Load FindZebra Summary
Disclaimer:
FindZebra Search conducts a search using our specialized medical search engine.
FindZebra Summary uses the text completions API
(subject to OpenAI’s API data usage policies)
to summarize and reason about the search results.
The search is conducted in publicly available information on the Internet that we present “as is”.
You should be aware that FindZebra is not supplying any of the content in the search results.
FindZebra Summary is loading...
-
Premature Chromatid Separation Trait
OMIM
MVA occurs when the offspring of 2 parents heterozygous for the PCS trait inherits both mutant BUB1B alleles. ... Several references listed in this entry (e.g., Rudd et al., 1983; Gabarron et al., 1986) have incorrectly used the designation PCD when referring to PCS. To avoid confusion, we have changed the designation to PCS in our discussion of these references. ... Petkovic (2007) reported a 3-generation family with autosomal dominant inheritance of PCS. Cytogenetic studies on peripheral blood lymphocytes demonstrated PCS frequency of 8.5% to 13.5% in 4 affected members. ... The father and mother had 6.5% and 16% PCS in cultured lymphocytes, respectively, consistent with heterozygosity for the PCS trait. Amniocentesis at 16 weeks in a subsequent pregnancy showed 4.5% cells in PCS and a nonmosaic 46,XY karyotype, also consistent with a heterozygous carrier of the PCS trait.
-
Pachyonychia Congenita
GeneReviews
Diagnosis/testing. PC is diagnosed by clinical findings and/or by the identification of a heterozygous pathogenic variant in one of the five keratin genes known to cause PC: KRT6A , KRT6B , KRT6C , KRT16 , and KRT17 . ... Nomenclature Based on data from the International Pachyonychia Congenita Research Registry (IPCRR), the most recent classification for pachyonychia congenita is by mutated gene [McLean et al 2011, Wilson et al 2011]: PC-K6a (caused by pathogenic variants in KRT6A ) PC-K6b (caused by pathogenic variants in KRT6B ) PC-K6c (caused by pathogenic variants in KRT6C ) PC-K16 (caused by pathogenic variants in KRT16 ) PC-K17 (caused by pathogenic variants in KRT17 ) The classification suggested for PC prior to the identification of the genetic basis of the disease was based solely on clinical findings. Historically, the two major subtypes of PC were based on subtle variable phenotypic features (primarily on the presence or absence of pilosebaceous cysts and natal or prenatal teeth) [Leachman et al 2005, Liao et al 2007]: PC-1 (Jadassohn-Lewandowski syndrome) PC-2 (Jackson-Lawler syndrome) With detailed clinical histories and pathogenic variants identified in an increasing number of people with PC, it became clear that the older classification of PC-1 and PC-2 was not applicable to the broader population of individuals with PC. ... The International PC Research Registry has identified 774 individuals in 419 families with genetically confirmed PC. ... Familial onychogryphosis without the associated palmoplantar keratoderma or other features of PC can be confused with PC. Individuals who have nail findings only are unlikely to have a pathogenic variant in one of the PC-related genes.
-
Posterior Cord Syndrome
Wikipedia
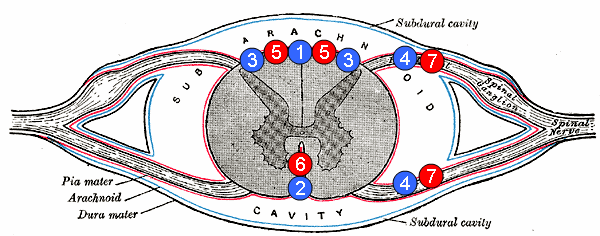
Human spinal cord disorder Posterior cord syndrome 5: posterior spinal arteries Posterior cord syndrome (PCS) , also known as posterior spinal artery syndrome (PSA), is a type of incomplete spinal cord injury . [1] PCS is the least commonly occurring of the six clinical spinal cord injury syndromes , with an incidence rate of less than 1%. ... In addition, the demographics of patients suffering from PCS are widespread as the onset of symptoms typically follows a traumatic event. Additionally, research has suffered setbacks because PCS is extremely rare with few documented cases, unlike anterior spinal cord injury. [1] [3] [9] However, ongoing research has helped in differentiating PCS from other brain injuries. Therefore, better therapies for PCS treatment can be developed. For instance, one study suggests that a tissue plasminogen activator (tPA) therapy intervention, commonly used in stroke patients, [10] may aid in treating patients with symptoms of PCS. [11] References [ edit ] ^ a b c d "Incomplete Spinal Cord Injury" . ... Archived from the original (PDF) on 28 March 2018 . Retrieved 27 March 2018 . ^ Sakurai, Takeo; Wakida, Kenji; Nishida, Hiroshi (2016).
