"Diagnosis and management of urinary infections in older people" . Clinical Medicine . 11 (1): 80–3. doi : 10.7861/clinmedicine.11-1-80 . ... The Journal of Pediatrics . 212 : 102–110.e5. doi : 10.1016/j.jpeds.2019.04.053 . ... Current Microbiology . 63 (5): 484–90. doi : 10.1007/s00284-011-0006-2 . ... Archives of Internal Medicine . 172 (13): 988–96. doi : 10.1001/archinternmed.2012.3004 . ... Nature Reviews. Urology (Review). 12 (2): 81–90. doi : 10.1038/nrurol.2014.361 .
Although it is very uncommon in the United States, more than 80% of induced abortions throughout the second trimester are labor-induced abortions in Sweden and other nearby countries. [74] Only limited data are available comparing this method with dilation and extraction. [74] Unlike D&E, labor-induced abortions after 18 weeks may be complicated by the occurrence of brief fetal survival, which may be legally characterized as live birth. ... The degree of force, if severe, can cause serious internal injuries without necessarily succeeding in inducing miscarriage . [79] In Southeast Asia, there is an ancient tradition of attempting abortion through forceful abdominal massage. [80] One of the bas reliefs decorating the temple of Angkor Wat in Cambodia depicts a demon performing such an abortion upon a woman who has been sent to the underworld . [80] Reported methods of unsafe, self-induced abortion include misuse of misoprostol and insertion of non-surgical implements such as knitting needles and clothes hangers into the uterus. ... Such methods are rarely used in countries where surgical abortion is legal and available. [81] Safety A likely illegal abortion flyer in South Africa The health risks of abortion depend principally upon whether the procedure is performed safely or unsafely. ... A 2007 study reported that "26% of all pregnancies worldwide are terminated by induced abortion," whereas "deaths from improperly performed [abortion] procedures constitute 13% of maternal mortality globally." [87] In Indonesia in 2000 it was estimated that 2 million pregnancies ended in abortion, 4.5 million pregnancies were carried to term, and 14-16 percent of maternal deaths resulted from abortion. [88] In the US from 2000 to 2009, abortion had a lower mortality rate than plastic surgery , and a similar or lower mortality rate than running a marathon. [89] Five years after seeking abortion services, women who gave birth after being denied an abortion reported worse health than women who had either first or second trimester abortions. [90] The risk of abortion-related mortality increases with gestational age, but remains lower than that of childbirth. [91] Outpatient abortion is as safe from 64 to 70 days' gestation as it before 63 days. [92] There is little difference in terms of safety and efficacy between medical abortion using a combined regimen of mifepristone and misoprostol and surgical abortion (vacuum aspiration) in early first trimester abortions up to 10 weeks gestation. [57] Medical abortion using the prostaglandin analog misoprostol alone is less effective and more painful than medical abortion using a combined regimen of mifepristone and misoprostol or surgical abortion. [93] [94] Vacuum aspiration in the first trimester is the safest method of surgical abortion, and can be performed in a primary care office , abortion clinic , or hospital. ... "Vaginal delivery may result in dissemination of neoplastic cells into lymphovascular channels, haemorrhage, cervical laceration and implantation of malignant cells in the episiotomy site, while abdominal delivery may delay the initiation of non-surgical treatment." [167] History and religion Main articles: History of abortion and Religion and abortion Bas-relief at Angkor Wat , Cambodia , c. 1150, depicting a demon inducing an abortion by pounding the abdomen of a pregnant woman with a pestle . [80] [168] "French Periodical Pills".
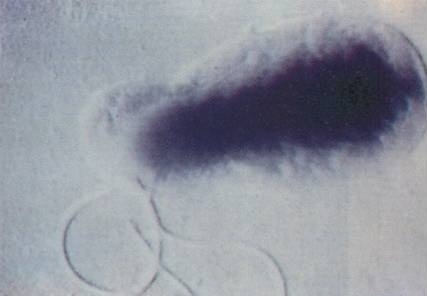
Bacillary angiomatosis Bartonella (bacterial species which causes this condition) Specialty Infectious disease Bacillary angiomatosis ( BA ) is a form of angiomatosis associated with bacteria of the genus Bartonella . [1] Contents 1 Symptoms 1.1 Presentation 2 Causes 3 Diagnosis 4 Treatment and prevention 5 History 6 See also 7 References 8 External links Symptoms [ edit ] Cutaneous BA is characterised by the presence of lesions on or under the skin. Appearing in numbers from one to hundreds, these lesions may take several forms: [ citation needed ] papules or nodules which are red, globular and non-blanching, with a vascular appearance purplish nodules sufficiently similar to Kaposi's sarcoma that a biopsy may be required to verify which of the two it is a purplish lichenoid plaque a subcutaneous nodule which may have ulceration, similar to a bacterial abscess While cutaneous BA is the most common form, it can also affect several other parts of the body, such as the brain, bone, bone marrow, lymph nodes, gastrointestinal tract, respiratory tract, spleen, and liver. Symptoms vary depending on which parts of the body are affected; for example, those whose livers are affected may have an enlarged liver and fever , while those with osseous BA experience intense pain in the affected area. [ citation needed ] Presentation [ edit ] BA is characterised by the proliferation of blood vessels, resulting in them forming tumour-like masses in the skin and other organs. [ citation needed ] Causes [ edit ] It is caused by either Bartonella henselae or B. quintana . [2] B. henselae is most often transmitted through a cat scratch or bite, [3] though ticks and fleas may also act as vectors. ... Neutrophilic infiltrate. [1] Treatment and prevention [ edit ] While curable, BA is potentially fatal if not treated. [1] BA responds dramatically to several antibiotics. ... "An atypical subcutaneous infection associated with acquired immune deficiency syndrome". Am J Clin Pathol . 80 (5): 714–8. doi : 10.1093/ajcp/80.5.714 .
