A, ADAM9, INS-IGF2, BCL2L2, CASP9, GAB2, PTK2B, PLCB1, ABCA1, GRN, CASP12, SQSTM1, FERMT2, HLA-DRB1, NFIC, CSF1R, APOB, MARK4, HSPG2, MS4A6A, CELF1, VCP, SYNJ1, ZCWPW1, MS4A4E, APH1B, APOC2, F13A1, EXOC3L2, PLXNA4, ADAMTS4, AKAP9, MADD, DST, PILRA, FRMD4A, LAMP1, SLC24A4, GLIS3, SPON1, CADPS2, IL34, COL18A1, TRIP4, SPI1, TGFB2, BCL3, MTHFD1L, AICDA, IL6R, DCHS2, MEGF10, SLC16A7, EPHX2, NDUFAF6, DSG2, OSBPL6, CELF2, UBE2L3, SPPL2A, MAPK7, CDH13, LAMA1, SGK1, SUCLG2, LUZP2, PTPRG, ST6GAL1, AP2A2, RBFOX1, SORCS3, TSPOAP1-AS1, TLN2, ZAP70, ALDH1A2, TCF7L2, FMN2, OTOF, EXOC4, HSD17B10, DNM1L, ALOX5, GULOP, HTR2A, AHCYL1, SDR42E2, HTR6, GTF2H1, GRM5, IAPP, AHSA1, STAG3, ALB, FARP1, TSHZ1, HRES1, AGFG2, DCAF7, SIGMAR1, BCKDK, RTN3, TPPP, HSPA4, HCLS1, HSP90AA1, G3BP1, PGAM5P1, BACE1-AS, KHDRBS1, ALDH2, CFH, HCRT, GPC6, ABCA8, GLP1R, NR3C1, TNRC6A, IL19, PARVB, DDX25, BZW2, FCN2, FGF10, GOLIM4, LINC00476, HPGDS, FANCD2, PDE7B, SIGLEC7, TSPAN16, TRPC4AP, POLDIP2, CNTNAP2, RNF19A, BACE2, UBQLN1, ITSN2, GRIN2B, ZGLP1, BCAS3, EPDR1, TMED9, ENO2, CYCS, ANXA1, EPHB2, EPO, RAPGEF6, APH1A, LARS1, EYA4, SNX9, ESR2, WAC, SLC8A1-AS1, DCTN4, RMDN1, QPCT, MTOR, SGK3, FBXL7, SZT2, GIP, ACSL6, WWC1, CLEC16A, KAZN, LINC01672, PRRC2C, COLGALT2, PLD3, HECW1, ZNF292, MYO16, DKK1, SIRT2, ACOT7, PSIP1, CLASRP, LINC00271, SIRT3, SIRT1, GFAP, NUP62, FYN, CBLC, INHCAP, TRIM51CP, GABPA, GABRA2, GABRG3, DAPK2, SMUG1, GAP43, GATA1, GCG, GCHFR, TARDBP, NCS1, GDNF, HDAC6, RAB3D, MVP, OGG1, LINC02268, LINC02325, SOAT1, ACTG2, ACTG1, SNCG, SNCA, SNCB, SNAP25, NOS1, ACTB, MEF2C-AS1, SLC6A4, NPC1, SLC6A3, GEMIN7-AS1, SLC1A2, LINC01508, LINC01725, NEFL, COX2, TNFRSF1B, STAG3L5P, TLR4, TLR2, MSH2, THY1, MT3, TH, SST, STAG3L5P-PVRIG2P-PILRB, TGFB1, TFF1, RNR2, LINC02653, LINC01712, TCF3, NRGN, CX3CL1, ELMO1, CCL2, PIK3CG, ABCA2, PLA2G1B, EIF2AK2, SERPINA3, PLG, MAPK8, MAPK1, PRKCB, PRKCA, PRKAB1, PRKAA2, PRKAA1, PMS2P1, POLD1, PTPA, PON1, PIK3CD, PIK3CB, PIK3CA, MOK, UPK3B, SORT1, ROS1, SERPINE1, REST, REN, RELB, RAC1, LINC01965, PVR, PVALB, PTPRA, LINC00972, ABCB1, LINC02008, MTCO2P12, TP53, OVCH1-AS1, MOBP, MNAT1, SLC4A8, AGT, INSIG1, AZIN1-AS1, EIF3E, FHL5, IRF2, GSTO1, ITM2B, GRAP2, LIPG, ADIPOQ, KL, MSC, LRAT, AP4M1, CCRL2, IL18, IL17A, IL12A, ST18, IFNG, ARL17B, AKT1, AIF1, KRBOX1, IGFALS, HDAC9, PHF14, IL10, IL1A, ALOX12-AS1, MICAL2, IL2RB, IL4, CLOCK, CXCL8, KCNN2, MPZL1, HERC2, VDR, TFEB, MAOA, COX10-AS1, ZNF232, YWHAZ, VLDLR, PARP1, UTRN, BCAM, UCHL1, UBB, TYROBP, AFF1, TTR, TRPM1, MMP9, AIMP2, LTBP2, KNG1, CRADD, CACNA1G, LAMC2, RPSA, CDK5R1, SUCLA2, LCN2, LDLR, BECN1, LPL, PDE5A, ABCB11, APOC4-APOC2, KHSRP, DENR, AGPS, LPA, CDKAL1, PPARA, MEIKIN, COL4A4, CRK, CEACAM22P, SCIMP, CREB1, CR1L, ZNF862, SH2D4B, CP, PNPLA7, SIMC1, GGACT, COMT, COL12A1, FAM181A, BRCA2, PPP1R37, GPR141, TENM3-AS1, CNR2, L3MBTL4, UBXN11, ACKR2, TMEM132C, CASTOR3, CLPTM1, STRADA, FNIP1, CDCA5, NLRP3, APOA1, CRMP1, KAT8, CSMD1, CLMN, PINK1, CYP8B1, AHNAK, MIR132, MIR107, CUX1, POTEM, PPP1R3B, FAS, SAP30L, ANKRD55, CTSD, CTSB, GEMIN7, EHMT1, LINC01184, CTNNA2, LINC01185, TMC5, THSD4, CCDC134, SP6, LINC01567, PDCD1LG2, SETD7, APOD, BHMG1, CSF2, HYI, BLOC1S3, TSPO, CHRNA4, CHRNA2, TAS2R62P, C3orf67, C9orf72, CCDC83, CCDC89, KDM1B, CD14, TGM6, ATXN7L1, RSPO4, ADGRF2, STH, TAS2R64P, CALHM1, RUNX1T1, PPP1R42, ALPK2, PCSK9, CAT, ANKRD31, CASP6, NKAIN3, TRIQK, CALB1, STEAP1B, CASP1, EPHA1-AS1, CAPN1, APOC4, FAM181A-AS1, NKPD1, SPRED2, CD36, SCARB1, PLPP4, MED12L, ARAP2, CHN2, CHI3L1, ACTBL2, C10orf71, MCIDAS, LRRK2, AKR1C4, ANO4, AGBL1, CEACAM20, ZNF813, RMDN3, CETP, CDR1, LINC00343, TCAM1P, APOC1P1, IGSF23, RMDN2, CDK1, SLC25A48, NKAIN2, FSIP1, CD68, BMPER, C3, CD40, CYP19A1, CRP, NIT2, ANO3, DLG4, ARHGAP20, RCAN1, WDR41, NDUFA12, STK32B, EDEM2, DSCAML1, RNF165, SH3RF1, DYRK1A, MIR29A, SYBU, AQP4, APBB1, DLX5, DBN1, PALM2AKAP2, CEACAM19, DPP4, IL6-AS1, ARVCF, CDC42SE2, DMXL1, TULP4, DAPK1, PMS2CL, POTEKP, MIR34A, VAT1L, OLR1, HDAC2, LRP8, GSN, CCL11, S100A9, COL25A1, POTEF, KLK6, BLMH, HSD17B7, P2RX7, COX8A, ABCB6, PRRT2, IL2, SORCS1, NR1I2, MAPK3, ITGAM, CASR, ATP7B, VDAC1, EGR1, PDE4A, RAB5A, SUMO1, NRG1, OXER1, NTRK1, TFCP2, ANK1, CSNK1D, DLST, APLP1, BLVRA, NFIB, IL1RN, HTT, ACAT1, PLA2G4A, NFIX, NLRP1, GPRC6A, HMGCR, PPID, LPAR3, FZD4, REG1A, MRGPRX1, LRP2, DBH, PSENEN, VPS35, ESCO1, HSD11B1, VN1