Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
2-Hydroxyglutaric Aciduria
MedlinePlus
The major types of this disorder are called D-2-hydroxyglutaric aciduria (D-2-HGA), L-2-hydroxyglutaric aciduria (L-2-HGA), and combined D,L-2-hydroxyglutaric aciduria (D,L-2-HGA). ... D-2-HGA and L-2-HGA have each been reported to affect fewer than 150 individuals worldwide. ... L-2-HGA results from mutations in the L2HGDH gene. Combined D,L-2-HGA is caused by mutations in the SLC25A1 gene. ... Mutations in either of these genes lead to a shortage of functional enzyme, which allows D-2-hydroxyglutarate or L-2-hydroxyglutarate to build up in cells.

-
Inclusion Body Rhinitis
Wikipedia
Find sources: "Inclusion body rhinitis" – news · newspapers · books · scholar · JSTOR ( September 2011 ) ( Learn how and when to remove this template message ) Suid betaherpesvirus 2 Virus classification (unranked): Virus Realm : Duplodnaviria Kingdom: Heunggongvirae Phylum: Peploviricota Class: Herviviricetes Order: Herpesvirales Family: Herpesviridae Genus: Roseolovirus Species: Suid betaherpesvirus 2 Synonyms [1] Suid herpesvirus 2 Pig cytomegalovirus Inclusion Body Rhinitis , also known as IBR or Cytomegalic Inclusion Disease , is a pig disease caused by Suid betaherpesvirus 2 (SuHV-2), which is a member of the herpesvirus family. ... It is not zoonotic but the risk to humans that receive pig organ transplants is currently under investigation. Contents 1 Clinical signs 2 Diagnosis 3 Treatment and control 4 References Clinical signs [ edit ] Clinical signs are normally only seen in either piglets less than 3 weeks old or pregnant sows. ... Treatment and control [ edit ] Often no treatment is required. However, as Suid betaherpesvirus 2 is a member of Herpesviridae it remains latent and sheds at times of stress. ... Retrieved 21 December 2018 . Suid herpesvirus 2 (SuHV-2) Suid herpesvirus 2 (Pig cytomegalovirus) Inclusion body rhinitis , reviewed and published by Wikivet at http://en.wikivet.net/Inclusion_Body_Rhinitis , accessed 08/09/2011. v t e Taxonomy of the Herpesvirales Higher taxonomy: Duplodnaviria > Heunggongvirae > Peploviricota > Herviviricetes > Herpesvirales Malacoherpesviridae Aurivirus AbHV-1 Ostreavirus OsHV-1 Alloherpesviridae Batrachovirus RaHV-1 2 3 Cyprinivirus AngHV-1 CyHV-1 2 3 Ictalurivirus AciHV-2 IcHV-1 2 Salmonivirus SalHV-1 2 3 Herpesviridae IgHV-2 α ChHV-6 Iltovirus GaHV-1 PsHV-1 Mardivirus AnHV-1 CoHV-1 GaHV-2 3 MeHV-1 SpAHV-1 Scutavirus ChHV-5 TeHV-3 Simplexvirus AtHV-1 BoHV-2 CeHV-2 HHV-1 2 LeHV-4 McHV-1 MaHV-1 2 PnHV-3 PaHV-2 PtHV-1 SaHV-1 Varicellovirus BoHV-1 5 BuHV-1 CaHV-1 CpHV-1 CeHV-9 CvHV-1 2 EHV-1 3 4 8 9 FeHV-1 HHV-3 MoHV-1 PhHV-1 SuHV-1 β CaHV-2 TuHV-1 Cytomegalovirus AoHV-1 CbHV-1 CeHV-5 HHV-5 McHV-3 8 MnHV-1 PnHV-2 PaHV-3 4 SaHV-4 Muromegalovirus MuHV-1 2 8 Proboscivirus ElHV-1 4 5 Roseolovirus HHV-6A 6B 7 McHV-9 MuHV-3 SuHV-2 γ EHV-7 PhHV-2 SgHV-1 Lymphocryptovirus CalHV-3 CeHV-14 GoHV-1 HHV-4 McHV-4 10 PnHV-1 PaHV-1 PoHV-2 Macavirus AlHV-1 2 BoHV-6 CpHV-2 HiHV-1 OvHV-2 SuHV-3 4 5 Percavirus EHV-2 5 FeHV-1 MusHV-1 PhHV-3 VeHV-1 Rhadinovirus AtHV-2 3 BoHV-4 CrHV-2 HHV-8 McHV-5 8 11 12 MuHV-4 7 SaHV-2 Unassigned species listed below parent taxon –– Source: ICTV –– Wikispecies Taxon identifiers Suid herpesvirus 2 Wikidata : Q18965187 Wikispecies : Suid betaherpesvirus 2 IRMNG : 11460896 Suid betaherpesvirus 2 Wikidata : Q24808743 NCBI : 1608255
-
D-2-Hydroxyglutaric Aciduria 2
OMIM
A number sign (#) is used with this entry because of evidence that D-2-hydroxyglutaric aciduria-2 (D2HGA2) is caused by heterozygous mutations in the mitochondrial isocitrate dehydrogenase-2 (IDH2; 147650) gene on chromosome 15q26. ... Inheritance The transmission pattern of D-2-hydroxyglutaric aciduria-2 in the families reported by Kranendijk et al. (2010) was consistent with autosomal dominant inheritance. Molecular Genetics Somatic mutation in the IDH1 (147700) or IDH2 genes had been shown to result in the enzyme's abnormal ability to convert 2-ketoglutarate (2-KG) to D-2-hydroxyglutarate (D2HG) (Yan et al., 2009; Ward et al., 2010). ... The higher excretion of D-2-hydroxyglutaric acid in type 2 patients compared to type 1 patients could best be explained by hyperproduction of this metabolite. The involvement of mitochondrial IDH2 is also consistent with the finding that D-2-hydroxyglutaric acid is derived from mitochondrial 2-KG.
