A possible explanation for increased mortality in the Cardiac Arrhythmia Suppression Trial (CAST)" . Circulation . 90 (6): 2843–52. doi : 10.1161/01.cir.90.6.2843 .
The disorder results from the combination of indoctrinations by the alienating parent and the child's own contributions to the vilification of the alienated parent. [3] He also stated that the indoctrination may be deliberate or unconscious on the part of the alienating parent. [12] [13] Gardner initially believed that parents (usually mothers) made false accusations of child abuse and sexual abuse against the other parent (usually fathers) in order to prevent further contact between them. [14] [15] While Gardner initially described the mother as the alienator in 90% of PAS cases, he later stated both parents were equally likely to alienate. [16] [17] [18] He also later stated that in his experience accusations of sexual abuse were not present in the vast majority of cases of PAS. [19] Characteristics [ edit ] Gardner described PAS as a preoccupation by the child with criticism and deprecation of a parent. [20] Gardner stated that PAS occurs when, in the context of child custody disputes, one parent deliberately or unconsciously attempts to alienate a child from the other parent. [21] According to Gardner, PAS is characterized by a cluster of eight symptoms that appear in the child.
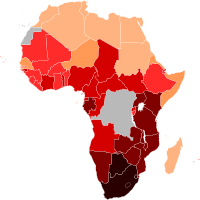
With the HIV infection, 77% of men, women, and children, develop AIDS, and die in Sub-Saharan Africa. Of those deaths, "more than 90% of AIDS orphans and children [were] infected with HIV". [41] Lack of money is an obvious challenge, although a great deal of aid is distributed throughout developing countries with high HIV/AIDS rates. ... For example, the philanthropist Inder Singh oversaw a program which reduced the cost of pediatric HIV/AIDS drugs by between 80 and 92 percent by working with manufacturers to reduce production and distribution costs. [58] Manufacturers often cite distribution and production difficulties in developing markets, which create a substantial barrier to entry. ... The number has remained virtually the same in Lesotho and Mozambique. [12] Zimbabwe's first reported case of HIV was in 1985. [77] There are widespread practices of sexual networking that involve multiple overlapping or concurrent sexual partners. [78] Men's sexual networks, in particular, tend to be quite extensive, [79] [80] a fact that is tacitly accepted or even encouraged by many communities. [81] Along with having multiple sexual partners, unemployment and population displacements resulting from drought and conflict have contributed to the spread of HIV/AIDS. [81] According to Susser and Stein (2000), men refuse to use condoms during intercourse with girls or women performing sex work (p. 1043-1044).
Men have a 100% chance of transmitting the mutation to a daughter and 0% chance to a son. [11] [12] [13] [14] Although males do not generally exhibit PCDH19 gene-related history such as cluster seizures, in a study involving four families with PCDH19 gene mutations, 5 of the fathers had obsessive and controlling tendencies. [14] The linkage of chromosome Xq22.1 to PCDH19 gene-related epilepsy in females was confirmed in all of the families. [14] The inheritance pattern is very unusual, in that men that carry the PCDH19 gene mutation on their only X-chromosome are typically unaffected, except in rare instances of somatic mosaicism. [11] [11] Alternatively, approximately 90% of women, who have the mutation on one of their two X-chromosomes, exhibit symptoms. [11] [12] [13] It has been suggested that the greater occurrence of PCDH19-epilepsy in females may relate to X-chromosome inactivation , through a hypothesized mechanism termed ‘‘cellular interference’’. [5] [11] A 2011 study found instances where patients had PCDH19 mutation, but their parents did not.
A number sign (#) is used with this entry because early infantile epileptic encephalopathy-9 (EIEE9), also known as epilepsy and mental retardation restricted to females (EFMR), is caused by mutation in the gene encoding protocadherin-19 (PCDH19; 300460) on chromosome Xq22. Description Epileptic encephalopathy-9 is an X-linked disorder characterized by seizure onset in infancy and mild to severe intellectual impairment. Autistic and psychiatric features have been reported in some individuals. The disorder affects heterozygous females only; transmitting males are unaffected (summary by Jamal et al., 2010). For a general phenotypic description and a discussion of genetic heterogeneity of EIEE, see EIEE1 (308350).