"Flail Chest in a Neonate Resulting from Nonaccidental Trauma". Southern Medical Journal . 99 (5): 536–538. doi : 10.1097/01.smj.0000216471.54786.e5 . ^ Granetzny A; Abd El-Aal, M; Emam, E; Shalaby, A; Boseila, A (2005).
More than 30% of cases of AF are considered to be 'lone AF,' unassociated with coronary artery disease, hypertension, valvular heart disease, hyperthyroidism, heart failure, or structural heart disease (summary by Wang et al., 2010). ... At 39 years of age, the first patient had onset of AF that eventually became persistent AF. ... Flecainide testing did not induce a Brugada-like ECG pattern. The second patient had onset of AF at 35 years of age, and had persistent AF. There was no family history of AF, but his mother had premature atrial complexes on ECG. This patient did not consent to a flecainide test. The third patient had onset of AF at 36 years of age, and had paroxysmal AF.
Penetrance Among individuals with an SCN5A pathogenic variant: Approximately 20%-30% have an ECG diagnostic of Brugada syndrome; Approximately 80% manifest the characteristic ECG changes when challenged with a sodium channel blocker (ajmaline) [Hong et al 2004b, Benito et al 2009].
A number sign (#) is used with this entry because of evidence that Brugada syndrome-6 (BRGDA6) is caused by heterozygous mutation in the KCNE3 gene (604433) on chromosome 11q13. Description Brugada syndrome is characterized by an ST segment elevation in the right precordial electrocardiogram leads (so-called type 1 ECG) and a high incidence of sudden death in patients with structurally normal hearts. The syndrome typically manifests during adulthood, with a mean age of sudden death of 41 +/- 15 years, but also occurs in infants and children (summary by Antzelevitch et al., 2005). For a general phenotypic description and discussion of genetic heterogeneity of Brugada syndrome, see BRGDA1 (601144). Clinical Features Delpon et al. (2008) studied a Danish pedigree in which the previously asymptomatic proband had cardiac arrest at age 36 years and was resuscitated.
A number sign (#) is used with this entry because of evidence that Brugada syndrome-9 (BRGDA9) is caused by heterozygous mutation in the KCND3 gene (605411) on chromosome 1p13. Description Brugada syndrome is characterized by ST segment elevation in the right precordial electrocardiogram leads (so-called type 1 ECG) and a high incidence of sudden death in patients with structurally normal hearts. The syndrome typically manifests during adulthood, with a mean age of sudden death of 41 +/- 15 years, but also occurs in infants and children (summary by Antzelevitch et al., 2005). For a discussion of genetic heterogeneity of Brugada syndrome, see BRGDA1 (601144). Clinical Features Giudicessi et al. (2011) reported 2 unrelated patients with Brugada syndrome and mutations in the KCND3 gene (see MOLECULAR GENETICS).
A number sign (#) is used with this entry because of evidence that Brugada syndrome-4 (BRGDA4) is caused by heterozygous mutation in the gene encoding the beta-2 subunit of the voltage-dependent L-type calcium channel (CACNB2; 600003) on chromosome 10p12. Description Brugada syndrome is characterized by an ST segment elevation in the right precordial electrocardiogram leads (so-called type 1 ECG) and a high incidence of sudden death in patients with structurally normal hearts. The syndrome typically manifests during adulthood, with a mean age of sudden death of 41 +/- 15 years, but also occurs in infants and children (summary by Antzelevitch et al., 2005). For a discussion of the genetic heterogeneity in Brugada syndrome, see BRGDA1 (601144). Clinical Features Antzelevitch et al. (2007) described a 25-year-old white male of European descent who presented with aborted sudden cardiac death and had a QTc of 330 ms on ECG, with coved-type ST segment elevation in V1 and V2 after ajmaline challenge.