R17P, SOX2, AGTR1, XBP1, MIR155, MRGPRX4, MRGPRX3, GAL, GPR151, IL13, PAEP, OGDH, GPR166P, STAT3, SET, NFIA, PLB1, AR, LGR6, DHRS11, ABCG2, C4A, KCNIP3, HSD17B13, ABCG1, TTBK1, NOTCH1, EIF2AK3, SLCO6A1, CHMP2B, RBM45, CD44, RIPK1, APBA1, GSTK1, ADNP, ICAM1, BRCA1, APCS, TNFRSF1A, NFKB1, CNTF, MMP3, KLC1, LBP, CTNNA3, SGSM3, FGF2, C4B, HIF1A, CREBBP, SERPINA1, TMEM106B, GRIA1, GRIA2, ECE1, C4B_2, GSAP, OGA, TFRC, PLA2G6, ST3GAL4, PAWR, MFAP1, KAT5, GSTM1, APRT, COX1, HP, NTF3, MIR206, FPR2, CDC42, FUS, MARK1, FGF1, PREP, C5AR1, PON2, MIR29C, CALB2, PDIK1L, SYK, S100A1, CH25H, SREBF2, COX5A, GRIN2A, VCAM1, TMED10, GSTP1, KLK8, PHF1, CXCL10, MEFV, SP1, GJA1, IGFBP3, SLC17A7, CYP3A4, FOXO3, HMGA1, SLC11A2, XPR1, MARK2, PPIF, CRHR1, SHANK3, MYC, CD40LG, CPOX, FKBP5, ANPEP, CAST, C1D, FKBP4, HSPA1A, FLT1, MIF, PLA2G2A, CX3CR1, CSF3, IFNB1, KALRN, PLTP, STXBP3, DDR1, PWAR1, PRDX2, TP63, VIM, IL23A, F2RL3, MMP14, MEF2C, TREM1, TMED10P1, NAT2, MIR342, SAMD9, RAB7A, PGR-AS1, TRPM2, EGFR, ADRB2, CLDN5, ETS2, SYT1, TIMP1, NME8, ELANE, F2R, CD59, EPHA4, CBS, MSMB, APLN, MMP2, MYCL, CALML5, SYN1, XRCC1, TGM2, EEF2, PLA2G7, ELAVL2, EDN1, TMEM97, HMGB1, MIR455, HTRA1, BPIFA2, SLC52A2, NQO1, TUBA1B, FOS, CRYAB, SLC2A1, GPR3, LGMN, SLC2A3, RIDA, FN1, ABCA4, HSPA1B, PECAM1, PTEN, HSPA8, HLA-A, PTPN1, HAMP, TXNIP, GRM2, P4HB, LIN28A, PSPH, GSTT1, CCN2, DECR1, CPLX1, BCYRN1, NES, POU5F1P4, MIR21, GRK5, POU5F1, NTSR1, MIR212, PRKN, LINC02210-CRHR1, HSPA5, DLD, DAB1, HTRA2, POU5F1P3, MIR137, DNAH8, MAPK10, GH1, SERPING1, ADAMTS2, EEF2K, GSTO2, ROCK2, NEDD9, SPTBN1, NTN1, CEBPD, GDF2, CEBPB, PWAR4, SYNM, IGFBP2, GLUL, ABCC9, ATM, PSPN, PTPRC, MIR29B1, RANBP9, NDRG2, CNR1, RTN4R, PTBP1, AQP1, PDK1, MIR106B, PDE4D, ARNTL, PRDX1, ADM, RENBP, POMC, PTPN4, MS, PDGFRB, P2RY2, MIR142, PDE9A, SSTR4, KLK3, BCL2A1, C2, SRPK2, NFATC2, ADCYAP1, LRRTM3, PLD1, NUBP1, MIR424, ATF4, BSG, MIR29B2, PPY, BMP4, TBP, SLC18A3, POLB, NOTCH3, SLC18A2, NPTX2, MTR, SI, MIR222, SH3GL2, ND2, NR4A2, SELENOP, CXCL12, CCL5, ATXN1, CALM3, ITGAX, IFNA13, DISC1, OPTN, HTR1F, HTR4, WNT3A, COL11A2, C20orf181, IFNA1, KEAP1, HDAC4, KLK4, SEMA6A, LRRC4, CRTC1, IL9, DAO, ALOX15, AGTR2, IDO1, SLC25A27, ABCG4, CD55, APOA4, MCOLN1, REM1, ATCAY, EBPL, HSPB2, HSPA9, PLK2, GAD1, NANOG, DCX, COASY, UBE2K, DDIT3, TREML2, APBB2, MAP1LC3B, SRRM2, GZMB, FXN, HNRNPA1, HPSE, RAB10, CIP2A, FOLH1, STIM2, DIO2, MMP24, CEBPZ, GBA, CDR2, ITPR3, CDKN2A, MELTF, SLC30A3, ADRA2B, FTO, FNDC5, GGA3, XPNPEP1, VGF, NR1H2, UGCG, MFGE8, MGAT3, CXCR4, TLR9, APOC3, GPT, ELK3, NEAT1, ADORA2A, MMEL1, TRPC6, EIF4E, CAMK2A, MS4A6E, SRR, HSPA14, IRS2, MGAM, C1orf52, HDAC3, PABPC4, ACKR3, LGALS3, FAM20C, WNK1, DRD4, CYP2C9, MBTPS1, DRD1, LRP6, GRK2, CYP2B6, OGT, LIPA, AD11, GORASP1, PTGDS, SPEN, MIR200B, NPTXR, DNMBP, MIR200A, NCOA6, MIR181C, RBP4, RELA, OPN1LW, EFHD2, MIR188, TPH1, HNRNPA1P10, CTNNBL1, SLC40A1, PNO1, CHCHD2, SDF4, RETN, GOLM1, PPP3R1, PYCARD, PAG1, CCR2, DDIT4, RCBTB1, SBNO1, PPARD, CD274, PCBP4, ACE2, PROS1, PRND, PPP1R15A, CIZ1, MIR26B, TPSG1, GGA1, CFAP97, MAP2K1, AATF, SHANK2, PRL, RBMS3, LOC107987479, MAP2K2, PAXIP1, CHCHD10, SBNO2, PTGES, SPHK1, LPAR2, PPIG, NRXN3, MED23, SPP1, MAPK8IP1, BAG3, APBA3, TAP2, PRDX6, CARTPT, SNAP91, SV2A, MALAT1, MAK16, SYVN1, GDF11, TAC1, TAT, SLC6A2, TRPV1, CISD3, TPT1, TSC2, TM7SF2, TXN, UBE2I, TLE1, TIMP2, XK, CNTN2, TGFBR2, YY1, GOLGA6A, ANP32A, TAM, TERT, DCP1B, TNFSF10, PPP1R1B, GPHN, RGS2, ADAM30, SAA1, METAP2, IMMT, SDS, ADAP1, RYR3, ECHDC3, RYR2, RXRA, SCD, RPS6KB1, CIT, RPS6, CTXN3, LMTK2, NLGN1, ROCK1, RGS4, ATXN2, SRL, STUB1, SHBG, LILRB2, AKR1A1, OLFM1, SKIL, SLC9A6, CREB3, PITRM1, SCGN, CXCR6, OCM, RIN3, SGCA, SFPQ, NCKAP1, CPLX2, TP73, EDAR, CCL3, NPS, BEST1, HPS1, CXCL1, GLO1, LIF, CDH1, LHCGR, CDK4, PCNA, PCK1, L1CAM, GPR42, GPX1, GRB2, ANGPT1, ANGPT2, CETN1, MYD88, GLB1, AKT2, LMNA, DNMT3B, TSC22D3, CD38, PER1, CD69, DRD3, LOX, CD74, LMNB1, DNM2, GAS6, AVP, GC, DMRT1, SARDH, GRIN1, ANXA5, BACH1, P2RY1, INPPL1, CSF1, CS, NEFM, NTS, APEX1, STS, HTC2, NEUROD1, NPTX1, NPPA, ATF2, HTR2C, ARRB2, IGFBP7, HMGCS2, CLK1, CKB, APBA2, CYP17A1, GSTM3, CHGA, CYBB, JUN, CTSS, ORM1, ITPR1, CHRM1, CHRM2, OPRK1, OPRD1, CTRL, HK1, ADRB1, LAMP2, CASP2, EDNRA, PLD2, CAPN2, F2RL1, ACO1, ERBB4, FAT1, FGFR3, CALM2, BRS3, CALCA, CAD, EGF, CASP4, FCGR3B, FDPS, LYZ, FCGR3A, FLNA, MECP2, FABP3, ADRA1A, PLXNA2, MBP, FAAH, ENPEP, F12, BAG1, MEOX2, HOMER1, ITGB2, DBA2, ITGAL, ITGB1, AIM2, AZIN2, CD80, ITIH4, CD46, VIP, CHRNA3, ATG5, TREML1, MCL1, GPRASP2, VWF, APOA5, TMEM119, KLF4, SOCS6, WNT1, XBP1P1, FOXQ1, C3AR1, OPN4, USF1, C1QA, CXCR2, VPS26A, MOGAT3, CCR3, IL6ST, IL5, IL1RAP, LMF2, IL1R1, CREB3L1, UNG, MCU, CLSTN3, SNPH, IL9R, NPEPPS, NAPSA, USF2, MEF2A, ING1, CGB8, FTMT, CHM, VAV1, IMPA1, C1R, ILK, ADAMTS3, IL16, MAP3K5, IL15, GDF15, CGB5, CA2, KCNB1, TSPOAP1, SMAD2, DPPA2, SGO1, LIG3, IFNL3, TNK1, CP20, APCDD1, HSD17B6, CDKN1A, TTBK2, CDKN1B, SLC2A14, CFLAR, STMN1, LIPC, TAB3, CHIT1, BRAP, SPARCL1, MLKL, PTCRA, CD47, LGR5, CD8A, CCT, NR4A3, USP9X, MSRB3, CDR3, CCK, LNPEP, CASP7, CHRFAM7A, CAMP, PER3, YES1, CGA, MARK3, CGB3, SYNGR1, CALCR, IL1RL1, ARHGEF2, PER2, SLC33A1, TPH2, CHEK1, RAB7B, NOG, MBL2, CFL2, HSPB6, AHSA2P, SLC30A1, CD200R1, SOCS3, KDR, KIF5A, HAP1, CALR, CES1, TRPA1, HSPB3, WASF1, SLC32A1, ARHGEF7, CAMK4, COL3A1, H3P40, SCRN1, PTCD1, SPHK2, ATN1, PPIL2, POU2F1, FOSB, GCA, FLT4, SH2B1, APPL1, DNMT1, FLG, FOXO1, DUSP1, DUSP6, HHAT, HSPB8, E2F1, PLXNA3, DOCK2, PNPLA2, ADI1, GMFB, GPI, GPC1, KIF21B, NMNAT2, TRIB3, ALS2, DLG2, DLG3, GLS, PDSS2, ASTN2, MCF2L, GLI2, KIDINS220, CBLIF, SYNE1, DMD, GGT1, FKBP1A, SIT1, SV2C, GDE1, FOXP3, ASCC1, TMED7, FIS1, PRLH, CRYL1, ADIPOR1, LSR, F11, MBL3P, SIRT6, TRMO, NRN1, LCMT1, PRRX2, ERN1, BIN2, UBR5, HEBP1, GEMIN4, PDCD4, TBK1, SLC25A38, FGF14, PCSK1N, TRPM7, DLL1, FLVCR1, AHI1, SETD2, ELK1, IL22, NCAPH2, ELN, PADI1, BPTF, NRBF2, FABP5, EP300, PLA2G3, GRHL3, CXCR3, NAV3, SIRPB1, FLOT1, MET, KCNMB2, CRISPLD2, ARHGAP24, HNMT, SNX27, NPL, BHLHB9, TRIM13, HMOX2, KLF2, CSNK1E, LPAL2, CPQ, PPP1R2C, RAPGEF3, TET1, CRYZ, SORBS3, CTBP1, BCL2L11, COL17A1, NCAPD2, COX10, UBASH3B, NR1I3, ACOT8, PTPN5, PPP1R9B, TOM1, MINDY4, CPE, HTR7, NR1H3, HTR1B, HTR1A, LRPPRC, PDIA6, RHBDD1, NAA25, HLA-C, TPX2, BCAN, ADAMTS13, MOAP1, TNMD, GRIA3, NEUROD6, CHEK2, PADI2, HRH3, PHB2, SIL1, MGLL, FFAR1, GADD45A, MMRN1, DEFA1, IL21, NLRC4, GPR17, AZI2, HHIP, CTF1, HCRTR2, HLA-B, CTNND2, HHEX, HGF, CTSG, HDAC1, CTCF, DHX40, PTGES3, STIP1, CTSZ, CXADR, CARD14, CYP1A2, PDE10A, LILRB1, EHMT2, PDIA2, UMOD, ANGPT4, MIR339, SYN2, MSD, ACP3, APEH, ST8SIA1, AZU1, PI4KA, NCAM1, MIR144, SMIM10L2B, MSI1, NAP1L1, PRSS3, PEBP1, MASP1, SIM2, TIA1, MIR15B, ATD, RPL29, ABCC1, NCAM2, MIR125A, TLR5, SERPINF2, TNFAIP1, CXADRP1, MAPK9, ZFHX3, GGTLC4P, AD10, SGCG, MIR451A, CDR1-AS, TPTEP2-CSNK1E, MIR384, ITSN1, CBSL, MPZ, NFATC4, PKM, NCL, NTF4, LOC643387, SLC1A3, APOA2, THAS, PSMB6, SERPINB6, PSMB9, RHOA, ARMCX5-GPRASP2, SMPD1, REG3A, ATP4A, MIR193B, RFC1, NOTCH4, SLPI, PGF, AEBP1, MIR214, MIR219A1, SLC19A1, MIR22, PCSK1, ALPP, AMD1, MTNR1A, TGFBR1, COX3, ATP12A, NM, ADD3, PSD, TGM1, ARR3, NPM1, PAK1, TMED7-TICAM2, PNP, MIR195, MTHFD1, ADH1B, AMPH, ND4, AMD1P2, ARG1, SULT2A1, RRAS, PDE7A, TTPA, TYK2, TXNRD1, PPP1R1A, PPP1R10, SPG7, SPAST, RAB4A, PPP2CA, OTC, PPP2R2B, MMP1, ARMS2, RAB3A, GGTLC3, RTL1, P2RX4, ST13, SPARC, ALAS1, PPP3CA, NEFH, SEL1L, TYR, RAB6A, MICB, PPIA, CCL4, BST1, TICAM2, BNIP3, MIR326, OPRL1, PON3, BMP6, OPRM1, AHSG, H3P17, SDC2, AGRN, TYRP1, PPP1CA, BMI1, TYRO3, PNMT, DEFA1B, GGT2, ORI6, SMIM10L2A, CISD2, ARSA, EIF2AK4, PRKAR1A, LRP1-AS, SRSF2, MIR98, MIRLET7B, CD200, PDCD1, RAP1A, GGTLC5P, CCND1, ANXA6, FXYD1, S100A6, NFATC3, PLK1, ABO, PTGER3, APC, S100A12, ASL, HSP90B2P, SETMAR, PRRX1, PZP, STAT1, ODC1, CFB, CDNF, ZBTB4, PARK16, SUGP1, DIO1, SORCS2, MAGEE1, ALDH1A1, MIR1306, DES, DIAPH1, LSM2, MIR1229, XPO5, HCN3, CFD, MIR664A, KIF17, WDR48, MTRNR2L12, PRX, DHFR, EPG5, SEPTIN1, FAS-AS1, RNF213, MIR320E, MIR1908, HECW2, LINC00672, NUFIP2, ABCD1, DLG1, QRFP, ZNF410, AOC2, NBEAL1, CYP11A1, OPN1MW2, AD6, P2RY12, NMNAT1, DEPTOR, TNS3, CYP2D7, FAM72A, NUCKS1, CLEC7A, CYP26A1, ARAP3, GREM2, CDKN2B-AS1, CYP2J2, UBE2Z, MIR1246, TSPY3, MIR632, CTSK, GTDC1, CTSL, MIR650, MIR660, SNORD118, TNFAIP8L2, LYNX1, MUL1, PAGR1, CYLD, MAPKAP1, APOF, PDCL3, CYP2C19, NOC3L, CYP27A1, DPEP2, MFT2, PROK2, HPSE2, AD14, AKR1C2, ALPI, TRPV4, NTN4, PRM3, PDF, JAM2, ALOX5AP, TSPY10, DEFB4A, DEFB4B, ALOX12, PTBP2, DCN, NECAB3, FKBPL, NEUROG2, DGKQ, SLC25A4, MIR873, MIR301B, CENPK, DAXX, GFRA4, GOLPH3, ERVK-6, MTUS1, DBI, MIR937, ANG, ACE3P, SOD2-OT1, ANK3, DRD2, ZNF608, NAT10, DYM, LOC102724334, TRIT1, EIF4G2, TET2, EIF5, SERPINB1, ELAVL4, PDP1, ACO2, THRA1/BTR, UGT1A1, CCHCR1, CPVL, SMOX, TOLLIP, TERF2IP, SNTG1, EMP1, LOC102723407, EIF4EBP1, MIR6845, EIF2S3, ADCY2, NUDT11, EGR2, MSTO1, ADARB1, SLC6A15, ADA, TAPBPL, TESC, MIR6840, ACVRL1, FOCAD, EIF4A1, CASZ1, QRICH1, PGPEP1, EIF4A2, NDE1, ASIC2, CTTN, ACADVL, GSKIP, LNCRNA-ATB, ATP6V1H, H3P7, TDP2, ERBB2, CINP, ZCCHC17, H3P13, DTL, GPRC5B, ERCC1, DCDC2, NAT8B, GULP1, H3P23, ERG, H3P28, H3P11, PPIL1, STIN2-VNTR, NANS, EPHA8, H2BS1, POLE3, ACACA, SLC29A1, FXYD6, LRP1B, CST12P, SIRT1-AS, INPP5K, MSRB1, ARID4B, EPOR, ABL1, AAVS1, NR2F6, ERVK-32, LOC110366354, MNS16A, EFNA5, SLC47A1, ALAD, EEF1A1, SNHG19, MICA, DNTT, SOX21-AS1, DOCK3, DPYSL3, MIR626, XAB2, MFF, DUSP22, ARNTL2, SPPL2B, MCCC1, TMX2-CTNND1, ANKS1B, DPYSL5, FXYD6-FXYD2, BARHL1, DSC1, TWSG1, TLE5, DNASE1, DNA2, OCLN, NLN, AMIGO1, AHR, PLEKHG5, SLC24A3, SPC25, TTC7A, PELI1, JAG1, TMEM159, RTN4, APMAP, CD177, CAMK1D, PLAAT1, NR0B1, TIGAR, P2RX5-TAX1BP3, PARD3, GKN1, ADH6, INAVA, CDK5RAP2, OGDHL, LINC01080, ATF7IP, IPO9, VAC14, DVL1, PPP4R3A, OPN1MW3, EBM, OTUB1, SOX6, SLC30A10, SMPD3, MEG3, PLIN2, FBXW7, TDP1, ADORA1, DSC3, ACSS2, BTNL2, KIAA1217, ZNF253, CFC1, MIR4668, DSG1, APOM, MYO5C, MIR4487, NOTCH2NLC, USE1, SELENOS, GDNF-AS1, DSPP, ADCY10, ADRA2A, ZNF415, LINC-ROR, NARS2, CSF2RB, MIR616, MIR20A, CDC25C, PROM2, ATP6V1E1, IL23R, GLIS1, PM20D1, PHF13, CDH2, ZNF569, MIR191, CDK9, MIR192, PRIMA1, CDKN2D, MIR196A1, OR2AG1, LAYN, PIWIL4, MIR19B1, GPBAR1, CDC25B, GDF7, ZDHHC15, MIR139, CD63, SGMS2, MIR140, TMPRSS6, RHBDL3, AVPR2, MIR15A, CBLL2, MIR186, PRUNE2, AMOTL1, CD81, MIR18A, SLC2A12, CDA, MIR181A2, ATP5PO, SESN3, ATP6V1B2, UBR1, ATP5PF, PPME1, MIR224, LYZL4, KCNH8, MTERF4, MIR23A, CPO, ACMSD, MIR23B, BHLHE23, MIR25, CFL1, OSCAR, SPNS2, SEZ6, SLC38A10, MSI2, CFTR, MIR27A, MIR223, ALDH7A1, MIR221, SELENOM, ATP5MC2, CACUL1, HECTD2, SREK1, CTCFL, CBLN4, CDSN, ATP5MC1, DEFB104A, OCIAD2, MAGEC3, MIR210, CEACAM5, CECR, PPARGC1B, ATP5F1B, IL31RA, GNPDA2, SCARB2, NSMCE1, SOCS4, UBE2L1, BNC1, CACNA1C, SLC25A20, SERPINA13P, BLM, SREK1IP1, MIF-AS1, C20orf203, SYPL2, ZNF763, CCL4L1, BID, BGN, ZSCAN1, ZADH2, SMIM20, MILR1, PGP, GOLGA6L2, TMEM189-UBE2V1, TMEM189, IL31, C4BPA, BTK, AMIGO2, HCN1, NHLRC2, ATP9B, SBSN, BMP1, OSTN, C5, CFAP410, BARHL2, NANOS3, C9, STING1, GADL1, ARMH1, VPS51, HCAR2, CAPG, LINC00639, TMEM201, LIN28B, CD5L, MIR127, CD19, KIF6, MS4A1, MS4A3, HYLS1, STOX1, FOLH1B, OR8J1, TRIML2, CD28, KHDRBS2, GAPT, CENPV, KLHDC8B, MIR134, CD86, PIKFYVE, SLC29A4, CCNC, KRIT1, PTF1A, DAOA-AS1, BDKRB2, HCA1, BRD3OS, ASPM, BCS1L, SGMS1, BCR, MIRLET7D, MIR122, RUNX1, ANKK1, BCL6, PHYHD1, BAK1, MIR10A, EBF3, CCKAR, MIR28, FRMD6, MIR613, ADAMTS10, TMEM175, XIAP, MAF1, BIRC3, SLA2, ANTXR1, ASCC2, CRHBP, EVA1A, QRFPR, MAGT1, NCALD, LOC646506, ROPN1L, L3MBTL2, GMNC, SCFV, CSE1L, RNF146, PHF6, HOOK3, BRSK1, MBOAT4, COX15, MIR497, MFSD2A, MIR501, ACCS, ARG2, FAM126A, CPB1, CPN1, ECSCR, MAP1LC3A, MIR484, AQP9, CPS1, SNORD35B, CPT1A, ABLIM2, FASLG, NETO1, DOCK8, RNFT2, C1QBP, TM2D3, ASRGL1, PTGES2, PANK2, MIR590, SCD5, CSNK2A1, VCAN, ZC3H14, CSPG4, CTBS, MIR592, MIR598, CAMKMT, CTNS, SLTM, PTCD2, CTNND1, MIR603, NUBPL, WDR26, SPHKAP, GSTT2B, TMEM163, NCF1, LBH, SPAG11A, SFTPA1, CSF3R, ZNF436, CSN2, NDFIP1, MIR545, SLC44A4, SLC19A3, FAM72B, AIRE, IQCJ, CSNK1G2, DNAJC5, CSNK1G3, LINGO1, ATG4C, ATP2B4, MIR346, SERPINC1, CISH, ASPA, CLC, UCN3, TMEM54, ASS1P1, CLCN3, NACC1, ASIP, STX1B, IFT43, MIR133B, MIR151A, CLK2, TP53INP1, MIR330, MIR335, MIR338, MIR93, SLC26A7, OMA1, CHGB, PLD4, TDRD9, CHD1, MIR299, ATIC, ATHS, H4-16, LRIG3, EXOSC6, CHRM3, MIR30B, MIR30E, AGAP2, MIR31, MIR34C, MIR9-1, GRIN3B, GRIN3A, ASAH1, MIR369, H2BC12, KPRP, LRSAM1, MIR429, H4C15, SHF, GADD45GIP1, COL11A1, ZNF628, MIR431, HNP1, NAV2, MIR409, SLC31A1, RPPH1, SNORD14E, SNORD14D, SNORD14C, SNORD14B, COX6B1, MIR485, CNTN1, DNM1P33, CNTFR, CHRDL1, CLN3, MIR361, MYOCD, PRDM6, DNER, SPECC1, MIR377, CLN5, EXOC3L4, CNP, MIR425, ARRB1, ZNF804A, BDNF-AS, NLRP12, CCR6, ABCC2, DEFB104B, LRRC15, POU3F4, HOOK1, TERC, NAT1, MCM2, EZR, MDH1, VEGFC, MDH2, MDM4, MEF2D, UVRAG, UROD, UQCRC1, UGT1A, SLC35A2, UCP2, UBTF, UBP1, MID1, UBE3A, UBE2V1, CXCL9, ATXN3, UBE2D2, UBE2A, UBC, MAP3K10, MC1R, WARS1, WAS, ZMYM2, MANF, SCG2, FZD5, SMAD7, MAG, MAP3K12, MAP1A, MAP1B, ZNF236, ZNF224, ZNF217, RNF112, WEE1, MZF1, ZIC1