-
Pachyonychia Congenita
Wikipedia
PC-K6b is caused by a mutation in the KRT6B gene and more commonly associated with an increased age of onset (>14 years). PC-K6c is caused by a mutation in the KRT6C gen e and is the least common sub-type. ... Pachyonychia Congenita Project is a non-profit dedicated to finding a cure for PC. The organization houses a genetic registry (the International PC Research Registry) and offers free genetic testing for individuals suspected to have PC. [11] Treatment [ edit ] There is currently no cure for pachyonychia congenita. ... ISBN 0-7216-2921-0 . ^ a b "International PC Research Registry (IPCRR)" . www.pachyonychia.org . ... Journal of the European Academy of Dermatology and Venereology . 28 (3): 279–285. doi : 10.1111/jdv.12098 .

-
Post-Concussion Syndrome
Wikipedia
Education about symptoms and details about expectation of recovery are important. The majority of PCS cases resolve after a period of time. ... However, studies have found some subtle physiological changes associated with PCS using more novel imaging modalities. Studies using positron emission tomography have linked PCS to a reduction in glucose use by the brain. ... Many of these PCS sufferers were misdiagnosed as having other unrelated conditions due to commonality of symptoms. (see diagnosis above). [70] Headache is one of the criteria for PCS, but it is notably undetermined where the headache comes from.
-
Mosaic Variegated Aneuploidy Syndrome 1
OMIM
See also premature chromatid separation (PCS; 176430), which can be caused by heterozygous mutation in the BUB1B gene. PCS is inherited as an autosomal dominant trait without phenotypic consequences. ... Another 10 relatives showed 0 to 1% cells with total PCS and so were judged negative for the total PCS trait. ... Plaja et al. (2001) studied 3 patients with MVA related to PCS, showed that the phenomenon is expressed in vivo, and found that PCS is a cancer-prone disorder. ... Cytogenetic analysis of 2 infants showed 48.5% and 83.2% lymphocytes in total PCS; their parents had 3.5 to 41.7% of their lymphocytes in total PCS.
-
Pancreatic Neuroendocrine Tumor
Wikipedia
PanNETs are a type of neuroendocrine tumor , representing about one third of gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Many PanNETs are benign , while some are malignant . ... However, morphological imaging alone is not sufficient for a definite diagnosis [14] [16] On biopsy , immunohistochemistry is generally positive for chromogranin and synaptophysin . [17] Genetic testing thereof typically shows altered MEN1 and DAXX / ATRX . [17] Staging [ edit ] The 2010 WHO classification of tumors of the digestive system grades all the neuroendocrine tumors into three categories, based on their degree of cellular differentiation (from well-differentiated "NET G1" through to poorly-differentiated "NET G3"). ... Combinations of several medicines have been used, such as doxorubicin with streptozocin and fluorouracil (5-FU) [12] and capecitabine with temozolomide. [ citation needed ] Although marginally effective in well-differentiated PETs, cisplatin with etoposide has some activity in poorly differentiated neuroendocrine cancers (PDNECs), [12] particularly if the PDNEC has an extremely high Ki-67 score of over 50%. [8] : 30 Several targeted therapy agents have been approved in PanNETs by the FDA based on improved progression-free survival (PFS): everolimus (Afinitor) is labeled for treatment of progressive neuroendocrine tumors of pancreatic origin in patients with unresectable, locally advanced or metastatic disease. [20] [21] The safety and effectiveness of everolimus in carcinoid tumors have not been established. [20] [21] sunitinib (Sutent) is labeled for treatment of progressive, well-differentiated pancreatic neuroendocrine tumors in patients with unresectable locally advanced or metastatic disease. [22] [23] Sutent also has approval from the European Commission for the treatment of 'unresectable or metastatic, well-differentiated pancreatic neuroendocrine tumors with