A number sign (#) is used with this entry because of evidence that Brugada syndrome-8 (BRGDA8) is caused by heterozygous mutation in the HCN4 gene (605206) on chromosome 15q24. Description Brugada syndrome is characterized by an ST segment elevation in the right precordial electrocardiogram leads (so-called type 1 ECG) and a high incidence of sudden death in patients with structurally normal hearts. The syndrome typically manifests during adulthood, with a mean age of sudden death of 41 +/- 15 years, but also occurs in infants and children (summary by Antzelevitch et al., 2005). For a discussion of genetic heterogeneity of Brugada syndrome, see BRGDA1 (601144). Clinical Features Ueda et al. (2009) studied a 41-year-old man who had recurrent episodes of syncope at rest and whose electrocardiogram (ECG) showed saddleback ST segment elevation in leads V1 and V2, incomplete right bundle branch block, and QT intervals at the upper limit of normal.
Overview Brugada (brew-GAH-dah) syndrome is a rare but potentially life-threatening heart rhythm condition (arrhythmia) that is sometimes inherited. People with Brugada syndrome have an increased risk of irregular heart rhythms beginning in the lower chambers of the heart (ventricles). Treatment of Brugada syndrome includes preventive measures such as reducing fever and avoiding medications that might trigger the arrhythmia. Some people with Brugada syndrome need a medical device called an implantable cardioverter-defibrillator (ICD). Symptoms Brugada syndrome often doesn't cause any noticeable symptoms.
A number sign (#) is used with this entry because of evidence that Brugada syndrome-2 (BRGDA2) is caused by heterozygous mutation in the GPD1L gene (611778) on chromosome 3p22. Description Brugada syndrome is characterized by an ST segment elevation in the right precordial electrocardiogram leads (so-called type 1 ECG) and a high incidence of sudden death in patients with structurally normal hearts. The syndrome typically manifests during adulthood, with a mean age of sudden death of 41 +/- 15 years, but also occurs in infants and children (summary by Antzelevitch et al., 2005). For a discussion of genetic heterogeneity of Brugada syndrome, see BRGDA1 (601144). Clinical Features Weiss et al. (2002) reported a large multigenerational family with a progressive conduction disease consistent with Brugada syndrome in which they identified 12 affected individuals with an autosomal dominant inheritance pattern characterized by incomplete penetrance that appeared to be dependent on age and sex.
Brugada syndrome is a heart condition that causes a disruption of the normal rhythm in the heart's lower chambers (ventricular arrhythmia). Signs and symptoms usually develop in adulthood but the diagnosis may be made at any age. Symptoms and complications often occur during rest or sleep, and may include fainting, seizures, difficulty breathing, or sudden death. The underlying genetic cause of inherited forms of Brugada syndrome is not known in most cases, but in up to 20-30% of people with Brugada syndrome, it is caused by a mutation in the SCN5A gene. A number of other genes have been reported to be associated with Brugada syndrome in the literature, but the role they play in causing Brugada syndrome remains to be clearly defined.
A number sign (#) is used with this entry because of evidence that Brugada syndrome-3 (BRGDA3) is caused by heterozygous mutation in the gene encoding the alpha-1C subunit of the L-type voltage-dependent calcium channel (CACNA1C; 114205) on chromosome 12p13. Description Brugada syndrome is characterized by an ST segment elevation in the right precordial electrocardiogram leads (so-called type 1 ECG) and a high incidence of sudden death in patients with structurally normal hearts. The syndrome typically manifests during adulthood, with a mean age of sudden death of 41 +/- 15 years, but also occurs in infants and children (summary by Antzelevitch et al., 2005). For a discussion of the genetic heterogeneity of Brugada syndrome, see BRGDA1 (601144). Clinical Features Antzelevitch et al. (2007) reported 2 probands with Brugada syndrome who also had shortened QT intervals on ECG.
Brugada syndrome is a condition that causes a disruption of the heart's normal rhythm . Specifically, this disorder can lead to irregular heartbeats in the heart's lower chambers (ventricles), which is an abnormality called ventricular arrhythmia. If untreated, the irregular heartbeats can cause fainting (syncope), seizures, difficulty breathing, or sudden death. These complications typically occur when an affected person is resting or asleep. Brugada syndrome usually becomes apparent in adulthood, although it can develop any time throughout life.
A cardiac disorder characterized on electrocardiogram (ECG) by ST segment elevation with a coved aspect on the right precordial leads, and a clinical susceptibility to ventricular tachyarrhythmias and sudden death occurring in the absence of overt myocardial abnormalities. Epidemiology Given that the ECG pattern diagnostic for Brugada Syndrome is fluctuant, unlike other inherited arrhythmogenic syndromes, the data regarding the prevalence of the disease are controversial. According to a recent metanalysis, the worldwide prevalence of Brugada syndrome is estimated at 1/2,000, but it varies according to region and ethnicity. Brugada syndrome is rare in Hispanic and Caucasian populations and non-rare in Asian populations. By region, the prevalence is estimated at 1/20,000 in North America and 1/10,000 in Europe.