Female restricted epilepsy with intellectual disability is a rare X-linked epilepsy syndrome characterized by febrile or afebrile seizures (mainly tonic-clonic, but also absence, myoclonic, and atonic) starting in the first years of life and, in most cases, developmental delay and intellectual disability of variable severity. Behavioral disturbances (e.g. autistic features, hyperactivity, and aggressiveness) are also frequently associated. This disease affects exclusively females, with male carriers being unaffected, despite an X-linked inheritance.
Molecular Genetic Testing Used in Ataxia with Vitamin E Deficiency View in own window Gene 1 Method Proportion of Probands with Pathogenic Variants 2 Detectable by Method TTPA Sequence analysis 3 >90% 4, 5 Gene-targeted deletion/duplication analysis 6 Unknown 7 1.
Familial isolated vitamin e deficiency Other names Ataxia With Vitamin E Deficiency Familial isolated vitamin e deficiency has an autosomal recessive pattern of inheritance . Specialty Neurology Familial Isolated Vitamin E Deficiency is a rare autosomal recessive neurodegenerative disease . [1] [2] [3] [4] Symptoms are similar to those of Friedreich ataxia . Contents 1 Cause 2 Diagnosis 3 Treatment 4 See also 5 References 6 External links Cause [ edit ] Familial Isolated Vitamin E Deficiency is caused by mutations in the gene for a- tocopherol transfer protein. [5] Diagnosis [ edit ] This section is empty. You can help by adding to it . ( July 2017 ) Treatment [ edit ] This section is empty. You can help by adding to it . ( July 2017 ) See also [ edit ] Vitamin E deficiency TTPA References [ edit ] ^ "Ataxia with vitamin E deficiency" . www.orpha.net .
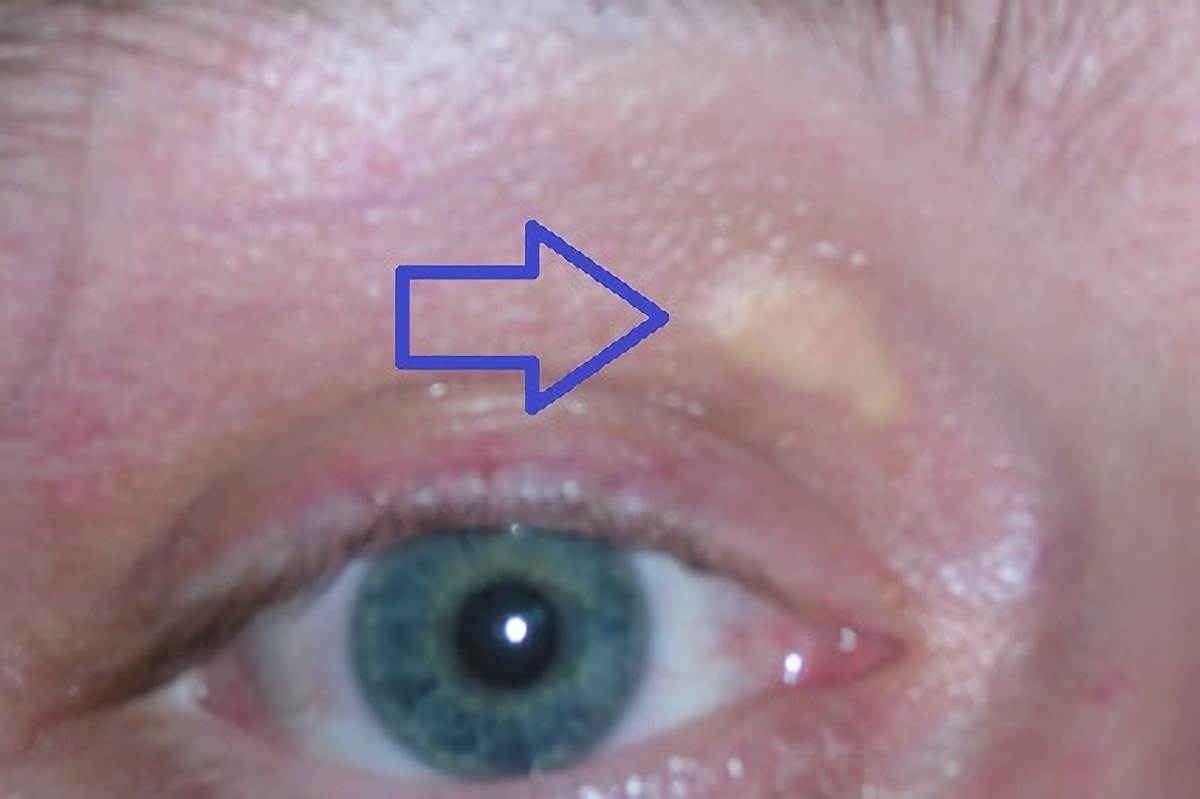
Ataxia with vitamin E deficiency is a disorder that impairs the body's ability to use vitamin E obtained from the diet. Vitamin E is an antioxidant, which means that it protects cells in the body from the damaging effects of unstable molecules called free radicals. A shortage (deficiency) of vitamin E can lead to neurological problems, such as difficulty coordinating movements (ataxia) and speech (dysarthria), loss of reflexes in the legs (lower limb areflexia), and a loss of sensation in the extremities (peripheral neuropathy). Some people with this condition have developed an eye disorder called retinitis pigmentosa that causes vision loss. Most people who have ataxia with vitamin E deficiency start to experience problems with movement between the ages of 5 and 15 years.