disease progression in adults'. [24] A phase III study of sunitinib treatment in well differentiated pNET that had worsened within the past 12 months (either advanced or metastatic disease) showed that sunitinib treatment improved progression-free survival (11.4 months vs. 5.5 months), overall survival , and the objective response rate (9.3% vs. 0.0%) when compared with placebo. [25] Genetics [ edit ] Pancreatic neuroendocrine tumors may arise in the context of multiple endocrine neoplasia type 1 , Von Hippel–Lindau disease , neurofibromatosis type 1 (NF-1) or tuberose sclerosis (TSC) [26] [27] Analysis of somatic DNA mutations in well-differentiated pancreatic neuroendocrine tumors identified four important findings: [28] [6] as expected, the genes mutated in NETs, MEN1 , ATRX , DAXX , TSC2 , PTEN and PIK3CA , [28] are different from the mutated genes previously found in pancreatic adenocarcinoma . [29] [30] one in six well-differentiated pancreatic NETs have mutations in mTOR pathway genes, such as TSC2 , PTEN and PIK3CA . [28] The sequencing discovery might allow selection of which NETs would benefit from mTOR inhibition such as with everolimus , but this awaits validation in a clinical trial . mutations affecting a new cancer pathway involving ATRX and DAXX genes were found in about 40% of pancreatic NETs. [28] The proteins encoded by ATRX and DAXX participate in chromatin remodeling of telomeres ; [31] these mutations are associated with a telomerase -independent maintenance mechanism termed ALT (alternative lengthening of telomeres) that results in abnormally long telomeric ends of chromosomes . [31] ATRX / DAXX and MEN1 mutations were associated with a better prognosis . [28] References [ edit ] ^ Burns WR, Edil BH (March 2012).MEN1, ATRX, DAXX, ELK3, TP53, EPHB1, SLC6A2, CEACAM5, CEACAM7, UQCRFS1, DHDDS, CHPT1, RALBP1, CIB1, SEMA4D, RIPK1, CXCR4, VEGFA, TTR, GNA12, TSC2, TFE3, CDKN1B, PSG2, POMC, MYCN, CEACAM3, GRN, MUC16

-
Neuroendocrine Tumor
Wikipedia
G1 and G2 neuroendocrine neoplasms are called neuroendocrine tumors (NETs) – formerly called carcinoid tumours. ... Unsourced material may be challenged and removed. ( November 2015 ) ( Learn how and when to remove this template message ) NETs from a particular anatomical origin often show similar behavior as a group, such as the foregut (which conceptually includes pancreas, and even thymus, airway and lung NETs), midgut and hindgut ; individual tumors within these sites can differ from these group benchmarks: Foregut NETs are argentaffin negative. ... Bone metastasis is uncommon. Hindgut NETs are argentaffin negative and rarely secrete 5-HT, 5-HTP, or any other vasoactive peptides. ... Not all cells are immediately killed; cell death can go on for up to two years. [ citation needed ] PRRT was initially used for low grade NETs. It is also very useful in more aggressive NETs such as Grade 2 and 3 NETs [83] [84] provided they demonstrate high uptake on SSTR imaging to suggest benefit. ... World Journal of Clinical Oncology . 2 (1): 28–43. doi : 10.5306/wjco.v2.i1.28 .MEN1, CDKN1B, SSTR2, DAXX, ATRX, BRAF, TYMS, PTHLH, SSTR3, SSTR1, BAP1, MTOR, SST, GAST, SLC6A2, INSM1, CTNNB1, RET, PIK3CA, DNMT3A, POMC, EPHB1, PIK3CG, PIK3CD, CHGA, ELK3, CHEK2, PIK3CB, GRN, CD274, SMUG1, AKT1, GNA12, TP53, SYP, VEGFA, CDKN2A, ASCL1, BCL2, ENO2, NCAM1, GCG, MYCN, EGFR, MGMT, KIT, RASSF1, VHL, SCLC1, SSTR5, FOLH1, NKX2-1, KRAS, CALCA, CCND1, TAC1, PTPRF, VIP, NTS, PAX5, RHBDF2, GRP, IGF1, SDHD, GOT1, MAP2K7, CCK, ERBB2, DLL3, PPY, CXCL12, TP63, SMAD4, MUC1, INS, GCGR, CKAP4, NEUROD1, ISL1, MYC, NGF, SATB2, GLP1R, HSP90AA1, H3P10, HRAS, CHGB, CALR, NTRK1, TEK, DLK1, CDK4, CDX2, TGFA, UCHL1, RPE65, PGR, PDGFRA, CARTPT, CRH, UVRAG, SLC5A5, CXCR4, IGF1R, OTP, IL6, PHLDA3, TTF1, PAX8, TACR1, STK11, TRIM21, PLA2G15, SCG2, SQLE, SLC18A2, TERT, HDAC9, SLC2A1, PROM1, BCL2L11, NTSR1, PAX6, NAMPT, NOCT, INA, PLCB3, CD200, MKI67, PDX1, MAPK1, NES, HPSE, PTEN, STMN1, ABO, RIPK1, RORC, RAF1, IL1B, TRPV1, GATA3, ANGPT2, FOXM1, PTK2B, SDHAF2, ACCS, BDNF, EPAS1, EGF, ACSS2, MIB1, DNMT1, CCN2, TRPM8, CLDN4, CPE, CD34, CD44, FLNA, CEACAM5, B3GAT1, GH1, GIP, GHSR, GIPR, ADCY2, ALB, H3P28, TPPP2, H4C5, GGH, MIR1290, TMEM209, ELOA3, H4C13, H4C14, GPR151, SRPX, LGR5, TNFSF11, PSMG1, DCBLD2, H4-16, NRP1, MRGPRX4, SOCS1, H4C2, MIR3137, MRGPRX3, TNFRSF25, H3P12, CYYR1, AZIN2, DNER, AK6, MLIP, LMLN, NRP2, GPR68, MIR1246, H4C8, MAFK, MIR150, MIR155, MBOAT4, H4C9, MIR21, POTEKP, VN1R17P, SNORD95, GPR166P, ARID1A, EID3, SLC7A5, MIR375, H4C15, FZD4, MIRLET7C, OXER1, H4C12, HMGA2, H4C3, ARX, ELOA3B, GPRC6A, H4C11, H4C6, C17orf97, POTEM, MRGPRX1, ARMH1, H4C1, GADL1, ACTBL2, H4C4, BRI3, SQSTM1, ISYNA1, GHRL, ACOT7, KLF12, KRT20, SLC27A4, TET2, BCOR, EBNA1BP2, RALBP1, PGRMC1, LAMTOR1, FBXW7, MEG3, MAML3, TMEM127, NTNG1, ATRAID, KHDRBS1, DCTN4, SNORD61, NUP62, SNORD48, NTSR2, LPAR3, MAPK8IP2, SRRM2, BRD4, TRAM1, SPINK4, XIST, PPWD1, RBMS3, SETD1B, ZHX2, TNFSF13B, USE1, MAK16, UBE2Z, ONECUT2, FHL5, GCM2, DCLK1, ZBED1, ARHGEF2, PALB2, ALG9, SNED1, TET1, PDCD1LG2, TMPRSS13, MTA1, RPAIN, H1-10, EEF1E1, LGR6, PRMT5, NEUROD4, YAP1, SCML2, LANCL1, PAK4, RABEPK, ZNF197, CTNNBL1, PNO1, INSL5, EPB41L5, HDAC5, AKT3, CD302, GBA3, DCAF1, ATAT1, SERPINA3, VCL, CGA, ESR1, ERBB4, EPHB2, E2F1, DUSP2, DSG3, DPT, DPP4, DMBT1, DDC, DAD1, VCAN, CREB1, CRABP1, KLF6, CLU, FOXN3, CEACAM7, CEACAM3, ESR2, ETFA, EZH2, GHRH, HSPA4, AGFG1, HMOX1, HMGA1, GTF2H1, GSN, GNAS, GNA15, GFRA1, F3, GDNF, FSHR, FLT4, FLII, FLI1, FOXO1, FHIT, FGFR4, CGB3, CFL1, UQCRFS1, CDKN2C, FAS, APRT, APLP1, XIAP, APC, SLC25A6, SLC25A4, ANGPT1, ALK, AKT2, AFP, PARP1, ADCYAP1R1, ADCYAP1, ACVRL1, ACTN4, ACTG2, ACTG1, ACR, AQP4, ARF1, ATM, CASP3, CDK6, CD40LG, CD36, CD33, CCNE1, CCKBR, SERPINA6, CAV1, CA9, ATOH1, VPS51, C5, BRS3, BRCA2, DST, BAX, AVP, ATP4A, HTC2, HTR2A, TNC, IAPP, SDC1, SCT, SORT1, RNASE3, RARB, PTPRZ1, PTPRM, PTBP1, PSMD7, PSG2, PRKAR1A, PPP4C, POU4F1, PNN, PKD2, PITX2, PCYT1A, SERPINA5, PAX4, SDCBP, SDHB, SDHC, ST2, UBE2I, TPM3, TPH1, TNF, TM7SF2, TERC, TAT, STAT3, SSTR4, SEMA3F, SSR2, SOX11, SOX4, SOX2, SLPI, SLC3A2, SLC1A5, SFRP1, PAK3, PAK1, TNFRSF11B, KIF11, MDK, MAOA, LCN2, RPSA, L1CAM, KRT19, KRT7, KRT5, IL12A, MET, IL9, CXCL8, IL2, IL1A, IGFBP1, IGF2, IFNA13, IFNA1, MDM2, MFAP1, ODC1, MUTYH, NTRK2, NT5E, NRAS, NOTCH3, NPY, NOTCH1, NFKB1, NEFM, MUC4, CD99, NUDT1, COX2, MTAP, MST1R, MST1, MSMB, MMP7, MLH1, PTPRC

-
Pancreatic Neuroendocrine Tumor
GARD
However in some cases, a pancreatic NET occurs outside of the pancreas. A NET arises from cells that produce hormones, so the tumor can also produce hormones. ... Pancreatic NETs are called either functional or nonfunctional. A functional pancreatic NET causes specific symptoms because it makes extra hormones, such as gastrin, insulin, or glucagon. ... Pancreatic NETs can be hard to diagnosis, often not identified until 5 to 10 years after they begin to grow. Most pancreatic NETs are not inherited and occur sporadically in people with no family history of NETs.MEN1, PCSK1, ATM, BRCA2, C11orf65, IGF2, SST, TP53, CDKN2A, SLC6A2, MTOR, EPHB1, POMC, GH1, GCGR, DAXX, ELK3, KRT19, SSTR2, CHGA, SSTR5, UCHL1, FZD4, GCM2, DLGAP1, DCLK1, SSTR4, INA, STK11, EIF2AK3, TFE3, THBD, CXCR4, PAX8, TSC1, TTR, TYMS, VEGFA, ABO, CNPY2, MRGPRX4, GPR166P, VN1R17P, MIR196A1, GADL1, MRGPRX1, GPRC6A, OXER1, GPR119, GPR151, MRGPRX3, SEMA3A, AZIN2, ACCS, STK33, LGR6, ACSS2, MEG3, NEUROG3, LPAR3, LILRB1, PLA2G15, RET, SLC2A3, INSM1, GRN, FFAR1, GHRH, GAST, FGFR4, F3, EGFR, DHCR24, CSF1, CRH, CHGB, CD44, CCK, CALCA, VPS51, ATRX, ASS1, ASCL1, ANGPT2, HSF1, PDX1, SLC2A2, KIT, SLC2A1, SEA, SDHB, SDHA, AKT1, PYGM, PTH, PTEN, PPY, PTPA, PGR, PCYT1A, PCNA, NFKB1, NEUROD1, MUC1, SMAD4, STMN1, KRAS, H3P10
-
Neuroendocrine Tumor
GARD