Crotti et al. (2012) analyzed 12 Brugada syndrome susceptibility genes in 129 unrelated patients with possible or probable Brugada syndrome and identified SCN5A mutations in 21 (16.3%) of the patients; only 6 (4.6%) of the patients carried a mutation in 1 of the other 11 genes. In a cohort of 91 SCN5A-negative Brugada syndrome patients and 91 European controls from the 1000 Genomes Project database, Di Resta et al. (2015) analyzed 158 arrhythmia- and cardiac defect-associated genes. A significant enrichment in Brugada syndrome samples was found only for the DSG2 gene (125671), with 6 (6%) of 91 patients having a rare functional variant compared to none of the 91 controls (p = 0.029). ... Alings and Wilde (1999) stated that only about 200 cases of Brugada syndrome had been reported. Over 90% of these cases had been in male patients, the mean age at first arrhythmic event ranging between 22 and 65 years. ... Veldkamp et al. (2003) concluded that sodium channel mutations displaying an I-pst or a negative shift in inactivation may account for the bradycardia seen in LQT3 patients, whereas SA node pauses or arrest may result from failure of SA node cells to repolarize under conditions of extra net inward current. Sudden unexplained nocturnal death syndrome (SUNDS), a disorder found in southeast Asia, is characterized by an abnormal electrocardiogram with ST segment elevation in leads V1 to V3 and sudden death due to ventricular fibrillation, identical to that seen in Brugada syndrome. ... The cumulative effect of the 3 loci on disease susceptibility was unexpectedly large (p trend = 6.1 x 10(-81)). Bezzina et al. (2013) concluded that the association signals at SCN5A-SCN10A demonstrated that genetic polymorphisms modulating cardiac conduction can also influence susceptibility to cardiac arrhythmia.
In Gussak I, Antzelevitch C, Wilde AA, Powell BD, Ackerman MJ, Shen WK (eds.). ... OCLC 841465583 . ^ a b c d e f g h i j k l Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. ... "High prevalence of concealed Brugada syndrome in patients with atrioventricular nodal reentrant tachycardia". Heart Rhythm . 12 (7): 1584–94. doi : 10.1016/j.hrthm.2015.03.015 . ... "Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men". Circulation . 96 (8): 2595–600. doi : 10.1161/01.CIR.96.8.2595 . PMID 9355899 . ^ Mizusawa Y, Wilde AA (June 2012). "Brugada syndrome" .
Most of the women who develop DCIS do not experience any symptoms. The majority of cases (80-85%) are detected through screening mammography. ... PMC 7251423 . PMID 32461218 . ^ a b Virnig BA, Shamliyan T, Tuttle TM, Kane RL, Wilt TJ (September 2009). ... The Cochrane Database of Systematic Reviews . 11 (11): CD000563. doi : 10.1002/14651858.CD000563.pub7 . PMID 24259251 . ^ Virnig, BA; Tuttle, TM; Shamliyan, T; Kane, RL (2010). ... National Institutes of Health. September 2009. ^ a b Virnig, BA; Shamliyan, T; Tuttle, TM; Kane, RL; Wilt, TJ (September 2009). ... They found women undergoing mastectomy were less likely than women undergoing lumpectomy plus radiation to experience local DCIS or invasive recurrence. Women undergoing BCS alone were also more likely to experience a local recurrence than women treated with mastectomy.
Overview Ductal carcinoma in situ (DCIS) is the presence of abnormal cells inside a milk duct in the breast. DCIS is considered the earliest form of breast cancer. DCIS is noninvasive, meaning it hasn't spread out of the milk duct and has a low risk of becoming invasive. DCIS is usually found during a mammogram done as part of breast cancer screening or to investigate a breast lump. While DCIS isn't an emergency, it does require an evaluation and a consideration of treatment options. Treatment may include breast-conserving surgery combined with radiation or surgery to remove all of the breast tissue.
One was a 61-year-old white man who had paroxysmal atrial fibrillation (AF) and hypertension. He was diagnosed with AF at 55 years of age, and electrocardiogram (ECG) showed saddleback-type ST segment elevation in the right precordial leads during sinus rhythm, with a prolonged PR interval of 220 ms. ... Echocardiography was normal. He had no family history of AF. The other patient was a 57-year-old white man with paroxysmal lone AF, who had saddleback-type ST segment elevation in the right precordial leads and slight left atrial enlargement on echocardiography. Both his father and mother had AF, as well as coronary artery disease. Molecular Genetics Watanabe et al. (2009) screened the 4 genes encoding sodium channel beta subunits, SCN1B (600235), SCN2B, SCN3B (608214), and SCN4B (608256), in 480 patients with atrial fibrillation, including 118 patients with lone AF and 362 patients with AF and other cardiovascular disease. They identified 2 unrelated male patients, 1 with AF and hypertension and 1 with lone AF, who had heterozygous missense mutations in the SCN2B gene, R28W (601327.0001) and R28Q (601327.0002), respectively.