-
2-Aminoadipic 2-Oxoadipic Aciduria
OMIM
Danhauser et al. (2012) stated that over 20 individuals with 2-aminoadipic 2-oxoadipic aciduria were known. ... The second individual presented with microcephaly, mild motor developmental delay (sitting alone at age 9 months and walking alone at the age 22 months), and prominent speech delay in early childhood. Her hearing was normal. At 2 years of age metabolic analysis of urine and plasma revealed the characteristic biochemical profile of 2-aminoadipic 2-oxoadipic aciduria (2-oxoadipate, 520-970 mmol/mol creatinine; 2-hydroxyadipate, 100-150 mmol/mol creatinine; 2-aminoadipate, elevated). ... Molecular Genetics Danhauser et al. (2012) performed whole-exome sequencing in a patient with 2-aminoadipic 2-oxoadipic aciduria and found her to be compound heterozygous for 2 mutations in the DHTKD1 (614984) gene. ... Functional assays showed that reduced DHTKD1 activity was responsible for the 2-aminoadipic and 2-oxoadipic aciduria in the affected individuals. History Barshop et al. (2000) reported a patient who presented with 2-oxoadipic aciduria and 2-aminoadipic aciduria at 2 years of age with manifestations typical of organic acidemia and episodes of ketosis and acidosis, progressive to coma.
-
Arterial Embolism
Wikipedia
Contents 1 Signs and symptoms 1.1 Commonly occluded sites 2 Risk factors 3 Pathophysiology 3.1 Materials 4 Diagnosis 5 Prevention 6 Treatment 7 Prognosis 8 Complications 9 Epidemiology 10 References 11 External links Signs and symptoms [ edit ] Symptoms may begin quickly or slowly depending on the size of the embolus and how much it blocks the blood flow. [2] Symptoms of embolisation in an organ vary with the organ involved but commonly include: Pain in the involved body part [2] Temporarily [2] decreased organ function Later symptoms are closely related to infarction of the affected tissue. ... Symptoms of limb infarction include coldness, [1] [2] decreased or no pulse beyond the site of blockage, [1] [2] pain, [1] [2] muscle spasm, [2] numbness and tingling, [1] [2] pallor [1] [2] and muscle weakness , [1] [2] possibly to the grade of paralysis [1] in the affected limb. ... Less common sites include the kidneys, intestines, and eyes. [2] Risk factors [ edit ] Risk factors for thromboembolism , the major cause of arterial embolism, include disturbed blood flow (such as in atrial fibrillation and mitral stenosis ), injury or damage to an artery wall, and hypercoagulability [1] (such as increased platelet count). [2] Mitral stenosis poses a high risk of forming emboli which may travel to the brain and cause stroke . [2] Endocarditis increases the risk for thromboembolism, [2] by a mixture of the factors above. ... Antiplatelet drugs may also be needed. [2] Treatment [ edit ] Treatment is aimed at controlling symptoms and improving the interrupted blood flow to the affected area of the body. [2] Medications include: Antithrombotic medication. ... Arterial embolism can be serious if not treated promptly. [1] [2] Without treatment, it has a 25% to 30% mortality rate . [1] The affected area can be permanently damaged, and up to approximately 25% [1] [2] of cases require amputation of an affected extremity.

-
Central Nervous System Primitive Neuroectodermal Tumor
Wikipedia
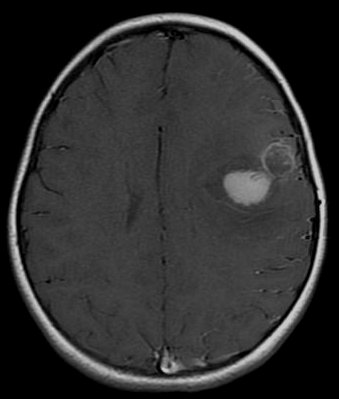
It is a rare disease occurring mostly among children, [1] [2] accounting for 1.9 to 7% of childhood brain tumors. [2] Symptoms involve emotional, visual, motor, and speech defects. [2] Magnetic resonance imaging (MRI) and computed tomography (CT) are used to diagnose PNETs. [2] Even though a universal treatment plan hasn't been stablished yet, common strategies involve chemotherapy and radiotherapy for individuals older than 3 years of age. [1] [2] Their efficacy, however, is still controversial. [2] Surgery can be used to remove mass affected by tumorous cells. [2] The prognosis of the disease is more positive for adults than for children, who have a higher probability of having sequelae from the tumor. [1] [2] It is important to note that this classification term has been removed from the latest WHO classification of CNS tumors as of 2016. Instead PNETs are now included into the category of "Embryonal Tumors with Multilayered Rosettes" along with ependymoblastoma and embryonal tumor with abundant neuropil and true rosettes (ETANTR). [5] Contents 1 Classification 1.1 PNET vs. medulloblastoma 2 Risk factors 3 Diagnosis 4 Treatment 5 Prognosis 6 References Classification [ edit ] Histology of Medulloepithelioma The World Health Organization has classified the central nervous system primitive neuroectodermal tumors into five subtypes: neuroblastoma , ganglioneuroblastoma , medulloepithelioma , ependymoblastoma, and not otherwise specified PNET. [1] The last one encompasses the PNETs with varying characteristics that hasn't been well defined yet. [1] Neuroblastomas are PNETS that involve the process of cell differentiation into neurons, [1] [2] while ganglioneuroblastomas are PNETs that involve ganglion cells . [1] Medulloepithelioma , on the other hand, are tumors involving the constant cell division on the epithelium tissue where bundle of neuron endings are located. [1] Such tissue will differentiate into a similar form as the embryonic neural tube, also known as the starting structure of the central nervous system . [1] [2] [3] Medulloepitheliomas also present a pattern known as rosettes, characterized by the arrangement of a bundle of cells into circular shapes and around a center or a neuropil. [1] Ependymoblastoma also present rosettes as well as a higher density of cells. [1] [3] It involves the process of differentiation into ependymal cells. [2] [3] Rosettes in Ependymoblastoma histology Further classification types have come up but not yet approved by the World Health Organization . [1] The term "embryonal tumor with abundant neuropil and true rosettes", or ETANTR, has been proposed as a sixth subtype of PNET. [1] However, the still unofficial term "embryonal tumor with multilayered rosettes" (ETMR) has been more frequently used and encompasses ETANTRs, medulloepitheliomas , ependymoblastomas, and variants of PNETs with presence of rosettes and with no well defined classification. [3] PNET vs. medulloblastoma [ edit ] The differentiation between primitive neuroectodermal tumor in the central nervous system and medulloblastoma is recent. [1] [2] According