, MARS1, MAS1, MAT1A, MAT2A, MAZ, XIST, WT1, WNT2B, WNT5A, UBA52, KMT2A, MLLT3, THBS1, TLR3, TLE3, MSH3, TKT, TIMP4, TIMP3, MSR1, MSRA, THRA, THOP1, THBS4, CYTB, MRE11, NUDT1, TGFBI, TGFB3, ND1, TFF3, TFDP1, MTNR1B, MTRR, TRNL1, MUC1, TERF2, TSPAN7, MRC1, TWIST1, TRAF2, TUBA4A, NR3C2, TTN, TSPY1, TSHR, TSG101, TSC1, MMP7, MMP8, TRPC1, TRH, NR2C2, TNFAIP6, MMP13, TPM1, MOG, MOV10, TP53BP2, TP53BP1, MPG, TNR, TNNI3, MPI, MPST, SLBP, REEP5, DEK, SNX3, TNFRSF6B, RAB11A, LDHA, LEPR, LGALS4, LGALS9, GPAA1, RNMT, GBF1, ADAM19, URI1, TRADD, LCK, B3GALT4, LIFR, LIG1, SOCS1, NUMB, LIMS1, AOC3, PDE8B, USO1, TNFSF11, STK16, LCT, CES2, KMO, KLRC1, BRSK2, KCNQ1, KIR2DL2, NOL3, ATP6V0E1, SELENBP1, USP13, KLKB1, CDK5R2, RAB29, MBD2, KIF11, TMEM11, ENDOU, EIF2S2, TAX1BP1, NAE1, KRT14, KRT18, LAMC1, LBR, PROM1, LCAT, SOCS2, PRKRA, DEGS1, DDX39B, PABPN1, EOMES, BAP1, LTF, H4C9, COLQ, DYSF, CHAF1B, LYN, NRIP1, COIL, SLC7A5, AD5, H4C1, FGF23, ADAM12, BRD3, PSCA, ARHGEF5, TFPI2, FZD3, GHS, M6PR, MARCKS, SMAD1, LTC4S, H4C4, BHLHE40, IRS4, MAPKAPK5, LMO4, LOXL1, CST7, DDO, DGKZ, GAS7, PIK3R3, PKP4, PPFIA1, LRPAP1, SORBS2, H4C6, CUL4A, GNPAT, LTB, H4C14, H4C13, H4C5, H4C2, H4C8, H4C3, H4C11, H4C12, TERF1, MUC4, KCNMA1, TDO2, PCP4, CDK18, RBM3, RBL2, RBBP6, RB1, RASGRF1, RASA1, RARRES2, RAN, RAF1, RAD52, PCYT1A, RAD23B, RAC2, PDB1, RAB27B, RAB27A, PDC, PDE2A, PURA, PDGFB, ENPP2, PDYN, PTPN13, PC, PAX6, PARN, RPL15, P2RX1, S100A8, P2RX3, P2RX5, P2RY4, RREB1, RPS23, RPS21, RPS6KB2, P2RY6, RPS3A, RPL13, RET, RPA1, PAFAH1B2, RORA, ROM1, SNORD15A, BRD2, RNASE1, RHO, RHD, PAK3, TRIM27, PTPN11, PENK, PTN, PMM2, PLAUR, PLCL1, PLEK, PRKCE, PRKCD, PLP1, PRKAR1B, PRKACB, PRKACA, PLXNB1, PML, PRG2, PLAT, PMP22, PRB1, PPT1, PPP2R5E, POLG, PPP2R1A, PPP1CB, PPL, PPIC, PPIB, POR, MAP2K3, PLAG1, PFDN5, PSMD2, PFKFB3, PTGER2, PTGER1, PGD, PTGDR, PTCH1, PGR, PHB, PSMD9, PSMD7, PSMD3, PSMB2, PITX2, SERPINE2, SERPINI1, KLK10, PIK3C3, PIK3R1, KLK7, PIK3R2, PRS, PROS2P, PIN4, PROC, S100A10, OXT, OXA1L, SQLE, STAR, ST14, ST2, NDUFA6, SSTR3, SSTR2, NDUFA9, NDUFB8, SRM, SRF, NEDD4, SEPTIN2, STC1, SP4, NEU1, SOX5, SOX3, SOS2, SOS1, SOD3, NFE2L1, NFKB2, SNRPG, SNRNP70, NDUFA5, STIM1, NME1, MYH9, TRBV20OR9-2, TCP1, TCN2, MMUT, MUTYH, TCF4, ELOC, TBX2, MX1, MYH6, TARBP2, MAP3K7, STK11, MYO6, TACR2, NACA, VAMP2, VAMP1, SURF1, ABCC8, SUOX, NDP, STXBP1, STX1A, NINJ2, NME2, SAA2, OMP, SFTPC, TRA2B, SRSF6, SRSF5, SRSF3, SRSF1, SFRP1, OCA2, MAP2K4, ODF1, SELE, OPA1, OAS3, CXCL11, CCL21, CCL20, CCL19, CCL8, CCL1, SCP2, SCN1A, ORM2, OSM, TSPAN31, OAT, SGSH, SUMO2, SLC8A3, SMPD2, SLN, SLIT3, SLC22A5, SLC22A2, NQO2, SLC18A1, SLC16A1, SLC12A3, SLC11A1, SLC10A2, SLC8A1, NUP98, SLC6A12, NPHP1, SLC6A1, SLC5A2, NRCAM, NRDC, SLC1A1, NRF1, PMEL, YBX1, NT5E, NAT8, SEMA5A, ESRRA, RAB31, MACF1, HEY2, BRD4, TRAM1, CBX5, ANGPTL2, OPN1MW, GRIP1, KCNH4, MSTN, GFER, GFRA1, SIRT5, COTL1, GFRA3, GHR, ZNF629, UBR4, GHSR, WASHC4, GLI1, NUP160, CLUH, GLI3, SCFD1, KCTD2, CLCF1, SEC14L2, NR5A1, SLC24A2, PRPF6, TFIP11, MAFF, EID1, RAB38, FSHR, TMEFF2, PLA2G15, SLC7A11, FTH1, SNHG1, TSPAN15, GAST, ACKR1, G6PD, GAB1, NTSR2, GABBR1, GAD2, GALNS, DDAH1, PADI4, GART, NBEAL2, GMPR, PLEKHM2, WDHD1, GPR39, CARD8, ARHGEF15, AAK1, SYNPO, GPX4, ECD, PARK7, KLF8, TREX1, WIF1, WDR45, ATF6, CORO1A, TBC1D8, SLC7A9, GRIA4, FAF1, GRIK4, RER1, GRM1, STMN2, RAPGEF4, ADRM1, MSRB2, RAB3GAP1, GNA12, ZNF423, ATG4B, UBXN4, RCOR1, SEPTIN8, STAB1, GNAI1, MAPK8IP3, GRAMD4, GNB3, NFASC, KIF1B, GOLGA2, GPER1, SETX, GOLGA4, PDZD2, SAMD4A, KDM1A, GPM6A, RAB21, GPR6, P2RX2, GPR20, SNW1, FRK, FBXO7, BRI3, SNX12, SOCS7, CD209, FABP6, TBX21, NOP53, FABP7, SLC2A8, UBQLN2, A1CF, PSAT1, FANCG, HOOK2, DELEC1, FASN, SNX8, NPC1L1, BLNK, MS4A2, FCGR1A, FCGR2A, MYLIP, SCG3, DROSHA, TMEM230, NOX4, PCA3, SPCS1, VRK3, ETFA, SLC25A37, TLR8, SLC22A17, ECSIT, CD320, EZH2, DNAJC27, CLEC1B, NT5C3A, MZB1, F2RL2, HP1BP3, F3, F9, IRAK4, SAR1B, UTP11, F13B, SH3GLB1, SIDT2, SHANK1, TRAT1, F11R, RGCC, TMEM176B, NOCT, FBXO2, NPTN, TPK1, VCX, GREM1, FKBP1AP2, FKBP1AP3, AGO1, SEZ6L2, FKBP1AP4, FGF21, CLDN17, NOC2L, SND1, FOXM1, EPC2, ATRNL1, LRP10, FLNB, FMR1, POU2F3, TXN2, FBXL2, FOLR1, FOLR2, FKBP1AP1, B3GAT1, IGHV1-68, TNFRSF21, FEB1, FES, FGF9, RABGEF1, PRPF19, FGF13, FGFR1, FGFR4, PDLIM3, RND1, KLHL20, COQ2, CACYBP, HCAR1, FHL2, VPS4A, IL37, GLS2, NAAA, CYTH4, DKK2, DKK3, BBC