A number sign (#) is used with this entry because of evidence that ataxia with vitamin E deficiency (AVED) is caused by homozygous or compound heterozygous mutation in the TTPA gene (600415) on chromosome 8q12. Clinical Features Harding et al. (1985) described a young woman with spinocerebellar degeneration thought to be due to a selective defect in vitamin E absorption. There was no evidence of fat malabsorption. Binder et al. (1967) suggested a relationship between neurologic dysfunction and vitamin E deficiency in patients with chronic steatorrhea. This was subsequently confirmed in patients with abetalipoproteinemia (200100), the most severe state of vitamin E deficiency known. When studied at age 23, the proband had no vitamin E in the serum. A progressive neurologic disorder comprising ataxia, areflexia and marked loss of proprioception developed at age 13.
A neurodegenerative disease belonging to the inherited cerebellar ataxias mainly characterized by progressive spino-cerebellar ataxia, loss of proprioception, areflexia, and is associated with a marked deficiency in vitamin E. Epidemiology Global prevalence is not known but population-based studies have been performed and prevalence can be extrapolated at approximately 1/300,000. AVED is the second most frequently inherited cerebellar ataxia in North Africa. As vitamin E deficiency might bring protection against malaria (see this term), it could explain a higher prevalence of AVED in Plasmodium infested areas. Clinical description AVED presents generally between ages 5 and 20 years with variable phenotype and severity.
Ataxia with vitamin E deficiency (AVED) is a progressive disease affecting motor control and movement. Symptoms of AVED include slurred speech (dysarthria), difficulty coordinating movements ( ataxia ), numbness in the hands and feet (peripheral neuropathy), and progressive leg weakness. Some affected individuals may experience vision loss due to damage to the back of the eye ( retinitis pigmentosa ). Symptoms typically begin during childhood or adolescence and worsen with age, resulting in the need for a wheelchair by early adulthood. AVED is caused by a mutation to the TTPA gene. When this gene is damaged, vitamin E cannot be distributed throughout the body.
Molecular Genetic Testing Used in Beta Propeller Protein-Associated Neurodegeneration (BPAN) View in own window Gene 1 Method Proportion of Probands with a Pathogenic Variant 2 Detectable by Method WDR45 Sequence analysis 3, 4 >99% 5, 6 Gene-targeted deletion/duplication analysis 7 See footnote 8 1.