to the World Health Organization , both tumors have the same histology but primitive neuroectodermal tumors occur outside the cerebellum . [2] Moreover, it has been documented that both have different genetic expression and mutations. [1] [2] Another essential difference between them is the location of their respective blood vessels within the brain. [2] It has also been theorized that PNETs influence mainly glia cells while medulloblastomas influence mainly neural behavior, however such theory hasn't been confirmed yet. [1] Medulloblastomas are more frequent than PNETs, representing 10% of all child deaths caused by cancer. [2] They also present better prognosis: children affected by medulloblastoma reach the 5 year survival mark in 70-80% of cases, while children affected by PNET reach the 5 year survival mark in less than 50% of cases. [1] Risk factors [ edit ] The rate of PNETs in not correlated with sex, but it shows a correlation with age. [1] Most cases occur in children around 5 years of age, having a very low frequency in adults. [1] Regarding genetic mutations, a specific type of gene alteration that directly leads to this tumor hasn't been defined yet. [1] However, a positive correlation between individuals with Li-Fraumeni syndrome with a mutation in the gene p53 and PNET has been reported. [2] A significant number of individuals with mutations on the rb tumor suppressor gene have also developed the tumor. [2] Such gene encodes for the protein Rb responsible for stopping the cell cycle at the G1 phase . [6] Another possible contributing factor are mutations in the CREB-binding protein , whose function includes activating transcription, [6] but this interaction still need to be studied further. [2] It has also been presumed that the tumor can arise from cranial irradiation. [2] Diagnosis [ edit ] Magnetic resonance image of PNET Most children that develop primitive neuroectodermal tumors are diagnosed early in life, usually at around 3-6.8 years of age. [2] Symptoms patients present at time of diagnosis include irritable mood, visual difficulties, lethargy , and ataxia . [2] The circumference of the patient's head might also suffer an enlargement and they might be subject to seizures, especially if they have less than one year of life. [2] Several analysis can be used to determine the presence of the disease. Physical examinations showing papilledema , visual field defects, cranial nerves palsy , dysphasia, and focal neurological deficits are evidences for possible tumor. [2] PNETs can also be spotted through computed tomography (CT) and magnetic resonance imaging (MRI). [2] In images produced by MRIs , an irregular augmentation among a solid mass will indicated the presence of tumor. [3] However, the results of MRIs are usually ambiguous in defining the presence for this specific tumor. [2] In CT scans , the presence of PNETs will be indicated by an elevated density and an increase in volume of the brain. [2] The CT scan can also show calcification , [3] which is present in 41-44% of PNET cases. [2] Since the tumor can be replicated in other parts of the nervous system through the cerebrospinal fluid (CSF), a CSF analysis can also be conducted. [2] A spinal MRI is a fourth type of analysis that is useful in investigating the level of tumor propagation to the spinal cord . [2] Treatment [ edit ] There is not a standardized procedure to treat primitive neuroectodermal tumors. [2] Common strategies involve risk-adapted radiotherapy combined with chemotherapy and stem cell rescue. [1] For patients younger than 2–3 years of age, treatment with radiation is not used, once they are in a more vulnerable phase and, thus, more prone to risks in development. [1] Examinations such as CSF analysis and spinal MRIs are used to investigate the effectiveness of treatment in preventing metastasis . [2] A method for eliminating tumorous mass is surgery, where the best outcome would be total resection, meaning the complete removal of the tumor. [2] Along with the surgery, several measures that contribute to a safe procedure can be taken: urine exams, transfusion, and the constant supervision of arterial pressure. [2] Possible problems that arise from the surgery include hemorrhage , brain edema , and hemiparesis . [2] MRIs are typically done after 1 or 2 days of postoperative in order to inspect the amount of tumor remaining. [2] Prognosis [ edit ] The probability of primitive neuroectodermal tumors to have recurrence and metastasize through cerebrospinal fluid is relatively high. [3] The outcome of PNET is more positive when the individual is an adult, independent of age subgroups, or an older child. [2] Less than 50% of children survive more than 5 years, [1] while the majority of adults live to 7 years. [2] The reason the prognosis for such tumor is worst in children is due to the higher probability of the tumor spreading to the rest of the nervous system through the cerebrospinal fluid and growing again. [2] Moreover, children have the probability of developing deficiencies in cognitive processes, problems in the endocrine system , and psychological obstacles after the disease. [2] Adults, on the other hand, don't show such propensity. [2] As a consequence, 37.7% of children affected by the tumor live to 4 years. [2] The effect of treatment strategies such as chemotherapy and radiation therapy on the prognosis of the disease is still controversial, with studies claiming either their benefits or their ineffectiveness. [2] The same holds true for the relationship between volume of tumor removed by surgery and survival. [2] Furthermore, factors such as tumor size, location of origin, race, and sex of individual don't show any influence on the outcome of the disease. [2] However, interactions of some factors such as tumor site, age, and treatment strategy can affect one's prognosis. [2] For instance, when younger children below the age of 3 suffering from tumors originating in places other than the pineal gland are treated with chemotherapy , they present better outcomes than those suffering from pineal tumors and treated with chemotherapy . [2] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab Karajannis, Matthias A.; Zagzag, David, eds. (2015). ... ISBN 0-87893-791-9 ". Biological Psychology . 52 (2): 185–186. doi : 10.1016/s0301-0511(99)00025-3 . ... The Quarterly Review of Biology . 78 (2): 225–226 . doi : 10.1086/377959 .