3, SDCBP2, GRM3, IL24, SPAG9, CXCL2, PCLAF, IGFBP1, ACAP1, IGFBP5, SART3, KDM4A, IGHG3, SDC3, SH3PXD2A, RGS6, SNCAIP, TCL1B, IL4R, IL7, BCAR1, CCL4L2, CXCR1, BAG2, IL12B, BAG5, TMEM59, TBPL1, GAL3ST1, AKAP5, STX8, PIEZO1, BMS1, PTDSS1, IDH1, SH2D3C, GNE, SH2B3, IRF8, SRA1, TANK, KCNE3, HNRNPDL, CCS, NUP153, MED12, HS3ST1, TOMM20, RBM8A, IDH2, CFI, SV2B, TECPR2, IFI27, TOMM70, KIAA0319, IFIT3, IFNAR1, IGBP1, INSRR, PCYT1B, IL27RA, CCNE2, NOLC1, TIAF1, JUND, ZMYM3, KCNC4, KCNJ13, HGS, SYNGR3, ATG12, CBFA2T2, RABEP1, P2RX6, RAB11B, SLC16A3, SLC16A4, ATP6V0D1, RGN, USP10, USP2, USP14, DNAJA3, CLDN1, CLDN8, ARTN, JUNB, GPR50, PICK1, ITPKB, IRAK1, HOMER2, IREB2, IRF3, IRF6, IRF7, ITGAV, CYP7B1, ITGB3, NRXN1, SLC22A8, ITPR2, AIMP1, JAG2, ITGBL1, LHX2, SLIT2, TAOK2, CD163, JAK2, GPR37L1, PIWIL1, MAPKAPK2, PDLIM7, IARS1, TNC, IL18BP, CCT2, HCK, NRG3, USP39, DCTN6, CD226, HDC, HDLBP, CAMKK2, HIP1, TXNRD2, NPC2, SLC35A1, HBG2, PRDX4, ZNRD2, HYOU1, HLA-DQA1, SEMA4D, HLA-DQB1, NXF1, HLA-DRA, ATP5PD, COG5, GPNMB, CHL1, HAS3, FAM3C, GYPA, GSK3A, COPS5, GSM1, GSTM2, EBNA1BP2, PRSS21, PRDX3, GSTZ1, GTS, GUSB, C1QL1, GYPB, HAS1, GYPC, CCL27, ALDH1L1, GYPE, HAGH, WASF3, HSPH1, GJB6, HARS1, ARPP19, DHS, HLA-DRB4, HLA-G, PQBP1, MPHOSPH6, STAM2, GLYAT, HOXA@, RABEPK, HPCA, CALCOCO2, HRC, OLIG2, DDX39A, TOPORS, EIF1, HSD17B1, RAMP2, WASF2, HSD17B4, HSPA2, HSP90AB1, DNAJB1, NAMPT, BCAP31, CTDSP2, TSPAN3, ACTR2, CERT1, ABCC4, HNRNPK, HMBS, NPM3, CFDP1, PRMT5, HMGB2, YAP1, IFITM3, SPAG11B, PEMT, RACK1, SYCP2, TUBB4B, SEMA3A, WARS2, HNRNPC, FOXA1, CCL26, TLR6, FOXA2, LAMC3, LANCL1, HNF4A, TCIRG1, APBB3, APC2, HNRNPA2B1, MTCH2

-
Angioedema Induced By Ace Inhibitors, Susceptibility To
OMIM
Clinical Features Blais et al. (1999) and Adam et al. (2002) reported significantly lower plasma aminopeptidase P (APP) activities in patients with a history of AEACEI. ... Measured genotype analysis strongly suggested that the linkage signal for APP activity at this locus was accounted for predominantly by the SNP association. ... There was a significant association between the -2399A allele and decreased serum APP activity in both men and women, but the APP activity was lower in men regardless of genotype. ... This haplotype was associated with decreased plasma APP activity and decreased luciferase gene expression compared to other haplotypes of these SNPs. Cilia La Corte et al. (2011) concluded that the ATG haplotype of XPNPEP2 is functional and contributes to the development of ACEi-angioedema through a reduction in APP activity.
-
Alzheimer Disease 18
OMIM
Q170H and R181G mutant mice showed significant attenuation of APP processing compared to wildtype, with a decrease in APP-CTF-alpha levels and an increase in sAPP-beta levels, indicating that the mutations attenuated Adam10 alpha-secretase activity on APP. Crossing these Adam10 mutant mice with the Tg2576 AD mouse model showed that the Adam10 mutations increased amyloidogenic APP processing, as manifest by a shift from the alpha-secretase to the amyloidogenic beta-secretase pathway. ... Collectively, these findings suggested that diminished alpha-secretase activity of ADAM10 on APP resulting from mutations in the ADAM10 prodomain can cause AD-related pathology.
-
Infantile Myofibromatosis
Orphanet
Clinical description Infantile myofibromatosis (IM) presents at birth or develops shortly thereafter, with 90% of cases occurring before the age of 2 years. IM is characterized by solitary or multiple nodules that are firm, flesh-colored to purple (''myofibroma''), and usually painless (except in case of compression of adjacent nerves). ... Etiology Most of these tumors are sporadic and isolated. Rare familial cases of IM have been described and 2 genes have been identified as disease causing: PDGFRB and NOTCH3 which encode PDGFRB and NOTCH3 respectively. ... Histopathology remains the gold standard for the diagnosis of IM. Biopsy reveals interlacing fascicles of spindle cells (myofibroblasts) in the periphery. ... Antenatal diagnosis Prenatal diagnosis is achieved by ultrasound examination and confirm by fetal MRI. Genetic counseling IM is mostly isolated and sporadic. In cases of familial and multifocal lesions, IM can be inherited as an autosomal recessive or dominant trait (incomplete penetrance and variable expressivity).