This theory helps to explain why negative life incidents precede depression in around 80 percent of cases, [71] [72] and why they so often strike people during their peak reproductive years. ... "Melancholia: A Historical Review" . Journal of Mental Science . 80 (328): 1–42. doi : 10.1192/bjp.80.328.1 . ... Archives of General Psychiatry . 41 (1): 72–80. doi : 10.1001/archpsyc.1984.01790120076010 . ... "Benzodiazepines: How They Work and How to Withdraw" . ^ Lydiard RB, Laraia MT, Ballenger JC, Howell EF (May 1987). "Emergence of depressive symptoms in patients receiving alprazolam for panic disorder". ... J Psychosom Res . 38 (Suppl 1): 113–23, discussion 118–23. doi : 10.1016/0022-3999(94)90142-2 . PMID 7799243 . ^ Ashton CH (March 1995).
Overview If you have a mood disorder, your general emotional state or mood is distorted or inconsistent with your circumstances and interferes with your ability to function. You may be extremely sad, empty or irritable (depressed), or you may have periods of depression alternating with being excessively happy (mania). Anxiety disorders can also affect your mood and often occur along with depression. Mood disorders may increase your risk of suicide. Some examples of mood disorders include: Major depressive disorder — prolonged and persistent periods of extreme sadness Bipolar disorder — also called manic depression or bipolar affective disorder, depression that includes alternating times of depression and mania Seasonal affective disorder (SAD) — a form of depression most often associated with fewer hours of daylight in the far northern and southern latitudes from late fall to early spring Cyclothymic disorder — a disorder that causes emotional ups and downs that are less extreme than bipolar disorder Premenstrual dysphoric disorder — mood changes and irritability that occur during the premenstrual phase of a woman's cycle and go away with the onset of menses Persistent depressive disorder (dysthymia) — a long-term (chronic) form of depression Disruptive mood dysregulation disorder — a disorder of chronic, severe and persistent irritability in children that often includes frequent temper outbursts that are inconsistent with the child's developmental age Depression related to medical illness — a persistent depressed mood and a significant loss of pleasure in most or all activities that's directly related to the physical effects of another medical condition Depression induced by substance use or medication — depression symptoms that develop during or soon after substance use or withdrawal or after exposure to a medication For most people, mood disorders can be successfully treated with medications and talk therapy (psychotherapy). When to see a doctor If you're concerned that you may have a mood disorder, make an appointment to see your doctor or a mental health professional as soon as you can.
George Washington was believed to have died of complications arising from quinsy, but is now thought to have died from epiglottitis . [22] James Gregory of the band The Ordinary Boys almost died from quinsy because it was left untreated for so long before emergency treatment was started. [23] Eiichiro Oda , author of the best-selling One Piece manga , was hospitalized due to complications. [24] Ian Maclaren died of complications from quinsy while on a lecture tour of the United States. [25] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae Galioto NJ (April 2017). ... S2CID 23505346 . ^ Sakae FA, Imamura R, Sennes LU, Araújo Filho BC, Tsuji DH (2006). "Microbiology of peritonsillar abscesses" . ... The Journal of Laryngology and Otology . 124 (4): 420–3. doi : 10.1017/S0022215109991939 . PMID 19930783 . ^ Chang BA, Thamboo A, Burton MJ, Diamond C, Nunez DA, et al. ... PMID 15908813 . S2CID 38122236 . ^ Hanna BC, McMullan R, Gallagher G, Hedderwick S (April 2006). ... Archived from the original on 2016-09-25. ^ Juvaini AA (1997). History of the World Conqueror .
A rare hemorrhagic disorder due to an acquired platelet anomaly characterized by hemolysis, elevated liver enzymes and thrombocytopenia that affects pregnant or post-partum women, and is frequently associated with severe preeclampsia. Symptoms are variable, typically including right upper quadrant or epigastric abdominal pain, nausea, vomiting, excessive weight gain, generalized edema, hypertension, general malaise, right shoulder pain, backache, and/or headache. Hepatic hemorrhage and rupture, renal failure, and pulmonary edema can result in maternal and/or fetal death.
HELLP syndrome is a life-threatening condition that can potentially complicate pregnancy. It is named for 3 features of the condition: H emolysis, E levated L iver enzyme levels, and L ow P latelet levels. It typically occurs in the last 3 months of pregnancy (the third trimester) but can also start soon after delivery. A wide range of non-specific symptoms may be present in women with HELLP syndrome. Symptoms may include fatigue; malaise; fluid retention and excess weight gain; headache; nausea and vomiting; pain in the upper right or middle of the abdomen; blurry vision; and rarely, nosebleed or seizures.
PMC 3288152 . PMID 21924324 . ^ Gazdar AF, Oie HK, Shackleton CH, Chen TR, Triche TJ, Myers CE, Chrousos GP, Brennan MF, Stein CA, La Rocca RV (September 1990). "Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis" . Cancer Research . 50 (17): 5488–96. PMID 2386954 . ^ a b Cote R, Suster S, Weiss L (2002). ... ISBN 0-07-139140-1 ^ Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. (May 2016). "Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma" . ... The New England Journal of Medicine . 356 (23): 2372–80. doi : 10.1056/NEJMoa063360 . hdl : 2318/37317 . ... The Journal of Clinical Endocrinology and Metabolism . 91 (6): 2027–37. doi : 10.1210/jc.2005-2639 .