-
Eosinophilic Cellulitis
Wikipedia
Eosinophilic cellulitis Other names Wells' syndrome, recurrent granulomatous dermatitis with eosinophilia [1] Initial rash in eosinophilic cellulitis Specialty Dermatology Symptoms Painful, red, raised, warm patches of skin [2] Usual onset Sudden and recurrent [2] Duration Few weeks [2] Causes Unknown [2] Differential diagnosis Vasculitis, cellulitis, anaphylaxis [2] [1] Medication Corticosteroids , antihistamines [2] [1] Prognosis Often goes away by itself [2] Frequency ~200 documented cases [1] Eosinophilic cellulitis , also known as Wells' syndrome (not to be confused with Weil's disease ), is a skin disease that presents with painful, red, raised, and warm patches of skin. [2] The rash comes on suddenly, lasts for a few weeks, and often repeatedly comes back. [2] Scar formation does not typically occur. [1] Eosinophilic cellulitis is of unknown cause. [2] It is suspected to be an autoimmune disorder . [2] It may be triggered by bites from insects such as spiders, fleas , or ticks , or from medications or surgery. [2] Diagnosis is made after other potential cases are ruled out. [1] Skin biopsy of the affected areas may show an increased number of eosinophils . [2] Other conditions that may appear similar include cellulitis , contact dermatitis , and severe allergic reactions such as anaphylaxis . [2] Treatment is often with a corticosteroids . [2] Steroids applied as a cream is generally recommended over the use of steroids by mouth. [3] Antihistamines may be used to help with itchiness. [1] Many times the condition goes away after a few weeks without treatment. [2] The condition is uncommon. [1] It affects both sexes with the same frequency. [2] It was first described by George Crichton Wells in 1971. [4] [1] Contents 1 Cause 2 Diagnosis 3 Treatment 4 References 5 External links Cause [ edit ] Eosinophilic cellulitis is of unknown cause. [2] It is suspected to be an autoimmune disorder . [2] It may be triggered by bites from insects such as mosquitos , [5] spiders, fleas , or ticks , or from medications or surgery. [2] Diagnosis [ edit ] Histology of a skin biopsy from acute phase eosinophilic cellulitis. ... Diagnosis requires ruling out other potential causes. [1] This includes ruling out vasculitis on skin biopsy . [1] Treatment [ edit ] Treatment is often with a steroids . [2] This can be either applied as a cream or taken by mouth. [3] As the condition tends to get better on its own taking steroids by mouth should generally only be tried if the rash covers a large area and it does not get better with other measures. [3] References [ edit ] ^ a b c d e f g h i j k Weins, AB; Biedermann, T; Weiss, T; Weiss, JM (October 2016). ... PMID 27357601 . ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set . St. Louis: Mosby. ISBN 1-4160-2999-0 . ^ Tatsuno K, Fujiyama T, Matsuoka H, Shimauchi T, Ito T, Tokura Y (June 2016).

-
Sulfhemoglobinemia
Wikipedia
It is a rare blood condition in which the hemoglobin molecule has the ability to bind irreversibly to any substance containing a sulfur atom . [1] When hydrogen sulfide (H 2 S) (or sulfide ions ) and ferric ions combine in the blood, the blood is incapable of carrying oxygen . Contents 1 Presentation 2 Causes 3 Treatment 4 Notable cases 5 References 6 External links Presentation [ edit ] Symptoms include a blueish or greenish coloration of the blood, skin , and mucous membranes , even though a blood count test may not show any abnormalities in the blood. ... It can be caused by phenazopyridine . [2] Treatment [ edit ] The condition generally resolves itself with erythrocyte (red blood cell) turnover, although blood transfusions can be necessary in extreme cases. ... S2CID 39437785 . ^ "Patient bleeds dark green blood" , BBC News , 8 June 2007 ^ "Dark Green Blood In The Operating Theatre" , Medical News Today , June 8, 2007 External links [ edit ] Cancer Web Project Online Medical Dictionary Classification D ICD - 10 : D74.8 ICD - 9-CM : 289.7 MeSH : D013436 External resources MedlinePlus : 003371 v t e Diseases of red blood cells ↑ Polycythemia Polycythemia vera ↓ Anemia Nutritional Micro- : Iron-deficiency anemia Plummer–Vinson syndrome Macro- : Megaloblastic anemia Pernicious anemia Hemolytic (mostly normo- ) Hereditary enzymopathy : Glucose-6-phosphate dehydrogenase deficiency glycolysis pyruvate kinase deficiency triosephosphate isomerase deficiency hexokinase deficiency hemoglobinopathy : Thalassemia alpha beta delta Sickle cell disease / trait Hereditary persistence of fetal hemoglobin membrane : Hereditary spherocytosis Minkowski–Chauffard syndrome Hereditary elliptocytosis Southeast Asian ovalocytosis Hereditary stomatocytosis Acquired AIHA Warm antibody autoimmune hemolytic anemia Cold agglutinin disease Donath–Landsteiner hemolytic anemia Paroxysmal cold hemoglobinuria Mixed autoimmune hemolytic anemia membrane paroxysmal nocturnal hemoglobinuria Microangiopathic hemolytic anemia Thrombotic microangiopathy Hemolytic–uremic syndrome Drug-induced autoimmune Drug-induced nonautoimmune Hemolytic disease of the newborn Aplastic (mostly normo- ) Hereditary : Fanconi anemia Diamond–Blackfan anemia Acquired: Pure red cell aplasia Sideroblastic anemia Myelophthisic Blood tests Mean corpuscular volume normocytic microcytic macrocytic Mean corpuscular hemoglobin concentration normochromic hypochromic Other Methemoglobinemia Sulfhemoglobinemia Reticulocytopenia v t e Proteins that contain heme ( hemoproteins ) Globins Hemoglobin Subunits Alpha locus on 16 : α HBA1 HBA2 pseudo ζ HBZ θ HBQ1 μ HBM Beta locus on 11 : β HBB δ HBD γ HBG1 HBG2 ε HBE1 Tetramers stages of development: Embryonic HbE Gower 1 (ζ 2 ε 2 ) HbE Gower 2 (α 2 ε 2 ) HbE Portland I (ζ 2 γ 2 ) HbE Portland II (ζ 2 β 2 ) Fetal HbF/Fetal (α 2 γ 2 ) HbA (α 2 β 2 ) Adult HbA (α 2 β 2 ) HbA 2 (α 2 δ 2 ) HbF/Fetal (α 2 γ 2 ) pathology: HbH (β 4 ) Barts (γ 4 ) HbS (α 2 β S 2 ) HbC (α 2 β C 2 ) HbE (α 2 β E 2 ) HbO (α 2 β O 2 ) Compounds Carboxyhemoglobin Carbaminohemoglobin Oxyhemoglobin / Deoxyhemoglobin Sulfhemoglobin Other human Glycated hemoglobin Methemoglobin Nonhuman Chlorocruorin Erythrocruorin Other human: Myoglobin Metmyoglobin Neuroglobin Cytoglobin plant: Leghemoglobin Other Cytochrome Cytochrome b Cytochrome P450 Methemalbumin see also disorders of globin and globulin proteins
-
