The diagnostic criteria were developed in pediatric populations and have not been validated for adult HLH patients. [20] Attempts to improve diagnosis of HLH have included use of the HScore , which can be used to estimate an individual's risk of HLH. [21] In adults, soluble IL-2 receptor has been found to be a very sensitive marker for HLH, demonstrating 100% sensitivity for ruling out HLH below a cutoff of 2400 U/mL and optimal cutoff for ruling in at 2515 U/mL (sensitivity, 100%; specificity, 72.5%), with 93% specificity at >10 000 U/mL. [22] Differential diagnosis [ edit ] The differential diagnosis of HLH includes secondary HLH and macrophage-activation syndrome or other primary immunodeficiencies that present with hemophagocytic lymphohistiocytosis, such as X-linked lymphoproliferative disease . [ citation needed ] Other conditions that may be confused with this condition include autoimmune lymphoproliferative syndrome . [23] As a syndrome of intense inflammation it needs to be differentiated from sepsis , what may be extremely challenging. [24] The diagnosis of acquired, or secondary, HLH is usually made in association with infection by viruses, bacteria, fungi, or parasites or in association with lymphoma, autoimmune disease, or metabolic disease.
Hemophagocytic syndrome (HPS) is a rare immune disease (see this term) and a potentially life-threatening disorder characterized by cytokine storm and overwhelming inflammation causing fever, hepatosplenomegaly, cytopenia, hypertriglyceridemia, hyperferritinemia, and hemophagocytosis in bone marrow, liver, spleen or lymph nodes. It can be either primary due to a genetic defect (primary hemophagocytic lymphohistiocytosis ; see this term), or secondary to malignancies, to infections, most commonly with viruses such as Epstein-Barr virus or cytomegalovirus, human immunodeficiency virus, or to autoimmune disorders such as systemic lupus erythematosus or adult-onset Still disease (secondary hemophagocytic lymphohistiocytosis) (see these termes).
Motor symptoms caused by dysfunction of the cerebellum Cerebellar ataxia Specialty Neurology Cerebellar ataxia is a form of ataxia originating in the cerebellum . [1] Non-progressive congenital ataxia (NPCA) is a classical presentation of cerebral ataxias. Cerebellar ataxia can occur as a result of many diseases and may present with symptoms of an inability to coordinate balance, gait, extremity and eye movements. [2] Lesions to the cerebellum can cause dyssynergia , dysmetria , dysdiadochokinesia , dysarthria and ataxia of stance and gait. [3] Deficits are observed with movements on the same side of the body as the lesion (ipsilateral). [2] Clinicians often use visual observation of people performing motor tasks in order to look for signs of ataxia. [2] Contents 1 Signs and symptoms 2 Causes 3 Treatment 3.1 Behavioral intervention 4 See also 5 References 6 External links Signs and symptoms [ edit ] Damage to the cerebellum causes impairment in motor skills and can cause nystagmus . Almost a third of people with isolated, late onset cerebellar ataxia go on to develop multiple system atrophy . [4] The cerebellum's role has been observed as not purely motor. It is combined with intellect, emotion and planning. [5] Causes [ edit ] Play media A male with gluten ataxia: previous situation and evolution after 3 months of gluten-free diet. There are many causes of cerebellar ataxia including, among others, gluten ataxia , [6] autoimmunity to Purkinje cells or other neural cells in the cerebellum, [7] CNS vasculitis , multiple sclerosis , infection, bleeding, infarction, tumors, direct injury, toxins (e.g., alcohol), genetic disorders and neurodegenerative diseases (such as progressive supranuclear palsy and multiple system atrophy ).
A number sign (#) is used with this entry because of evidence that Parkinson disease-17 (PARK17) is caused by heterozygous mutation in the VPS35 gene (601501) on chromosome 16q11. Description Parkinson disease-17 is an autosomal dominant, adult-onset form of the disorder. It is phenotypically similar to idiopathic Parkinson disease (summary by Wider et al., 2008). For a general phenotypic description and a discussion of genetic heterogeneity of Parkinson disease (PD), see 168600. Clinical Features Wider et al. (2008) reported a large family of Swiss origin with autosomal dominant levodopa-responsive Parkinson disease.
To test the validity of this generalization, Yamamoto et al. (2000) analyzed the data of serum and urinary calcium collected from 85 patients with idiopathic hypoparathyroidism and 15 with activating CASR mutations.