-
Alzheimer Disease
OMIM
See also APP-related cerebral amyloid angiopathy (CAA; 605714), which shows overlapping clinical and neuropathologic features. ... Genetic analysis identified a mutation in the APP gene (V717I; 104760.0002). Farlow et al. (1994) reviewed the clinical characteristics of the disorder in the AD family reported by Murrell et al. (1991) in which affected members had a mutation in the APP gene (V717F; 104760.0003). ... Rovelet-Lecrux et al. (2006) estimated that in their whole cohort of 65 ADEOAD families, the frequency of the APP locus duplication was roughly 8% (5 of 65), which corresponds to half of the contribution of APP missense mutations to ADEOAD. ... Revesz et al. (2003) reviewed the pathology and genetics of APP-related CAA and discussed the different neuropathologic consequences of different APP mutations. ... Further studies indicated that suppression of PPARGC1A in hyperglycemia resulted in activation of the FOXO3A (602681) transcription factor, which inhibits nonamyloidogenic secretase processing of APP and promotes amyloidogenic processing of APP.TOMM40, TREM2, ABCA7, APP, APOE, PSEN2, PSEN1, MAPT, SORL1, PRNP, CASP3, BACE1, GSK3B, NCSTN, IDE, IL1B, HFE, A2M, ACE, DHCR24, BIN1, ESR1, ADAM10, ADAMTS1, PGRMC1, VEGFA, ARC, CYP46A1, SLC30A4, VSNL1, PICALM, HMOX1, HLA-DRB5, IGF1R, IGF1, INPP5D, IGF2, MPO, NPY, NOS3, PLAU, PLCG2, PPARG, RELN, MTHFR, PYY, NECTIN2, SLC2A4, IGF2R, SOD2, MAOB, TF, LEP, TFAM, INSR, INS, TNF, TPI1, EPHA1, F2, ENO1, CR1, CASS4, ATP5F1A, CLU, CHRNB2, CHRNA7, MIR766, CD33, IQCK, EIF2S1, MIR505, APOC1, CALM1, MIR100, MIR146A, BDNF, BCL2, MIR375, MIR296, BCHE, MIR708, TPP1, SLC30A6, SNAR-I, DPYSL2, ACHE, CD2AP, GAPDHS, PCDH11X, CYP2D6, MIR4467, CRH, MIR3622B, BAX, AMFR, ABI3, CST3, MS4A4A, WWOX, BRCA2, FANCD2, TFF1, TAS2R64P, CTNNB1, SUCLA2, SNCA, CTSD, RNR2, NEFL, TAS2R62P, SOD1, ITPR3, ITPR2, ITPR1, FLAD1, PSENEN, TP53, CDK5R1, EIF2AK3, UBQLN1, ALG3, PIK3CG, PIK3CA, PIK3CD, SERPINA3, PIK3CB, DOCK3, APLP1, OGDH, CREB1, NOTCH1, CASP6, NGF, CCND1, FOS, DLX4, DLG4, DDIT3, RABGEF1, PEBP1, PYCARD, DAPK2, KCNIP3, CTSB, CSF2, CRMP1, CTSG, EHMT2, ENO2, ERBB4, TMED10, TERF2IP, PTK2B, FCN2, PTGES3, FGF2, ACKR1, DNM1L, SDC3, G6PD, GCHFR, ITM2B, CREBBP, MAP3K8, TRPM7, ADI1, MTCO2P12, UPK3B, ACTB, AKT1, AKT2, ANXA1, APBB1, DNLZ, STS, MIR34A, BRCA1, MIR137, C5AR1, DDR1, CAMK4, TMED10P1, MPEG1, C9orf72, ESCO1, CDCA5, PRRT2, MAP1LC3B, CAT, EHMT1, CNR2, SPPL2B, RAB9A, NRXN3, GFAP, SYNJ1, SERPINB5, CD99, MME, MNAT1, CCL2, RRAS, RPS27, RPS21, RAP1A, PYCR1, COX2, PTS, PTGS2, MTHFD1, MMUT, NCAM1, NFIA, NFIB, MAPK8, MAPK3, PRKCB, PRKCA, PPBP, MED1, NFIC, PPARA, NFIX, PKD1, NOTCH3, NRGN, MEOX2, MEF2A, SPRR2A, TTC3, GRIN2A, DENR, GRIN2B, RAB7A, LRP8, HPRT1, HSP90AA1, VIM, IDUA, UTRN, SUMO1, UBE2I, TTK, TPT1, SULT1E1, IL1A, IL6, IL12A, TSPAN6, TIE1, TGFB1, TG, KNG1, LAMC2, LGALS3, TERT, TERC, STIM1, H3P17
-
Estrogen And Neurodegenerative Diseases
Wikipedia
In addition, estrogen deprivation is likely to initiate or enhance degenerative changes caused by oxidative stress , and to reduce the brain's ability to maintain synaptic connectivity and cholinergic integrity leading to the cognitive decline seen in aged and disease-afflicted individuals. [5] There is sufficient evidence that estradiol is a powerful neuroprotectant which might have use against AD, stroke and Parkinson's disease both in women and men. [5] Estrogen and Alzheimer's disease [ edit ] This figure shows how APP cleavage produces toxic Abeta in Alzheimer's disease. Amyloid plaques formed by amyloid-β (Aβ) deposition and neurofibrillary tangles formed by tau protein phosphorylation are dominant physiological features of Alzheimer's disease. Amyloid precursor protein (APP) proteolysis is fundamental for production of Aβ peptides implicated in AD pathology. [6] By using a cell line that contains high levels of estrogen receptors, scientists found that treatment with physiological concentrations of 17 beta-estradiol is associated with accumulation in the conditioned medium of an amino-terminal cleavage product of APP (soluble APP or protease nexin-2), indicative of non-amyloidogenic processing. [7] Estrogen and Parkinson's disease [ edit ] Recommendations on the use of postmenopausal hormonal replacement therapy in women with Parkinson's disease or those genetically at risk. [8] But another group of scientists found a positive association between estrogen use and lower symptom severity in women with early PD not yet taking L-dopa . [9] Estrogen and Huntington's disease [ edit ] Huntington's disease (HD) is a polyglutamine disorder based on an expanded CAG triplet repeat [10] leading to cerebral and striatal neurodegeneration. [11] Potential sex differences concerning the age of onset and the course of the disease are poorly defined, as the difficulties of matching female and male HD patients regarding their CAG repeat lengths limit comparability. [12] Estrogen and Amyotrophic lateral sclerosis [ edit ] ALS occurs more commonly in men than in women, and women get the disease later in life compared to men. [13] This suggested the possible protective role of estrogen in ALS. ... "Estrogen and neurodegeneration". Neurochemistry Research . 28 (7): 1003–8. doi : 10.1023/A:1023246921127 . ^ Zhang, She-Qing; et al. ... "Octyl Gallate Markedly Promotes Anti-amyloidogenic Processing of APP through Estrogen Receptor-Mediated ADAM10 Activation" .
-
Congenital Disorder Of Glycosylation, Type Im
OMIM
A number sign (#) is used with this entry because of evidence that congenital disorder of glycosylation type Im (CDG1M), also known as dolichol kinase deficiency, is caused by homozygous mutation in the DOLK gene (610746), which encodes the enzyme responsible for the final step of the de novo biosynthesis of dolichol phosphate, on chromosome 9q34. ... Helander et al. (2013) reported 2 sibs, born of consanguineous Syrian Turkish parents, with CDG type Im. The patients presented at age 4 months with severe intractable seizures and hypsarrhythmia, consistent with a clinical diagnosis of West syndrome (see 308350). ... In 2 sibs, born of consanguineous Syrian Turkish parents, with CDG type Im and a purely neurologic phenotype, Helander et al. (2013) identified a homozygous mutation in the initiating methionine codon of the DOLK gene (610746.0006). ... Kranz et al. (2007) suggested that since dolichol kinase deficiency can be detected by isoelectric focusing (IEF) of serum transferrin, the disorder could be included in the CDG I group with the designation CDG Im. INHERITANCE - Autosomal recessive GROWTH Height - Normal birth length Weight - Normal birth weight Other - Failure to thrive HEAD & NECK Head - Normal birth head circumference - Microcephaly, acquired Eyes - Sparse eyebrows - Sparse eyelashes CARDIOVASCULAR Heart - Dilated cardiomyopathy SKIN, NAILS, & HAIR Skin - Ichthyosis Hair - Sparse eyebrows - Sparse eyelashes - Minimal hair growth NEUROLOGIC Central Nervous System - Hypotonia, profound muscular (in some patients) - Seizures (in some patients) - Hypsarrhythmia (in some patients) METABOLIC FEATURES - Hypoketotic hypoglycemia (in some patients) LABORATORY ABNORMALITIES - Abnormal transferrin isoelectric focusing (IEF) - Increased disialo- and asialotransferrin - Decreased lipid-linked oligosaccharides (LLO) MISCELLANEOUS - Death in early infancy (in some patients) - Some patients present with apparent nonsyndromic dilated cardiomyopathy in early childhood MOLECULAR BASIS - Caused by mutation in the transmembrane protein 15 gene (TMEM15, 610746.0001 ) ▲ Close

-
Cerebral Amyloid Angiopathy
Wikipedia