Hemoglobin Barts
Wikipedia
In this situation, a fetus will develop hydrops fetalis and normally die before or shortly after birth, unless intrauterine blood transfusion is performed. [2] Since hemoglobin Barts is elevated in alpha thalassemia, it can be measured, providing a useful screening test for this disease in some populations. [3] The ability to measure hemoglobin Barts makes it useful in newborn screening tests. ... Deletion of four alpha globin genes was previously felt to be incompatible with life, but there are currently 69 patients who have survived past infancy. [4] Genotypes Alpha-Globin Gene Deletions Clinical Component αα/αα 0 Normal - α/αα 1 Silent Carrier --/ αα or -α/-α 2 Alpha-Thalassemia Trait --/- α 3 Hb H Disease --/-- 4 Fetal Hydrops Table 1: α represents the presence of α-globin gene and- represents the deletion of α-globin gene. [5] The chance of a fetus developing Hemoglobin Bart's hydrops fetalis is dependent upon if one or both parent carries the alpha-thalassemia trait. ... "Homozygosity for the Mediterranean a-thalassemic deletion (hemoglobin Barts hydrops fetalis)" . Annals of Saudi Medicine . 30 (2): 153–5. doi : 10.4103/0256-4947.60523 . ... "Predictive value of cord blood hematological indices and hemoglobin Barts for the detection of heterozygous alpha-thalassemia-2 in an African-Caribbean population" . ... PMID 25770840 . v t e Proteins that contain heme ( hemoproteins ) Globins Hemoglobin Subunits Alpha locus on 16 : α HBA1 HBA2 pseudo ζ HBZ θ HBQ1 μ HBM Beta locus on 11 : β HBB δ HBD γ HBG1 HBG2 ε HBE1 Tetramers stages of development: Embryonic HbE Gower 1 (ζ 2 ε 2 ) HbE Gower 2 (α 2 ε 2 ) HbE Portland I (ζ 2 γ 2 ) HbE Portland II (ζ 2 β 2 ) Fetal HbF/Fetal (α 2 γ 2 ) HbA (α 2 β 2 ) Adult HbA (α 2 β 2 ) HbA 2 (α 2 δ 2 ) HbF/Fetal (α 2 γ 2 ) pathology: HbH (β 4 ) Barts (γ 4 ) HbS (α 2 β S 2 ) HbC (α 2 β C 2 ) HbE (α 2 β E 2 ) HbO (α 2 β O 2 ) Compounds Carboxyhemoglobin Carbaminohemoglobin Oxyhemoglobin / Deoxyhemoglobin Sulfhemoglobin Other human Glycated hemoglobin Methemoglobin Nonhuman Chlorocruorin Erythrocruorin Other human: Myoglobin Metmyoglobin Neuroglobin Cytoglobin plant: Leghemoglobin Other Cytochrome Cytochrome b Cytochrome P450 Methemalbumin see also disorders of globin and globulin proteins This biochemistry article is a stub .
-
Equine Proximal Enteritis
Wikipedia
Salmonella has not been consistently found in all horses with DPJ, although one study cultured toxigenic Clostridial species in 100% of affected horses. [2] Other potential causes include Fusarium infection and recent increase in dietary concentrate levels, which can alter the microbial population within the intestinal lumen. [2] Inflammation of the intestine leads to the secretion of a large amounts of electrolytes, primarily sodium and chloride, into its lumen, resulting in the osmotic movement of water. [2] The production of fluid is thought to be due to active hypersecretion, passive secretion of proteins secondary to damage to epithelium of the mucosa and capillaries, and a functional ileus which prevents removal of this fluid. [2] Massive fluid production results in extensive reflux, usually produced at a rate of 50–100 mL/min, [3] in addition to distention of the proximal small intestine, dehydration, and possible shock secondary to hypovolemia. [2] Proximal enteritis can also occur with inflammation of other organs in the gastrointestinal tract, including gastritis, ileitis, typhlitis , and colitis. [2] Clinical signs [ edit ] Signs include acute onset of moderate to severe pain, large volumes of gastric reflux (4–20 L per decompression) [2] which is usually orange-brown and fetid, distended small intestine (up to 5–7 cm in diameter) [2] palpable on rectal examination, fever, depression, increased heart rate (>60 bpm), [2] increased respiratory rate, prolonged CRT, and darkened mucous membranes. [2] After gastric decompression, the horse may show signs of malaise and act lethargic, but pain level usually improves. [2] Abdominocentesis usually reveals a yellow, turbid fluid with an increased white blood cell count (usually 5,000–10,000 cells/microliter) and protein level (>3.5 g/dl), [2] although the fluid may be serosanginous in severe cases. [2] [3] A chemistry panel will often show electrolyte abnormalities ( hypokalemia , hyponatremia , hypochloremia ) due to electrolyte loss into the lumen of the intestine. ... Horses with proximal enteritis usually have an intestinal diameter that is narrower, but wall thickness is often greater than 6mm, [3] containing a hyperechoic or anechoic fluid, with normal, increased, or decreased peristalsis. [2] However, obstructions that have been present for some time may present with thickened walls and distention of the intestine. [2] DPJ can only be definitively diagnosed during surgery or at necropsy , when its gross appearance of the small intestine may be evaluated. [2] Treatment [ edit ] Proximal enteritis usually is managed medically. This includes nasogastric intubation every 1–2 hours to relieve gastric pressure secondary to reflux, [3] [4] which often produces to 2–10 L, [2] as well as aggressive fluid support to maintain hydration and correct electrolyte imbalances. ... Horses are withheld food until reflux returns to less than 1–2 L of production every 4 hours, and gut sounds return, often requiring 3–7 days of therapy. ... It is suspected to improve healing and shorten the duration of the illness, since horses often become cachexic due to the protein losing enteropathy associated with this disease. [2] Surgery may need to be performed to rule out colic with similar presenting signs such as obstruction or strangulation, [4] and in cases that are long-standing (> 7 days) to perform a resection and anastomosis of the diseased bowel. [3] However, some horses have recovered with long-term medical support (up to 20 days). [2] Complications and survival [ edit ] Horses may develop pharyngitis , laryngitis , or esophagitis secondary to indwelling nasogastric tube. [2] Other complications include thrombophlebitis, laminitis (which subsequently reduces survival rate), and weight loss. [2] Horses are also at increased risk of hepatic injury. [5] Survival rates for DPJ are 25–94%. [2] Horses that survive the incident rarely have reoccurrence. [2] References [ edit ] ^ a b c d Edwards, G.