A number sign (#) is used with this entry because of evidence that autosomal dominant hypocalcemia-2 (HYPOC2) is caused by heterozygous mutation in the GNA11 gene (139313) on chromosome 19p13. For a discussion of genetic heterogeneity of autosomal dominant hypocalcemia, see HYPOC1 (601198). Clinical Features Hunter et al. (1981) reported a large 4-generation family of English Canadian origin segregating what they designated 'autosomal dominant hypoparathyroidism.' There were 8 affected members of the family, 2 of whom were asymptomatic. The proband was evaluated at 6 years old for 'febrile convulsions' and found to have positive Trousseau and Chvostek signs, possible laryngospasm, and low serum calcium (6.3 mg/dl) and phosphorus (7.8 mg/dl) levels, with a parathyroid hormone (PTH) level in the low-normal range (177 pg Eq/ml).
A number sign (#) is used with this entry because of evidence that X-linked hypoparathyroidism (HYPX) is caused by an interstitial deletion/insertion on chromosome Xq27.1, which may have a position effect on expression of SOX3 (313430). Clinical Features Peden (1960) reported a family in which multiple males had neonatal idiopathic hypoparathyroidism in an X-linked pattern of inheritance. No affected males reproduced. Peden (1960) suggested that most familial cases with early onset are of the X-linked type, the autosomal type (146200) having a later onset. Whyte and Weldon (1981) performed extensive studies of a second kindred from Missouri (where Peden's family also lived) with neonatal or infantile onset of X-linked isolated hypoparathyroidism. No ancestor common to the 2 kindreds could be identified. Whyte et al. (1986) reported the autopsy findings in a member of the family of Peden (1960) who had died as a teenager after an automobile crash.
A number sign (#) is used with this entry because familial isolated hypoparathyroidism can be caused by mutation in the parathyroid hormone gene (PTH; 168450), or in the GCM2 gene (603716), a homolog of the Drosophila glial cells missing gene. There is also an X-linked form of hypoparathyroidism (307700). Description Garfield and Karaplis (2001) reviewed the various causes and clinical forms of hypoparathyroidism. They noted that hypoparathyroidism is a clinical disorder characterized by hypocalcemia and hyperphosphatemia. It manifests when parathyroid hormone (PTH; 168450) secreted from the parathyroid glands is insufficient to maintain normal extracellular fluid calcium concentrations or, less commonly, when PTH is unable to function optimally in target tissues, despite adequate circulating levels. Congenital absence of the parathyroid and thymus glands (III and IV pharyngeal pouch syndrome, or DiGeorge syndrome, 188400) is usually a sporadic condition (Taitz et al., 1966).
Familial isolated hypoparathyroidism (FIH) is a rare heterogeneous group of metabolic disorders characterized by abnormal calcium metabolism due to deficient secretion of parathormone (PTH), without other endocrine disorders or developmental defects. Clinical description It can occur at any age (from the newborn period to adulthood) but generally starts within the first decade of life. Diagnosis is made when hypocalcemia, hyperphosphoremia, and low or undetectable PTH levels are observed. The clinical signs are mainly those of hypocalcemia: myopathy, muscular weakness, cramps, tetany, lenticular cataracts, teeth anomalies and short stature. Etiology FIH may be due to an activating mutation of the calcium-sensing receptor ( CASR ) gene.
There are 2 classic types of the disorder. In type I, representing 85% of patients, serum levels of C1NH are less than 35% of normal (Cicardi and Agostoni, 1996; Bowen et al., 2001).
Although somatotropinomas predominated among FIPA families with AIP mutations, mixed GH/prolactin-secreting tumors, prolactinomas, and nonsecreting adenomas were also found. Approximately 85% of the FIPA cohort and 50% of those with familial somatotropinomas were negative for AIP mutations.
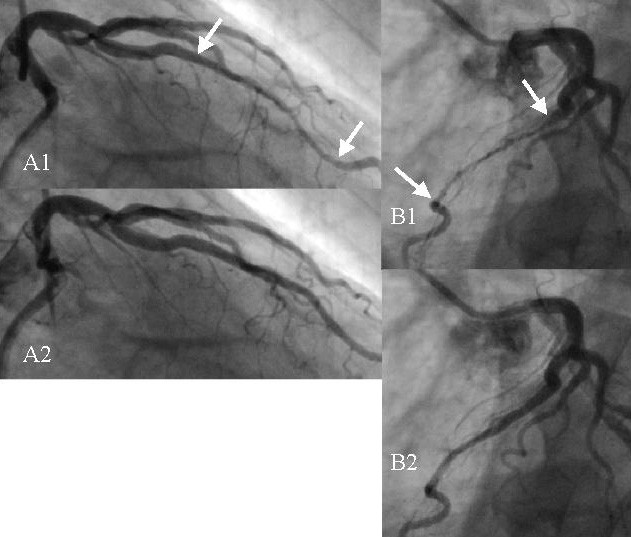
This is because arteries are sturdy and pliable, so after being compressed they are very slow to reopen, remaining in some level of semi-compression for most if not all of the diastolic period i.e. the other 85% of the heartbeat cycle (hence the critical need for dFFR testing in diagnosing myocardial bridges).