A neuroendocrine tumor (NET) is a rare type of tumor that arises from specialized body cells called neuroendocrine cells . ... Pancreatic neuroendocrine tumors (also called islet cell tumors) - NETs that typically arise in the pancreas, although they can occur outside the pancreas. A p heochromocytoma is another, rarer type of NET that usually develops in the adrenal gland , but can also arise in other parts of the body. ... Functional NETs produce a specific set of symptoms due to the production of excess hormones, while non-functional NETs generally do not cause specific symptoms. In many cases, a person has no symptoms until the tumor spreads to the liver and/or impairs the function of an organ or system. This can make NETs very hard to diagnose. The majority of NETs are not inherited and occur sporadically in people with no family history of NETs.
-
Pyruvate Carboxylase Deficiency
OMIM
Robinson et al. (1984) concluded that the 2 subtle types of PC deficiency result from 2 different mutations in the PC gene, 1 that synthesizes an inactive protein and 1 that results in lack of protein expression. ... Three families with the French presentation had absence of immunoreactive PC protein and PC mRNA; however, another 3 families with the French presentation had evidence of protein production as well as PC mRNA. ... Diagnosis Prenatal Diagnosis Tsuchiyama et al. (1983) reported a patient with PC deficiency and PC activity of about 5% of normal. ... Monnot et al. (2009) identified 9 novel mutations in the PC gene (see, e.g., 608786.0007-608786.0009) in 5 unrelated patients with PC deficiency: 3 had the more severe type B PC, and 2 had type A. PC activity in cultured fibroblasts was undetectable in all patients.
-
Parotid Proline-Rich Salivary Protein Pc
OMIM
Karn et al. (1985) identified a new polymorphism, Pc, in human salivary proteins. Two proteins, Pc1 and Pc2, determined by alleles Pc(1) and Pc(2), showed autosomal codominant inheritance. ... By in situ hybridization, Mamula et al. (1985) regionalized the salivary protein gene complex to 12p13.2. Although Pc is clearly a proline-rich salivary protein coded by the gene cluster on 12p, its relation to the 6 gene loci identified there is not known (Azen, 1990).
-
Familial Pancreatic Carcinoma
Orphanet
Familial pancreatic carcinoma is defined by the presence of pancreatic cancer (PC) in two or more first-degree relatives. Epidemiology The annual incidence has been estimated at approximately 1-10/1,000,000, representing 5-10% of all PC cases. Clinical description In familial cases, disease onset occurs before 50 years of age, earlier than for the other forms of PC. ... Smoking represents a significant risk factor associated with familial PC. PC can arise from the exocrine (95%) or endocrine portions of the pancreas. ... Etiology Mutations in the KRAS , CDKN2A , TP53 , and SMAD4 genes have been shown to play a role in the etiology of PC. However, they are still not clinically useful for screening or for diagnosing the disease. ... Even after complete resection of the tumor, recurrence rates remain high. Patients with a family history of PC should be strongly advised to avoid or cease smoking.
-
Postural Orthostatic Tachycardia Syndrome Due To Net Deficiency
Orphanet
A rare, genetic, primary orthostatic disorder characterized by dizziness, palpitations, fatigue, blurred vision and tachycardia following postural change from a supine to an upright position, in the absence of hypotension. A syncope with transient cognitive impairment and dyspnea may also occur. The norepinephrine transporter deficiency leads to abnormal uptake and high plasma concentrations of norepinephrine.

-
Neuroendocrine Neoplasm Of Esophagus
Orphanet
A group of esophageal epithelial neoplasms characterized by neuroendocrine differentiation, comprising well-differentiated neuroendocrine tumors (NETs), poorly differentiated neuroendocrine carcinomas (NECs), and mixed neuroendocrine-non-neuroendocrine neoplasms, an umbrella category including mixed adenoneuroendocrine carcinoma. ... NECs may also arise in other parts of the esophagus. On endoscopy, NETs usually appear as small polypoid or nodular submucosal masses, while NECs are large, infiltrative, and ulcerated. Patients most commonly present with dysphagia, pain, weight loss, and sometimes melena. Metastatic NETs may be associated with carcinoid syndrome.