-
Crigler-Najjar Syndrome Type 2
GARD
Crigler-Najjar syndrome type 2 (CN-2) is a rare disorder that causes elevated levels of bilirubin in the blood (hyperbilirubinemia). ... The main symptom of CN-2 is persistent jaundice, which is yellowing of the skin, mucous membranes and whites of the eyes. ... CN-2 is caused by mutations in the UGT1A1 gene and inheritance is autosomal recessive. CN-2 responds to treatment with phenobarbital ; however during an episode of severe hyperbilirubinemia, phototherapy may be needed. Not all people with CN-2 require treatment, but routine monitoring is still recommended.
-
Complement Component 2 Deficiency
MedlinePlus
Complement component 2 deficiency is a disorder that causes the immune system to malfunction, resulting in a form of immunodeficiency. ... Between 10 and 20 percent of individuals with complement component 2 deficiency develop SLE. Females with complement component 2 deficiency are more likely to have SLE than affected males, but this is also true of SLE in the general population. ... Causes Complement component 2 deficiency is caused by mutations in the C2 gene. ... The most common C2 gene mutation, which is found in more than 90 percent of people with complement component 2 deficiency, prevents the production of complement component 2 protein. ... It is unclear how complement component 2 deficiency leads to an increase in autoimmune disorders.
-
Infrapatellar Fat Pad Syndrome
Wikipedia
Infrapatellar fat pad syndrome Other names Hoffa's disease [1] Cross section of the human knee Specialty Orthopedics , sports medicine Symptoms Pain in the front of the knee [2] Causes Trauma, surgery [1] Differential diagnosis Patellar tendinopathy , infrapatellar bursitis [2] Treatment Steroid injections, physical therapy , surgery [2] [1] Frequency Relatively common (athletes) [2] Infrapatellar fat pad syndrome , also known as Hoffa's disease , is when pain in the front of the knee occurs due to problems with the infrapatellar fat pad . [2] Pain is generally just below the kneecap . [2] Symptoms may worsen if the knee is overly straightened or bent for too long a period. [2] Complications may include an inability to fully straighten the knee. [2] The underlying mechanism may involve bleeding, inflammation, or insufficient space for the fat pad. [2] This may occur as a result of trauma or surgery to the knee. [1] Diagnosis may be supported by magnetic resonance imaging (MRI). [2] Treatment is generally by steroid injections and physical therapy . [2] [1] If this is not effective surgery removal may be tried. [2] While overall it is an uncommon condition, [3] it is relatively common in athletes. [2] Treatment [ edit ] Treatment is generally by steroid injections and physical therapy. [2] [1] If this is not effective surgery removal may be tried. [2] High quality evidence for surgery is lacking as of 2015. [3] References [ edit ] ^ a b c d e f Dragoo, JL; Johnson, C; McConnell, J (1 January 2012).

-
Trisomy 2 Mosaicism
GARD
Trisomy 2 mosaicism is a rare chromosome disorder characterized by having an extra copy of chromosome 2 in a proportion, but not all, of a person’s cells. Many cases of trisomy 2 mosaicism result in miscarriage during pregnancy. In infants born with trisomy 2 mosaicism, severity as well as signs and symptoms vary widely. ... However, children with trisomy 2 mosaicism with no significant medical problems have been reported (although long-term follow-up was not available). ... When trisomy 2 mosaicism is detected during early pregnancy with chorionic villus sampling (CVS), the affected cells may be confined only to the placenta, and not present in the fetus.
-
Dens Evaginatus
Wikipedia
The extra cusp can cause occlusal interference, displace of the affected tooth and/or opposing teeth, irritates the tongue when speaking and eating and decay the developmental grooves. [2] Temporomandibular joint pain could be experienced secondarily due to occlusal trauma caused by the tubercle . [1] [2] This cusp could be worn away or fractured easily. [1] [4] [2] In 70% [4] of the cases, the fine pulpal extension were exposed which can lead to infection, [4] pulpal necrosis and periapical pathosis . Associated anomalies [ edit ] Additional tubercules [2] Aesthetic and/or occlusion problems [2] Agenesis [3] Bifid cingula [2] Exaggerated cusp of Carabelli [2] Gemination [3] Impaction [2] Labial drifting [2] Labial groove [2] Mesiodens [3] Megadont [2] Odontoma [2] Peg-shaped lateral incisor [2] Prominent marginal ridge [2] Shallow groove in the lateral incisor [2] Shovel-shaped incisor [2] Supernumerary [2] Cause [ edit ] The cause of DE is still unclear. [2] There is literature indicating that DE is an isolated anomaly. ... International Endodontic Journal . 30 (2): 79–90. doi : 10.1111/j.1365-2591.1997.tb00679.x . ... International Journal of Morphology . 28 (2): 375–378. doi : 10.4067/S0717-95022010000200006 . ... Journal of Endodontics . 27 (8): 540–2. doi : 10.1097/00004770-200108000-00010 .