Molecular Genetics Varley (2003) found that over 80% of a cohort of children with ADCC unselected for family history had a germline TP53 (191170) mutation; in addition, all 12 LFS or LFS-like families that they studied in which there was a case of ADCC had a germline TP53 mutation.
Adrenocortical carcinoma is a rare cancer affecting the outside of the adrenal glands ( adrenal cortex ). These glands are on top of each kidney and are responsible for producing certain hormones and keeping blood pressure at normal levels. Adrenocortical carcinoma is relatively frequent in children compared to many other cancers, although the cancer may also affect adults. Girls are more often affected than boys. Symptoms of adrenocortical carcinoma may include pain in the abdomen, hypertension, weight gain, frequent urination and possibly deepening of the voice. These symptoms are due to the tumors causing excess secretion of hormones from the adrenal glands.
Family evaluation was negative except for a great-grandparent who developed AF at an advanced age, and thus it was not considered to represent familial AF. Villafane et al. (2014) reported 2 girls with short QT intervals, AF, and bradycardia. The first was a 6.5-year-old girl who presented at birth with AF and a ventricular rate of 60 bpm, who was diagnosed with SQT at 4 months of age (QTc, 292 ms). ... Holter monitoring at 11 months of age showed AF with a slow ventricular response at 35 to 70 bpm. AF was refractory to DC cardioversion and multiple antiarrhythmic agents in both patients. ... In 2 unrelated girls with short QT intervals, AF, and bradycardia, Villafane et al. (2014) identified heterozygosity for the V141M mutation in the KCNQ1 gene.
CS1 maint: multiple names: authors list ( link ) ^ Vakis AF, Koutentakis DI, Karabetsos DA, Kalostos GN (2006). ... "Suprasellar arachnoid cysts: endoscopy versus microsurgical cyst excision and shunting". Br J Neurosurg . 21 (3): 276–80. doi : 10.1080/02688690701339197 . ... "Endoscopic management of intracranial cysts" . Neurosurg Focus . 19 (6): E7. doi : 10.3171/foc.2005.19.6.8 . PMID 16398484 . ^ Van Beijnum J, Hanlo PW, Han KS, Ludo Van der Pol W, Verdaasdonk RM, Van Nieuwenhuizen O (May 2006). ... "Arachnoid cysts: case series and review of the literature" . Neurosurg Focus . 22 (2): E7. doi : 10.3171/foc.2007.22.2.7 . PMID 17608350 . ^ Weber F, Knopf H (January 2006). "Incidental findings in magnetic resonance imaging of the brains of healthy young men". J. Neurol. Sci . 240 (1–2): 81–4. doi : 10.1016/j.jns.2005.09.008 .
Aarabi et al. (1979) reported father and daughter with surgically proven thoracic intradural arachnoid cysts. Two brothers of the father and another daughter were suspected by history to be affected also. One of these was deceased. The other two, with long-term, progressive, painless paraplegia, refused medical evaluation. The cysts were not associated with increased interpediculate distances or with distichiasis and lymphedema. The authors knew of no similar family. Spine - Thoracic intradural arachnoid cysts Neuro - Progressive, painless paraplegia Inheritance - Autosomal dominant ▲ Close
A disorder with extraparenchymal cysts, intra-arachnoidal collections of fluid, the composition of which is close to that of cerebrospinal fluid. They are often asymptomatic. Epidemiology Studies carried out since the advent of MRI and CT suggest that the prevalence is higher than previously thought, perhaps as high as 1 per 5000. In neurosurgery case series, cysts are more commonly located in the temporo-sylvian fossae. Temporal cysts are significantly more frequent in males than in females (4:1), while cysts in other locations do not show preponderance for a specific gender. A significant predominance of left-sided temporal cysts is found in males (2:1).
Description Arachnoid cysts are extraparenchymal, nonneoplastic accumulations of fluid with density similar to that of cerebrospinal fluid. They account for approximately 1% of intracranial space-occupying lesions, although studies since the advent of CT scanning suggest a higher percentage. A striking male preponderance has been observed (summary by Wilson et al., 1988). Clinical Features Handa et al. (1981) reported 2 brothers, aged 10 months and 3 years, with macrocrania and bilateral arachnoid cysts in the middle cranial fossa. They cited a Japanese report of 2 affected brothers. Pomeranz et al. (1991) described 2 brothers and a sister with intracranial arachnoid cysts.
Arachnoid cysts are sacs filled with cerebrospinal fluid (CSF) that are located between the brain or spinal cord and the arachnoid membrane, one of the three membranes that cover the brain and spinal cord. Arachnoid cysts can be primary or secondary. Primary arachnoid cysts are congenital (present at birth), resulting from abnormal development of the brain and spinal cord during early pregnancy. Secondary arachnoid cysts are less common, and result from head injuries, meningitis, tumors, or as a complication of brain surgery. Signs and symptoms depend on the location and size of the cyst and may include headache, nausea and vomiting, seizures, hearing and visual disturbances, vertigo , and difficulties with balance and walking. Although many affected individuals develop symptoms in the first year of life, some never develop symptoms.