Balint syndrome is a rare neurologic disease characterized by the triad of optic ataxia, ocular apraxia and simultanagnosia due to posterior parietal lobe lesions. Patients report ophthalmologic difficulties in the absence of underlying ophthalomologic anomalies and present severe visual and spatial disabilities in locating and reaching objects, initiating voluntary eye movements and perceiving more than one object at a time.
Of the 94 maltreated toddlers in foster care, 35% were identified as having ICD RAD and 22% as having ICD DAD, and 38% fulfilled the DSM criteria for RAD. [34] This study found that RAD could be reliably identified and also that the inhibited and disinhibited forms were not independent. ... Attachment theory focuses on the tendency of infants or children to seek proximity to a particular attachment figure (familiar caregiver), in situations of alarm or distress, behavior which appears to have survival value. [90] This is known as a discriminatory or selective attachment. ... Infants become attached to adults who are sensitive and responsive in social interactions with the infant, and who remain as consistent caregivers for some time. [91] Caregiver responses lead to the development of patterns of attachment, that in turn lead to internal working models which will guide the individual's feelings, thoughts, and expectations in later relationships. [92] [93] For a diagnosis of reactive attachment disorder, the child's history and atypical social behavior must suggest the absence of formation of a discriminatory or selective attachment. ... ISBN 978-0-7817-5033-2 . ^ a b Chaffin et al. (2006), p. 80. The APSAC Taskforce Report ^ Rutter M (2002). ... Archived 3 February 2008 at the Wayback Machine American Academy of Child & Adolescent Psychiatry, Facts for Families, No. 85; Updated December 2002. Retrieved on 13 February 2008. ^ For examples see Reactive Attachment Disorder Archived 28 December 2007 at the Wayback Machine , DCFS, State of Illinois and DBHS Practice Protocol: Disturbances and Disorders of Attachment (PDF), Arizona Department of Health Services, 2 October 2006.
Overview Reactive attachment disorder is a rare but serious condition in which an infant or young child doesn't establish healthy attachments with parents or caregivers. Reactive attachment disorder may develop if the child's basic needs for comfort, affection and nurturing aren't met and loving, caring, stable attachments with others are not established. With appropriate treatment, children who have reactive attachment disorder may develop more stable and healthy relationships with caregivers and others. Treatments for reactive attachment disorder include learning how to create a stable, nurturing environment and providing positive child and caregiver interactions. Parent or caregiver counseling and education can help. Symptoms Reactive attachment disorder usually starts in infancy.
Westport, CT: Praeger ISBN 0-275-97675-0 , pp. 98–103. ^ Mercer (2006), pp. 64–70. ^ Marshall, P.J.; Fox, N.A. (2005). ... ISBN 978-1-59385-470-6 . ^ Prior & Glaser (2006) p186-187 ^ Chaffin (2006) p 82 ^ Prior & Glaser (2006) p 262 ^ Chaffin et al. 2006, p. 79–80. The APSAC Taskforce Report. ^ Chaffin et al. (2006) p 79 ^ Boris 2003 ^ Mercer, Sarner & Rosa 2003 ^ Zeanah 2003 ^ Chaffin et al. (2006) ^ "ATTACh White paper on coercion" (PDF) .
"Munchausen by Internet: detecting factitious illness and crisis on the Internet". South. Med. J . 93 (7): 669–72. doi : 10.1097/00007611-200093070-00006 .
Suggestive Findings TRIO -related ID should be considered in individuals with the following clinical findings: Delayed speech and fine/gross motor development AND/OR mild (IQ 50-70) to borderline (IQ 70-85) ID; AND One or more of the following: Microcephaly; occipital-frontal circumference <2 SD Minor hand anomalies including short tapering fingers / brachydactyly, broad proximal interphalangeal joints, and clinodactyly of the fifth finger Dental anomalies including dental crowding and delayed or failed tooth eruption Facial features including facial asymmetry and/or micrognathia Other, less specific features: Neonatal feeding problems that may persist into infancy Behavioral problems including autistic traits or autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD), and/or aggression Spinal deformities including scoliosis and/or kyphosis Establishing the Diagnosis The diagnosis of TRIO -related ID is established in a proband by identification of a heterozygous pathogenic variant in TRIO on molecular genetic testing (see Table 1).
Penetrance was age-dependent and incomplete even in older mutation carriers (85% after 40 years). Six percent of 238 mutation carriers were asymptomatic, while 20% of carriers were unaware of their symptoms.
Spastic paraplegia type 4 (also known as SPG4) is the most common of a group of genetic disorders known as hereditary spastic paraplegias. These disorders are characterized by progressive muscle stiffness (spasticity) in the legs and difficulty walking. Hereditary spastic paraplegias are divided into two types: pure and complex. The pure types generally involve only spasticity of the lower limbs and walking difficulties. The complex types involve more widespread problems with the nervous system; the structure or functioning of the brain; and the nerves connecting the brain and spinal cord to muscles and sensory cells that detect sensations such as touch, pain, heat, and sound (the peripheral nervous system).
Spastic paraplegia 4 (SPG4) is the most common type of hereditary spastic paraplegia (HSP) inherited in an autosomal dominant manner. Disease onset ranges from infancy to older adulthood. SPG4 is characterized by slowly progressive muscle weakness and spasticity (stiff or rigid muscles) in the lower half of the body. In rare cases, individuals may have a more complex form with seizures, ataxia, and dementia. SPG4 is caused by mutations in the SPAST gene. Severity of symptoms usually worsens over time, however some individuals remain mildly affected throughout their lives. Medications, such as antispastic drugs and physical therapy may aid in stretching spastic muscles and preventing contractures (fixed tightening of muscles).
A rare form of hereditary spastic paraplegia with high intrafamilial clinical variability, characterized in most cases as a pure phenotype with an adult onset (mainly the 3rd to 5th decade of life, but that can present at any age) of progressive gait impairment due to bilateral lower-limb spasticity and weakness as well as very mild proximal weakness and urinary urgency. In some cases, a complex phenotype is also reported with additional manifestations including cognitive impairment, cerebellar ataxia, epilepsy and neuropathy. A faster disease progression is noted in patients with a later age of onset.
Viral hepatitides such as Hepatitis B virus and Hepatitis C virus can be vertically transmitted during birth via contact with infected blood. [34] [35] According to a 2012 NICE publication, "about 85% of hepatitis B infections in newborns become chronic". [36] In occult cases, Hepatitis B virus is present by HBV DNA , but testing for HBsAg is negative. [37] High consumption of alcohol can lead to several forms of liver disease including alcoholic hepatitis , alcoholic fatty liver disease , cirrhosis , and liver cancer . [38] In the earlier stages of alcoholic liver disease, fat builds up in the liver's cells due to increased creation of triglycerides and fatty acids and a decreased ability to break down fatty acids . [39] Progression of the disease can lead to liver inflammation from the excess fat in the liver .
Overview The liver is an organ about the size of a football. It sits just under your rib cage on the right side of your abdomen. The liver is essential for digesting food and ridding your body of toxic substances. Liver disease can be inherited (genetic). Liver problems can also be caused by a variety of factors that damage the liver, such as viruses, alcohol use and obesity. Over time, conditions that damage the liver can lead to scarring (cirrhosis), which can lead to liver failure, a life-threatening condition. But early treatment may give the liver time to heal. Symptoms Liver disease doesn't always cause noticeable signs and symptoms.
Revue Internationale de Recherches de Réadaptation . 26 (2): 93–9. doi : 10.1097/00004356-200306000-00003 . ... S2CID 23055506 . ^ Price CI, Pandyan AD (February 2001). "Electrical stimulation for preventing and treating post-stroke shoulder pain: a systematic Cochrane review" .
Paresis Specialty Neurology Symptoms Loss of motor skills Causes Stroke This article is about the physical malady. For the mental disorder, see general paresis of the insane . Not to be confused with Paruresis or Parecis . Look up paresis in Wiktionary, the free dictionary. In medicine, paresis ( / p ə ˈ r iː s ɪ s , ˈ p æ r ə s ɪ s / ) is a condition typified by a weakness of voluntary movement, or by partial loss of voluntary movement or by impaired movement. When used without qualifiers, it usually refers to the limbs, but it can also be used to describe the muscles of the eyes ( ophthalmoparesis ), the stomach ( gastroparesis ), and also the vocal cords ( Vocal cord paresis ). Neurologists use the term paresis to describe weakness, and plegia to describe paralysis in which all voluntary movement is lost.
Annals of the Royal College of Surgeons of England . 93 (4): 331. doi : 10.1308/rcsann.2011.93.4.331a . ... British Dental Journal . 195 (9): 489–93. doi : 10.1038/sj.bdj.4810659 . PMID 14610554 . ^ Peterson's principles of oral and maxillofacial surgery .
"HLA class II DPB1 and DRB1 polymorphisms associated with genetic susceptibility to beryllium toxicity". Occup Environ Med . 68 (7): 487–93. doi : 10.1136/oem.2010.055046 . PMID 21186201 . ^ "IARC Monograph, Volume 58" .