-
Genitourinary Tract Injury
Wikipedia
Find sources: "Genitourinary tract injury" – news · newspapers · books · scholar · JSTOR ( April 2020 ) ( Learn how and when to remove this template message ) Genitourinary tract injury Specialty Urology The kidney is the most commonly injured urinary tract organ. [1] Injuries occur commonly after automobile- or sports-related accidents. [1] A blunt force is involved in 80-85% of injuries to the kidney. [1] Major decelerations can result in major vascular injuries near the kidney's hilum. [1] Gunshots and knife wounds commonly are the cause of penetrating injuries. [1] Fractured ribs can result in penetrating injuries to the kidney. [1] Injuries to the urethra occur most commonly in men after pelvic fractures or straddle-type falls. [1] Contents 1 Presentation 1.1 Comorbidity 2 Diagnosis 2.1 Hematuria in Patients Presenting After Trauma 2.1.1 Foley Catheter 2.1.2 Abdominal Imaging 2.1.3 Retrograde Urethrography (RUG) 2.1.4 Retrograde Cystography 2.1.5 Angiography 3 Management 3.1 Genitourinary Trauma 3.1.1 Urethral Injuries 3.1.1.1 Posterior Urethra Injuries 4 References Presentation [ edit ] Comorbidity [ edit ] In 90% of bladder injuries there is a concurrent pelvic fractures. [1] Pelvic bone fragments penetrate and perforate the bladder. [1] Perforations can be either extraperitoneal or intraperitoneal . [1] Intraperitoneal perforations allow for urine to enter the peritoneal cavity. Symptoms typically develop immediately if the urine is infected. [1] Otherwise sterile urine may take days to cause symptoms. [1] Diagnosis [ edit ] Hematuria in Patients Presenting After Trauma [ edit ] Blood in the urine after abdominal trauma suggests a urinary tract injury. [2] Renal injuries are suggested by lower rib fractures. [2] Bladder and urethral injuries are suggested by pelvic fractures. [2] Foley Catheter [ edit ] The urethral meatus should be examined after trauma. [2] Blood at the urethral meatus precludes insertion of a foley catheter into the bladder. [2] Erroneously placing a foley in this situation can result in infections of periprostatic and perivesical hematomas or conversion of a partial transection to a complete urethral transections. [2] Blood at the urethral meatus suggests an injury to the urethra. [2] Otherwise a foley catheter can be placed into the bladder and hematuria can be assessed for. [2] Abdominal Imaging [ edit ] Hemodynamically-stable individuals should undergo further radiographic assessment. [2] Abdominal computed tomography (CT) with contrast can detect retroperitoneal hematomas, renal lacerations, urinary extravasation, and renal arterial and venous injuries. [2] A repeat scan ten minutes after the first is recommended. [2] Retrograde Urethrography (RUG) [ edit ] The purpose of this study is to identify and characterize injuries to the urethra. [2] The tip of a small (12F) foley catheter is placed in the urethral meatus. [2] The catheter remains fixed after 3 mL of water are instilled into the foley catheter's balloon. [2] Radiographic films are taken as 20 mL of water-soluble contrast material are injected [2] This outlines the urethra from the urethral meatus to the bladder neck. [2] If injuries exist, the location can be determined. [2] Retrograde Cystography [ edit ] The purpose of this study is to identify bladder perforations. [2] The bladder needs to be adequately distended with contrast medium. [2] 300 mL or more are generally recommended. [2] The study has two films. One film is taken when the bladder is adequately distended and filled with contrast. [2] The next film is taken after the bladder is emptied without the assistance of a foley catheter. [2] Angiography [ edit ] Helpful in identifying injuries to the kidney's parenchyma and vasculature. [2] Management [ edit ] Genitourinary Trauma [ edit ] Urethral Injuries [ edit ] Management depends on what part of the urethra was injured and to what extent. [3] The two broad anatomical separations are the posterior and anterior urethra. [3] The posterior urethra includes the prostatic and membranous urethra. [3] The anterior urethra includes the bulbous and pendulous portion. [3] Posterior Urethra Injuries [ edit ] The membranous urethra can be separated from the prostate's apex after blunt trauma. [3] The urethra should not be catheterized. [3] Initial management should be the creation of a suprapubic cystostomy for urine drainage. [3] The bladder should be opened in the midline so to facilitate inspection of bladder lacerations. [3] Perforations can be closed with absorbable sutures. [3] The suprapubic cystostomy remains in place for three months. [3] Incomplete urethral disruptions heal spontaneously and the suprapubic cystostomy can be removed after three weeks for these injuries. [3] Before removing a cystostomy, a voiding cystourethrography should demonstrate no urine extravasation. [3] Delayed urethral reconstruction may be performed within 3 months. [3] This typically entails a direct excision of the now strictured area and anastomosis of the bulbous urethra to the prostate's apex. [3] A urethral catheter and suprapubic cystostomy should be left in place. [3] These are removed within a month. [3] References [ edit ] ^ a b c d e f g h i j k l McAnich, Jack; Lue, Tom (2013).
-
Multiple Endocrine Neoplasia, Type 2 (Men 2)
Mayo Clinic
Overview Multiple endocrine neoplasia, type 2, also called MEN 2, is a rare condition. ... People diagnosed with medullary thyroid cancer are screened regularly for MEN 2 . Complications MEN 2 can cause the parathyroid glands to put too much calcium into the blood. ... Prevention Genetic testing is used to find out if someone has a changed gene that causes MEN 2 . Children of someone who has this changed gene could inherit it and develop MEN 2 . ... This is because MEN 2 can be treated or managed by removing the thyroid gland early in life. ... Diagnosis To diagnose multiple endocrine neoplasia, type 2, also called MEN 2, your health care provider will do a physical exam.
-
Cuffitis
Wikipedia
Cuffitis Different regions of Ileal pouch-anal anastomosis Specialty General surgery Cuffitis is inflammation at the anal transition zone or "cuff" created as a result of ileal pouch-anal anastomosis (IPAA). [1] It is considered a variant form of ulcerative colitis that occurs in the rectal cuff. [2] Cuffitis is a common complication of IPAA, particularly when a stapled anastomosis without mucosectomy procedure has been used. [2] Contents 1 Signs and symptoms 1.1 Complications 2 Diagnosis 3 Treatment 4 References Signs and symptoms [ edit ] Symptoms of cuffitis mimic those of pouchitis . [2] In addition, patients with cuffitis often present with small volume bloody bowel movements. [2] Often, cuffitis can produce the appearance of bright red blood on tissue. [1] Complications [ edit ] Surgery-associated ischemia may contribute inflammation at the anal transitional zone. [2] Patients whose cuffitis is refractory to mesalamine and/or corticosteroids should be evaluated for other disease in the cuff area, such as fistula or anastomotic leaks. [2] Cuffitis that is refractory to medication can also be a sign of Crohn's disease of the pouch. [2] Chronic cuffitis can also contribute to the development of anastomotic stricture . [2] Cuffitis that is refractory, Crohn's-related, or is associated with surgical complications can contribute to pouch failure. [2] Diagnosis [ edit ] Definitive diagnose of cuffitis is obtained by endoscopy . [2] Treatment [ edit ] Cuffitis is treated with mesalamine suppositories or topical application of lidocaine or corticosteroid medications. [2] Systemic medications are rarely used. [2] References [ edit ] ^ a b "Pouchitis: Causes, Symptoms & Treatment | Cleveland Clinic" . my.clevelandclinic.org .