The pyruvate dehydrogenase complex is made up of multiple copies of several enzymes called E1, E2, and E3, each of which performs part of the chemical reaction that converts pyruvate to acetyl-CoA. In addition, other proteins included in the complex ensure its proper function. One of these proteins, E3 binding protein, attaches E3 to the complex and provides the correct structure for the complex to perform its function. ... Mutations in the gene that provides instructions for making E1 alpha, the PDHA1 gene, are the most common cause of pyruvate dehydrogenase deficiency, accounting for approximately 80 percent of cases. These mutations lead to a shortage of E1 alpha protein or result in an abnormal protein that cannot function properly. ... Mutations in the genes that provide instructions for E1 beta (the PDHB gene), the E2 enzyme (the DLAT gene), E3 binding protein (the PDHX gene), and pyruvate dehydrogenase phosphatase (the PDP1 gene) have been identified in people with this condition.
They cannot, however, supply ATP to these cells and, therefore, phenotype depends largely on the nature/severity of the mutation. [5] [6] More rarely, mutations occur in the E2 ( dihydrolipoyl transacetylase ) or the E3 ( dihydrolipoyl dehydrogenase ) subunits of the PDC enzymatic complex, DLAT and DLD genes respectively. ... The DLAT gene is responsible for the coding of the E2 subunit, and the PDP1 is responsible for producing the PDH phosphatase catalytic subunit that catalyzes PDH dephosphorylation. ... The final gene that could be responsible for this disease is the PDHX gene, which codes for the E3 binding protein which is responsible for binding E3 dimers to the E2 subunit of the complex [8] Epidemiology [ edit ] Pyruvate dehydrogenase deficiency is extremely rare, with ~500 reported cases in the medical literature.
Overview Angelman syndrome is a genetic disorder. It causes delayed development, problems with speech and balance, intellectual disability, and, sometimes, seizures. People with Angelman syndrome often smile and laugh frequently, and have happy, excitable personalities. Developmental delays, which begin between about 6 and 12 months of age, are usually the first signs of Angelman syndrome. Seizures may begin between the ages of 2 and 3 years old. People with Angelman syndrome tend to live close to a normal life span, but the disorder can't be cured. Treatment focuses on managing medical, sleep and developmental issues.
Angelman syndrome is a genetic disorder that primarily affects the nervous system. Characteristic features of this condition include developmental delay, intellectual disability, severe speech impairment, problems with movement and balance (ataxia), epilepsy, and a small head size . Individuals with Angelman syndrome typically have a happy, excitable demeanor with frequent smiling, laughter, and hand-flapping movements. Many of the characteristic features of Angelman syndrome result from the loss of function of a gene called UBE3A . Most cases of Angelman syndrome are not inherited, although in rare cases a genetic change responsible for Angelman syndrome can be inherited from a parent.
Thirty-nine percent were hypopigmented compared to their family members. Frequent smiling was present in 96%. King et al. (1993) concluded from the study of 6 individuals with AS that hypopigmentation characterized by light skin, reduced retinal pigment, low hairbulb tyrosinase activity, and incomplete melanization of melanosomes is part of the phenotype of AS, and is similar to that found in Prader-Willi syndrome. ... Head circumference at birth was normal in all but skewed in distribution, with 62.5% at the 10th centile. Epilepsy was present in 96% with onset during the third year of life in 20 of 26 patients. ... Ametropia (refractive error) greater than 1 diopter (D) was present in 97% of cases: myopia in 9%, hyperopia in 76%, and astigmatism in 94%. Myopia and anisometropia (unequal refractive errors) were found only in the genetic deletion group. ... Pointing out that the diagnosis of Angelman syndrome can be confirmed by a genetic laboratory in only about 80% of cases, Williams et al. (2001) reviewed several mimicking conditions, including microdeletions or microduplications. ... Chan et al. (1993) presented a series of 93 Angelman syndrome patients, showing the relative contribution of the various genetic mechanisms. Sporadic cases accounted for 81 AS patients, while 12 cases came from 6 families.
A neurogenetic disorder characterized by severe intellectual deficit and distinct facial dysmorphic features. Epidemiology Prevalence of AS is estimated to be 1/10,000 to 1/20,000 worldwide. Clinical description Patients with AS appear normal at birth. In the first 6 months of the neonatal period, feeding difficulties and hypotonia may occur, followed by developmental delay between 6 months and 2 years of age. Generally from 1 year of age, the typical features of AS develop: severe intellectual deficit, absent speech, outbursts of laughter with hand flapping, microcephaly, macrostomia, maxillary hypoplasia, prognathia and neurological problems with a puppet-like gait, ataxia and epileptic seizures with specific electroencephalogram (EEG) abnormalities (triphasic delta activity with a maximum over the frontal regions). Other signs that have been described include a happy demeanor, hyperactivity without aggression, short attention span, excitability and sleeping problems with decreased need to sleep, increased sensitivity to heat, attraction to and fascination with water.