-
Pachyonychia Congenita
MedlinePlus
Researchers used to distinguish pachyonychia congenita as one of two types, PC-1 or PC-2, based on the genetic cause and pattern of signs and symptoms. ... When pachyonychia congenita is caused by mutations in the KRT6A gene, it is classified as PC-K6a. Similarly, KRT6B gene mutations cause PC-K6b, KRT6C gene mutations cause PC-K6c, KRT16 gene mutations cause PC-K16, and KRT17 gene mutations cause PC-K17.
-
Malaria
Wikipedia
The sporozoites are injected into the skin, in the saliva, when the mosquito takes a subsequent blood meal. [28] Only female mosquitoes feed on blood; male mosquitoes feed on plant nectar and do not transmit the disease. ... The mosquitoes remain on the wall until they fall down dead on the floor. Insecticide treated nets [ edit ] A mosquito net in use. Mosquito nets help keep mosquitoes away from people and reduce infection rates and transmission of malaria. Nets are not a perfect barrier and are often treated with an insecticide designed to kill the mosquito before it has time to find a way past the net. Insecticide-treated nets are estimated to be twice as effective as untreated nets and offer greater than 70% protection compared with no net. [73] Between 2000 and 2008, the use of ITNs saved the lives of an estimated 250,000 infants in Sub-Saharan Africa. [74] About 13% of households in Sub-Saharan countries owned ITNs in 2007 [75] and 31% of African households were estimated to own at least one ITN in 2008. ... That number increased to 20.3 million (18.5%) African children using ITNs in 2007, leaving 89.6 million children unprotected [76] and to 68% African children using mosquito nets in 2015. [77] Most nets are impregnated with pyrethroids , a class of insecticides with low toxicity .ICAM1, FCGR2B, HBB, CD36, NOS2, FCGR2A, TNF, CR1, G6PD, CRP, HP, ACKR1, GYPA, SLC4A1, GYPB, NCR3, TIRAP, GYPC, LTBR, CISH, IFNG, HMOX1, PKLR, ABO, ANK1, AQP4, ATP2B4, HBG2, CYTB, ENOSF1, MSMB, MST1, ZNF536, LINC00944, SMARCB1, DHODH, PDR, TREML4, ZNF804A, OR51F1, OR51B5, CDH13, PROCR, SPATA3, OR51N1P, DHFR, DDT, RECQL4, FAM155A, IGHG3, IL4, MMP26, IL6, IL10, TLR9, HLA-DRB1, CSMD1, HBE1, DNAJC5, TMPRSS13, KLHL3, HDGFL2, TLR4, ATAD1, LMLN, TENM3-AS1, MECP2, POMGNT2, MBL2, TFRC, TGFB1, MIF, HLA-B, HAMP, DHPS, SERPINA3, TLR2, IL1B, FOXP3, FHL5, ACOT7, POTEKP, POTEM, GEM, KIR3DL1, RN7SL263P, ACTG2, ACTG1, ACTB, ACTBL2, HBA2, CYP2B6, HSPA4, LSAMP, TRAP, FCGR3B, HSP90AA1, IL1A, LAMP3, CD81, OR10A4, CCL5, ABCB1, FAS, CD40LG, TEP1, CXCL8, IARS1, HLA-G, CTLA4, HBA1, INSRR, ANGPT2, TYMS, CFH, GSTP1, IFNAR1, AGT, GYPE, FCGR3A, TXN, IL13, HSPB3, APOE, MTCO2P12, ISYNA1, FCGR2C, FYB1, VDR, HLA-A, GSTM1, GSR, ATR, MBL3P, LAIR1, PNP, IL12B, MNAT1, IL1RN, CYP2D6, IGF1, CD55, ACHE, DECR1, COX2, IL3, CCL2, MAPK1, NLRP3, FBXW7, HAVCR2, THBD, VPS51, EMP1, ITGA2B, PTGS2, ANC, IL10RA, XPO1, VNN1, PLEK, UMPS, IL2, IL2RA, TPPP, VWF, ISG20, ADAMTS13, IRF1, IL7R, AIMP2, IL12RB1, CLEC11A, METAP2, CDK5R1, ING1, IL18R1, PGD, HAP1, H6PD, PRDX5, GRAP2, CXCL9, MMP9, MPO, TAP1, CCL4L2, COX1, EBI3, ITGAX, COX3, TLR6, CXCL11, MTHFR, NFKB2, NFYA, NOS1, TBC1D9, ORC1, MCF2, AKAP13, RNF19A, TLR7, NT5C3A, IRAK4, KIR2DS1, CCL4, KIR3DL2, ICOS, COQ2, PSIP1, PECAM1, TPT1, RNASE3, ARTN, TP53, POLDIP2, PDCD1, TLR1, AHSA1, UBL4A, AQP3, AGRP, H3C9P, CYP2C8, CYP2C19, GTF2H4, CRK, RNA18SN5, ANXA2, H3P37, CASP1, NANP, CCL4L1, MAPK14, CXCR3, GNAS, GLO1, FCN2, SMIM10L2B, FKBP4, CD27, FOXO3, RBM45, HM13, IL33, HK1, CCR5, IFNA13, IFNA1, H3P42, DNAJB1, CHIT1, CYP3A4, SMIM10L2A, EGF, CHI3L1, CAT, EPHA2, NSFL1C, ADRB2, MYMX, COX8A, GAPDH, ABCB6, NR1I3, TREML1, PUM3, FMN1, TICAM2, TRIM13, BMS1, FZD4, RABEPK, LANCL1, FUT9, TNFSF13B, DCTN6, CXCR6, ARL6IP5, MRGPRX1, ZNRD2, ASPM, KAT5, RAB7B, CIB1, SEMA3C, ARMH1, STING1, CFDP1, CPQ, MYLK4, DLC1, AKR1A1, PIEZO1, TMPRSS11D, HDAC9, CARTPT, DEFB4B, TIMELESS, SPHK1, TMED7-TICAM2, PSC, VNN2, PROM1, UPK3B, H3P23, H3P28, TNFRSF11A, TNFRSF18, TP63, PDXK, CNTNAP1, DHX16, STK24, H3P19, LOH19CR1, WASHC1, WASH6P, LPAR2, MIR146A, APOBEC3B, SPAG6, CLOCK, ATG5, MIR142, AIM2, ABCG2, PCSK9, MIR155, NCF1, PPIG, MIR29A, VN1R17P, GPR166P, CD163, MIR451A, CXADRP1, ARHGEF2, CERS1, SPINK5, MASP2, GEMIN4, ACD, TLR8, MPPE1, MCPH1, HSPA14, RNF34, TMED7, ARMC9, PPP1R2C, IL22, TRAF3IP2, A1CF, PDCD1LG2, SLC44A4, SGSM3, MCAT, HPGDS, B3GAT1, ROPN1L, PHGDH, RAB14, IL23A, ABCG4, IFIH1, CFC1, BTNL2, MARCHF1, POLE4, CMC2, TMED9, ACKR3, PDXP, RHOF, AICDA, POLD4, RBM25, TOLLIP, TREM1, LGR6, ADA2, BACH2, ERAP1, GOLPH3, PARS2, KRT88P, TRIM5, IL17RE, CHP1, GPR151, NRSN1, EIF5AL1, CD160, APCDD1, ERFE, OXER1, DNAJB1P1, DSTN, GPRC6A, CCNI, ADIRF, EBNA1BP2, TMED2, EHD1, RNPS1, HPSE, SEPTIN9, SCLT1, NT5C2, SLC25A21, LEO1, NLRP12, TIMD4, CDCA5, DBA2, CARD16, PTPMT1, CGAS, RAB39B, TADA1, MRGPRX3, MRGPRX4, PGLS, PANX1, SPO11, LPAR3, CBX5, POFUT2, SPPL3, NBEAL2, LUC7L, PTPRC, FGF23, EIF5, FLT3LG, FLT1, FECH, FBN2, FBN1, FANCD2, F3, EPO, ENO2, ADGRE1, ELK4, ELF4, EIF5A, EIF4G2, CXADR, EGR3, EDNRA, EDN1, S1PR3, RCAN1, ATN1, DNMT1, DEFB4A, DHX9, ACE, DBP, CYP1A2, CYC1, GABPA, GCHFR, GDF1, GPR42, IL4R, IL1R1, IGFBP1, IFNGR1, IFNB1, IFNA2, IFI27, IDE, HTN3, HSPA9, HSD11B1, HRES1, HPRT1, HPR, HPGD, HMGB1, HLA-DOA, UBE2K, HGF, SERPIND1, HBG1, GTF3A, GSTT1, GSN, GPX1, GPT, GRK5, CYBB, CTSL, IL9, ANXA1, C3, BSG, BRS3, BRCA2, PRDM1, BCL2, BAX, ASPA, ASIP, ARR3, NUDT2, ANXA7, ANXA4, ANPEP, CSH2, AMBP, ALOX5, ALB, AHR, AFP, ADSL, ADRA2B, ADRA1A, ADORA2A, ADH1B, ADA, ACP1, ACACA, CAST, CASR, CD1B, CD1C, CSH1, CSF1R, CSF1, CS, CRYZ, CREM, CR2, CLDN4, CPB1, CNTF, CCR4, CLU, ERCC8, CTSC, CEL, CDC25C, CD69, CD68, CD40, ENTPD1, CD34, CD28, CD19, CD14, CD9, CD1E, CD1D, IL5, IL12A, FOSL1, SELE, SPTA1, SPP1, SPINK1, SPG7, SOD3, SOD1, SMN1, SLC16A1, SLC11A1, SLC6A7, SLC2A1, SGCG, SET, SEA, ABCA1, SDC1, CXCL5, CCL22, CCL18, CCL3L1, CCL3, CCL1, SAFB, SORT1, RPS19, RBP2, RANBP2, PEX19, SSR2, SSTR4, DENND2B, STAT6, DDX39B, PRRC2A, PFBI, RAB7A, CXCR4, MOGS, ZBTB16, TRPV1, VCP, USP1, TYRP1, TTR, TTPA, TRPC1, TRP-AGG2-5, TPO, TPH1, TNFRSF1B, TLR3, TGFB2, TRBV20OR9-2, TCN2, HNF1A, TADA2A, ADAM17, TAC1, STK3, PTPRH, PTHLH, IL15, KIR3DS1, MAL, MAF, LTB, LTA, LMAN1, LEPR, LDLR, LCN2, LBR, RPSA, LAG3, KRT13, KNG1, KIR2DS5, PSMD9, KIR2DL3, KIR2DL2, KDR, KCNG1, KARS1, ITPA, ITGB2, ITGAM, ITGAL, CXCL10, IDO1, ILF3, IL18, MAP2, MAP6, MEFV, MVD, PSMD7, PSMD2, PSMB9, PSEN1, PSAP, PRSS1, PROC, MAP2K1, PRKG1, PRKAR1A, PPP1R1A, PPARG, SEPTIN4, PLP1, PGM1, PGAM1, P2RX7, SLC22A18, TNFRSF11B, OMD, ODC1, NOS3, NQO2, NFE2L2, NEK2, MYD88, MYC, H3P5
-
West Nile Fever
Wikipedia
Archived from the original on 18 October 2017 . Retrieved 28 October 2017 . ^ "Final Cumulative Maps and Data | West Nile Virus | CDC" . www.cdc.gov . 24 October 2017. Archived from the original on 27 October 2017 . Retrieved 28 October 2017 . ^ Gompf, Sandra. "West Nile Virus" . Medicine Net . MedicineNet Inc . Retrieved 15 January 2019 . ^ "Symptoms, Diagnosis, & Treatment" . ... Retrieved 2018-11-28 . ^ Tyler KL, Pape J, Goody RJ, Corkill M, Kleinschmidt-DeMasters BK (February 2006). ... PMID 21837806 . ^ a b "Prevention | West Nile Virus | CDC" . www.cdc.gov . 2018-09-24 . Retrieved 2018-11-28 . ^ American Academy of Pediatrics (8 August 2012).CCR5, ERVK-32, ROBO3, MAVS, DDX58, PLAAT4, IFIT2, ERVK-6, STAT1, SPP1, OAS1, IL1B, IFNB1, RNASEL, CASP8, HLA-DRB1, PELI1, SELENBP1, ARHGEF2, LRRFIP1, NAMPT, TRAIP, RIPK3, SEC14L2, CSF1R, LAMP3, ERVW-1, FOXP3, ZMYND10, DDX56, CCR7, VCP, CDKN2A, IFIH1, DHX58, ZBP1, HAVCR2, PIK3IP1, NLRP3, TNFRSF13C, TRIM6, RBM45, CCR2, ERVK-20, ERVK-18, VAMP8, TNFRSF1A, IFNA1, TNF, IFNA13, HLA-DQA1, IL1A, HLA-C, IL10, IL17A, IL18, IRF3, IRF5, KIR2DL2, KIR3DL1, KIR3DS1, LSAMP, CD180, SMAD4, MMP9, HLA-A, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PZP, GLS, CASP1, SNCA, GEM, DDX3X, TAP1, TLR3, ATF4

-
Familial Gastric Type 1 Neuroendocrine Tumor
Orphanet
A rare neoplastic disease characterized by occurrence of atypical and aggressive gastric type 1 neuroendocrine tumors (NET) in early adulthood. The tumors often show nodal infiltration requiring total gastrectomy. ... Patients present high serum gastrin concentrations and iron-deficiency anemia (rather than megaloblastic anemia, which is a typical feature in patients with sporadic gastric type 1 NET, where the tumor usually arises on the background of autoimmune atrophic gastritis).
-
Uric Acid Concentration, Serum, Quantitative Trait Locus 1
OMIM
Evidence for both an increased rate of uric acid synthesis and an impaired net elimination of uric acid by the kidney has been advanced. ... Genomewide Association Studies Kottgen et al. (2013) reported the identification and replication of 28 genomewide-significant urate concentration-associated loci, 18 of which were novel, using genomewide association study (GWAS) (26 loci) and pathway (2 loci) approaches. ... Autosomal dominant form Lab - Increased rate of uric acid synthesis - Impaired net elimination of uric acid by the kidney - Hyperuricemia Skin - Urate tophi ▲ Close