Abnormalities in each of these identified clearance pathways have been linked to CAA. [12] [13] In familial forms of CAA, the cause of Aβ build up is likely due to increased production rather than poor clearance. [14] Mutations in the amyloid precursor protein (APP), Presenilin (PS) 1 and PS2 genes can result in increased rates of cleavage of the APP into Aβ. ... The Boston Criteria require evidence of multiple lobar or cortical hemorrhages to label a patient as probably having CAA. [26] Susceptibility weighted imaging has been proposed as a tool for identifying CAA-related microhemorrhages. [28] Imaging [ edit ] Cerebral amyloid angiopathy can be presented with lobar intracerebral hemorrhage or microbleeds in the brain. ... ISBN 9780199710041 . ^ Godefroy, Olivier (2013-02-28). The Behavioral and Cognitive Neurology of Stroke . ... PMC 6059159 . PMID 28334869 . Retrieved 28 August 2020 . Further reading [ edit ] Chao, Christine P.; Kotsenas, Amy L.; Broderick, Daniel F. ... External links [ edit ] Classification D ICD - 10 : I68.0 MeSH : D016657 DiseasesDB : 32874 External resources MedlinePlus : 000719 eMedicine : neuro/628 Scholia has a topic profile for Cerebral amyloid angiopathy . v t e Amyloidosis Common amyloid forming proteins AA ATTR Aβ2M AL Aβ / APP AIAPP ACal APro AANF ACys ABri Systemic amyloidosis AL amyloidosis AA amyloidosis Aβ2M/Haemodialysis-associated AGel/Finnish type AA/Familial Mediterranean fever ATTR/Transthyretin-related hereditary Organ-limited amyloidosis Heart AANF/Isolated atrial Brain Familial amyloid neuropathy ACys+ABri/Cerebral amyloid angiopathy Aβ/Alzheimer's disease Kidney AApoA1+AFib+ALys/Familial renal Skin Primary cutaneous amyloidosis Amyloid purpura Endocrine Thyroid ACal/Medullary thyroid cancer Pituitary APro/Prolactinoma Pancreas AIAPP/Insulinoma AIAPP/Diabetes mellitus type 2 v t e Medicine Specialties and subspecialties Surgery Cardiac surgery Cardiothoracic surgery Colorectal surgery Eye surgery General surgery Neurosurgery Oral and maxillofacial surgery Orthopedic surgery Hand surgery Otolaryngology ENT Pediatric surgery Plastic surgery Reproductive surgery Surgical oncology Transplant surgery Trauma surgery Urology Andrology Vascular surgery Internal medicine Allergy / Immunology Angiology Cardiology Endocrinology Gastroenterology Hepatology Geriatrics Hematology Hospital medicine Infectious disease Nephrology Oncology Pulmonology Rheumatology Obstetrics and gynaecology Gynaecology Gynecologic oncology Maternal–fetal medicine Obstetrics Reproductive endocrinology and infertility Urogynecology Diagnostic Radiology Interventional radiology Nuclear medicine Pathology Anatomical Clinical pathology Clinical chemistry Cytopathology Medical microbiology Transfusion medicine Other Addiction medicine Adolescent medicine Anesthesiology Dermatology Disaster medicine Diving medicine Emergency medicine Mass gathering medicine Family medicine General practice Hospital medicine Intensive care medicine Medical genetics Narcology Neurology Clinical neurophysiology Occupational medicine Ophthalmology Oral medicine Pain management Palliative care Pediatrics Neonatology Physical medicine and rehabilitation PM&R Preventive medicine Psychiatry Addiction psychiatry Radiation oncology Reproductive medicine Sexual medicine Sleep medicine Sports medicine Transplantation medicine Tropical medicine Travel medicine Venereology Medical education Medical school Bachelor of Medicine, Bachelor of Surgery Bachelor of Medical Sciences Master of Medicine Master of Surgery Doctor of Medicine Doctor of Osteopathic Medicine MD–PhD Related topics Alternative medicine Allied health Dentistry Podiatry Pharmacy Physiotherapy Molecular oncology Nanomedicine Personalized medicine Public health Rural health Therapy Traditional medicine Veterinary medicine Physician Chief physician History of medicine Book Category Commons Wikiproject Portal OutlineCST3, ITM2B, APP, CTSD, APOE, ACTB, PTPRC, TSHZ1, SYP, SPP1, SDC2, HSPG2, SMAD2, ITGAX, GFAP, FN1, FAP, CD68, HSPB8

-
Analgesic Nephropathy
Wikipedia
UpToDate . Waltham, MA. ^ Boström, IM; Nyman, G; Hoppe, A; Lord, P (January 2006). ... PMID 16412133 . ^ Frendin, JH; Boström, IM; Kampa, N; Eksell, P; Häggström, JU; Nyman, GC (December 2006). ... American Journal of Veterinary Research . 67 (12): 1967–73. doi : 10.2460/ajvr.67.12.1967 . PMID 17144795 . ^ Boström, IM; Nyman, GC; Lord, PE; Häggström, J; Jones, BE; Bohlin, HP (May 2002). ... "Relevance of animal models to analgesic-associated renal papillary necrosis in humans". Kidney Int . 28 (4): 605–13. doi : 10.1038/ki.1985.172 . ... "Combination analgesic-induced kidney disease: the Australian experience". Am. J. Kidney Dis . 28 (1 Suppl 1): S39–47. doi : 10.1016/S0272-6386(96)90568-5 .

-
Coronary Artery Anomaly
Wikipedia
AA = antero-left; AR = antero-right; Cx = circumfles artery; IM = intramural; IS = intraseptal; LAD = left anterior descending artery; M = mitral valve; P = posterior; PP = prepulmonic; RA = retroaortic; RC = retrocardiac; RCA = right coronary artery; T = tricuspid valve. R-ACAOS-IM [2] is observed in a higher percentage of cases (0.35% of adolescents) than L-ACAOS-IM [3] but is less likely to be associated with sudden cardiac death in athletes. ... Assessment of severity of stenosis is best achieved by intravascular ultrasound (IVUS) imaging and it should be considered in known carriers of ACAOS-IM or that have symptoms or positive stress test results or are involved in competitive exercises. ... Importantly, untreated carriers of significant ACAOS should not generally engage in competitive sports or strenuous activities. Treatment options for ACAOS-IM include both catheter-based procedures ( percutaneous coronary intervention [PCI]) and surgical interventions. PCI consists of stent angioplasty of the proximal, intramural segment by placing a thin metal tube (a stent) in order to keep open the narrowed artery. PCI of R-ACAOS-IM is feasible and quite successful, but further experience is needed in L-ACAOS-IM since few cases have been treated percutaneously, while surgery is the recommended treatment in this subpopulation, at this time.
-
Pustular Psoriasis
Wikipedia
This skin eruption is often accompanied by a fever , muscle aches , nausea , and an elevated white blood cell count . [1] Annular pustular psoriasis (APP), a rare form of GPP, is the most common type seen during childhood. [6] APP tends to occur in women more frequently than in men, and is usually less severe than other forms of generalized pustular psoriasis such as impetigo herpetiformis. [6] This form of psoriasis is characterized by ring-shaped plaques with pustules around the edges and yellow crusting. [6] APP most often affects the torso, neck, arms, and legs. [6] Diagnosis [ edit ] Classification [ edit ] Pustular psoriasis is classified into two major forms: localized and generalized pustular psoriasis . [1] Within these two categories there are several variants: Classification of Localized and Generalized Pustular Psoriasis Localized pustular psoriasis Palmoplantar pustulosis (acute and chronic) Acrodermatitis continua (of Hallopeau) Generalized pustular psoriasis (von Zumbusch) acute generalized pustular psoriasis Acute generalized pustular psoriasis of pregnancy ( impetigo herpetiformis ) Infantile and juvenile Subacute circinate and annular Management [ edit ] injection of methotrexate This section is empty.IL36RN, IL1B, CARD14, IL1A, IL1RL2, IL17A, PI3, TNF, IL22, AP1S3, TLR3, TNFAIP3, MOG, TNIP1, LDHA, IL23A, NOD2, IL1F10

-
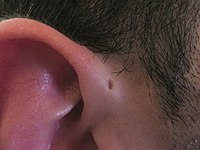
Preauricular Sinus And Cyst
Wikipedia
See also [ edit ] Branchial cleft cyst Thyroglossal cyst Lachiewicz Sibley syndrome List of cutaneous conditions References [ edit ] ^ Freedberg IM, Fitzpatrick TB (2003). Fitzpatrick's Dermatology in General Medicine (6th ed.). ... Acta Otorhinolaryngologica Italica . 28 (6): 302–5. PMC 2689545 . PMID 19205595 . ^ "Preauricular Sinus" . ... Acta Otorhinolaryngologica Italica . 28 (6): 302–5. PMC 2689545 . PMID 19205595 .