-
Biliary Sludge
Wikipedia
Biliary sludge Other names Gallbladder sludge, Microcrystalline disease, Biliary sediment, Thick bile, Biliary sand Abdominal ultrasonography showing biliary sludge and gallstones Specialty Gastroenterology Biliary sludge refers to a viscous mixture of small particles derived from bile . [1] [2] These sediments consist of cholesterol crystals, calcium salts, calcium bilirubinate , mucin , and other materials. [1] [2] [3] Contents 1 Signs and symptoms 1.1 Complications 2 Cause 3 Pathophysiology 4 Diagnosis 5 Treatment 6 Prognosis 7 Epidemiology 8 See also 9 References 10 External links Signs and symptoms [ edit ] Complications [ edit ] Biliary sludge may cause complications such as biliary colic , acute cholecystitis , acute cholangitis , and acute pancreatitis . [1] [2] Cause [ edit ] Biliary sludge has been associated with pregnancy , rapid weight loss, total parenteral nutrition , drugs such as ceftriaxone and octreotide , solid organ transplantation , and gastric surgery. [1] [2] In many of these conditions, it is thought that the impairment in the contractility of the gallbladder leads to the formation of the sludge. [2] Pathophysiology [ edit ] The pathophysiology of biliary sludge formation is likely related to gallbladder dysmotility . [2] It is presumed that because the gallbladder is unable to effectively empty, the biliary sludge can start to accumulate. [2] Diagnosis [ edit ] Gallbladder hepatization, which is biliary sludge filling the entire gallbladder, giving it an echogenicity similar to the liver (seen at left). ... Biliary sludge is typically diagnosed by CT scan or transabdominal ultrasonography . [1] [2] Endoscopic ultrasonography is another more sensitive option. However, the gold standard is considered to be direct microscopy of aspirated gallbladder bile. [1] [2] This method is much more sensitive, although it is less practical. [2] Treatment [ edit ] For patients without symptoms, no treatment is recommended. If patients become symptomatic and/or develop complications, cholecystectomy is indicated. [1] For those who are poor surgical candidates, endoscopic sphincterotomy may be performed to reduce the risk of developing pancreatitis . [1] Prognosis [ edit ] The clinical course of biliary sludge can do one of three things: (1) it can resolve completely, (2) wax and wane, or (3) progress to gallstones . [1] [2] [3] If the biliary sludge has a cause (e.g. pregnancy), it oftentimes is resolved when the underlying cause is removed. [3] Epidemiology [ edit ] The prevalence of biliary sludge is low in the general population. [2] It has been reported that the prevalence ranges from 0-0.20% in men and 0.18-0.27% in women. [2] However, in patients with certain conditions, the prevalence may be higher. [2] See also [ edit ] Biliary microlithiasis References [ edit ] ^ a b c d e f g h i Shaffer, E. ... Current Gastroenterology Reports . 3 (2): 166–73. doi : 10.1007/s11894-001-0015-6 .

-
Alk+ Large B-Cell Lymphoma
Wikipedia
ALK+ large B-cell lymphoma is a type of lymphoma . [1] [2] : 378 It was first reported in 1997. [2] : 378 [3] [4] It is a rare, aggressive large B-cell process that shows ALK expression. [2] : 378 [3] [5] It is distinct from anaplastic large cell lymphoma , a T-cell lymphoma. [2] : 564 [3] [6] Contents 1 Biology 2 Treatment 3 See also 4 References Biology [ edit ] The median age of diagnosis is approximately late thirties to early forties. [2] : 378 [3] [5] The estimates of childhood disease vary (8%, [7] 15%, [3] 30% [2] : 378 ) but it can be seen at any age. [5] [8] : 306 The disease usually arises in lymph nodes , particularly the neck, but extranodal involvement, including in the gastrointestinal tract , nasal cavity, ovary and brain, has been described. [3] [5] Morphologically, there are large immunoblast -like cells with large central nucleoli , often cellular clusters, with a predilection for the lymph node sinuses [2] : 378 [4] [8] : 306 in a cohesive pattern that can suggest carcinoma cells. [2] : 378 [8] : 306 Upregulation of ALK is mainly due to chromosomal translocation t(2;17), resulting in a fusion gene of CLTC with ALK, [4] [9] but can rarely be due to t(2;5), fusing NPM1 with ALK; [2] : 378 the later is the usual finding in anaplastic large cell lymphoma (ALCL). [4] [9] The t(2;17) translocation occurs in less than 1% of cases of ALK+ ALCL, but has been identified in inflammatory myofibroblastic tumors . [3] There is no association with Epstein–Barr virus [2] : 378 [6] or HHV8 , [6] or immunosuppression . [2] : 378 The cells are CD20 and CD30 negative, [8] : 306 showing weak focal expression in 3% and 6% respectively. [2] : 378 They are EMA and CD138 positive, [8] : 306 showing 100% expression respectively. [2] : 378 Treatment [ edit ] Multiagent chemotherapy is given, and can result in long term success, particularly in childhood [8] : 306 but prognosis is generally poor, [2] : 378 [3] [5] [7] [9] particularly in higher stage disease. [7] See also [ edit ] Lymphoma Anaplastic lymphoma kinase References [ edit ] ^ Swerdlow, Steven H.; International Agency for Research on Cancer; World Health Organization (2008). ... World Health Organization classification of tumours. 2 (4th ed.). International Agency for Research on Cancer. ... "A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2; 5 translocation". Blood . 89 (5): 1483–90. ... "ALK-positive diffuse large B-cell lymphoma of the duodenum: A case report and review of the literature" . Exp Ther Med . 8 (2): 409–412. doi : 10.3892/etm.2014.1786 . ... "ALK-positive diffuse large B-cell lymphoma: report of four cases and review of the literature" . J Hematol Oncol . 2 : 11. doi : 10.1186/1756-8722-2-11 .