Therefore, molecular genetic testing (methylation analysis and UBE3A sequence analysis) identifies alterations in approximately 90% of individuals. The remaining 10% of individuals with classic phenotypic features of AS have the disorder as a result of an as-yet unidentified genetic mechanism and thus are not amenable to diagnostic testing. ... DNA methylation analysis identifies approximately 80% of individuals with AS. Note: Most commercially available DNA methylation analysis tests cannot distinguish between AS resulting from a deletion, UPD, or an ID. ... Testing Used in Angelman Syndrome (AS) View in own window Method Genetic Mechanism Detected 1 Total Proportion of AS Detected by Method 2 15q11.2-q13del UPD ID UBE3A seq UBE3A del/dup DNA methylation analysis 3, 4 X X X 5 ~80% MS-MLPA 6 X X X ~80% FISH 7 X ~68% CMA 8 X X 9 ~68% UPD study 10 X ~7% AS IC deletion analysis 11, 12 X ~3% UBE3A sequence analysis X 13 ~11% 14 UBE3A gene-targeted deletion/duplication analysis 11, 15 X Rare IC = imprinting center; ID = imprinting defect; UPD = uniparental disomy 1. ... Will not distinguish genetic mechanism 5. 80%-90% of IDs are thought to be epigenetic pathogenic variants occurring during maternal oogenesis or in early embryogenesis [Buiting 2010]. ... Low copy repeat elements (LCRs) are located within these breakpoint regions (see text for details). Approximately 90% of chromosome deletions resulting in Angelman (more...)
Angelman syndrome is a complex genetic disorder that primarily affects the nervous system. Characteristic features of this condition include delayed development, intellectual disability, severe speech impairment, and problems with movement and balance (ataxia). Most affected children also have recurrent seizures (epilepsy) and a small head size (microcephaly ). Delayed development becomes noticeable by the age of 6 to 12 months, and other common signs and symptoms usually appear in early childhood. Children with Angelman syndrome typically have a happy, excitable demeanor with frequent smiling, laughter, and hand-flapping movements.
One of the most common causes of night sweats in women over 40 is the hormonal changes related to menopause and perimenopause . [3] This is a very common occurrence during the menopausal transition years. Over 80% of women experience hot flashes , which may include excessive sweating, during menopause. [4] Night sweats range from being relatively harmless to a sign of underlying disease . ... S2CID 24179827 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af Viera, Anthony J.; Bond, Michael M.; Yates, Scott W. (1 March 2003).
The role of HPV in the remaining 25-30% is not yet clear. [43] Oral sex is not risk free and results in a significant proportion of HPV-related head and neck cancer. [44] Positive HPV16 status is associated with improved prognosis over HPV-negative OSCC. [45] HPV can induce tumor by several mechanisms: [46] E6 and E7 oncogenic proteins. Disruption of tumor suppressor genes . ... A total of 11,170 people died of their disease in 2006. [80] The worldwide incidence exceeds half a million cases annually. ... DNA Repair . 9 (5): 542–50. doi : 10.1016/j.dnarep.2010.02.004 . hdl : 2115/43021 . PMID 20197241 . ^ Nemec AA, Wallace SS, Sweasy JB (October 2010). ... Journal of the National Cancer Institute . 91 (8): 726–8. doi : 10.1093/jnci/91.8.726a . hdl : 2434/520105 . ... The New England Journal of Medicine . 340 (23): 1773–80. CiteSeerX 10.1.1.460.1056 . doi : 10.1056/NEJM199906103402301 .
Among women who experience a third or fourth degree tear, 60-80% are asymptomatic after 12 months. [21] Faecal incontinence, faecal urgency, chronic perineal pain, pain with sex, and fistula formation occur in a minority of people, but may be permanent. [22] The symptoms associated with perineal tear are not always due to the tear itself, since there are often other injuries, such as avulsion of pelvic floor muscles, that are not evident on examination. [23] There are claims that sometimes the perineum is excessively repaired after childbirth, using a so-called " husband stitch " and that this can increase vaginal tightness or result in pain during intercourse. [24] References [ edit ] ^ a b Elharmeel, Suzan MA; Chaudhary, Yasmin; Tan, Stephanie; Scheermeyer, Elly; Hanafy, Ashraf; van Driel, Mieke L (2011-08-10). ... American Journal of Obstetrics and Gynecology . 200 (5): 573.e1–573.e7. doi : 10.1016/j.ajog.2008.11.022 . ... Third degree obstetric anal sphincter tears: risk factors and outcome of primary repair" . BMJ . 308 (6933): 887–91. doi : 10.1136/bmj.308.6933.887 . ... Obstetrics and Gynecology . 107 (6): 1261–8. doi : 10.1097/01.aog.0000218693.24144.bd . PMID 16738150 . S2CID 23901136 . ^ Lammers, K; Prokop, M; Vierhout, ME; Kluivers, KB; Fütterer, JJ (August 2013).