A number sign (#) is used with this entry because neurodegeneration with brain iron accumulation-5 (NBIA5) is caused by de novo heterozygous or hemizygous mutation in the WDR45 (300526) gene on chromosome Xp11. Description NBIA5, sometimes referred to as 'static encephalopathy of childhood with neurodegeneration in adulthood (SENDA),' is an X-linked neurodegenerative disorder characterized by global developmental delay in early childhood that is essentially static, with slow motor and cognitive gains until adolescence or early adulthood. In young adulthood, affected individuals develop progressive dystonia, parkinsonism, extrapyramidal signs, and dementia resulting in severe disability. Brain MRI shows iron accumulation in the globus pallidus and substantia nigra. A characteristic finding is T1-weighted hyperintensity surrounding a central band of hypointensity in the substantia nigra.
Beta-propeller protein-associated neurodegeneration (BPAN) , also known as static encephalopathy of childhood with neurodegeneration in adulthood (SENDA), is a hereditary neurologic disorder. It is part of the group of disorders known as neurodegeneration with brain iron accumulation . This disorder presents with global developmental delay in childhood which becomes progressive in early adulthood. Symptoms include dystonia (a movement disorder resulting in muscular spasms, twisting and repetitive movements) spasticity, parkinsonism (slurred or slow speech, stiffness of the muscles, slow movement, and visible tremors), and cognitive decline. BPAN is caused by mutations in the WDR45 gene. It is inherited in a dominant X-linked manner.
Beta-propeller protein-associated neurodegeneration (BPAN) is a disorder that damages the nervous system and is progressive, which means that it gradually gets worse. Affected individuals develop a buildup of iron in the brain that can be seen with medical imaging. For this reason, BPAN is classified as a type of disorder called neurodegeneration with brain iron accumulation (NBIA), although the iron accumulation may not occur until late in the disease. Many people with BPAN have recurrent seizures (epilepsy) beginning in infancy or early childhood. Several different types of seizures can occur in this disorder, even in the same individual.
Beta-propeller protein-associated neurodegeneration (BPAN), also known as static encephalopathy of childhood with neurodegeneration in adulthood, is a rare form of neurodegeneration with brain iron accumulation (NBIA) characterized by early-onset developmental delay and further neurological deterioration in early adulthood.
Evidence for slow progression, carrier fundus findings, and possible genetic linkage with glucose-6-phosphate dehydrogenase locus". Arch. Ophthalmol . 99 (3): 468–472. doi : 10.1001/archopht.1981.03930010470016 .
A number sign (#) is used with this entry because autosomal dominant hyper-IgE recurrent infection syndrome-1 (HIES1) is caused by heterozygous mutation in the STAT3 gene (102582) on chromosome 17q21. Description Hyper-IgE recurrent infection syndrome is a primary immunodeficiency disorder characterized by chronic eczema, recurrent Staphylococcal infections, increased serum IgE, and eosinophilia. Patients have a distinctive coarse facial appearance, abnormal dentition, hyperextensibility of the joints, and bone fractures (Buckley et al., 1972; Grimbacher et al., 1999). Genetic Heterogeneity of Hyper-IgE Recurrent Infection Syndrome See also HIES2 (243700), caused by mutation in the DOCK8 gene (611432), HIES3 (618282), caused by mutation in the ZNF341 gene (618269), and HIES4 (618523), caused by mutation in the IL6ST gene (600694). Clinical Features Davis et al. (1966) reported 2 unrelated girls with lifelong histories of indolent Staphylococcal abscesses.
Hyper IgE syndromes (HIES) are rare primary immune deficiencies characterized by elevated serum IgE , skin inflammation (dermatitis) and recurrent skin and lung infections. There are two forms of HIES, which have the above characteristics in common but otherwise have distinct presentations, courses and outcomes: autosomal dominant HIES (AD-HIES) and autosomal recessive HIES (AR-HIES). Click on the embedded links to learn more about autosomal dominant HIES (or Job syndrome) and autosomal recessive HIES.
Autosomal dominant hyper-IgE syndrome (AD-HIES), formerly known as Job syndrome, is a condition that affects several body systems, particularly the immune system. Recurrent infections are common in people with this condition. Affected individuals tend to have frequent bouts of pneumonia, which are caused by certain kinds of bacteria that infect the lungs and cause inflammation. Inflammation is a normal immune system response to injury and foreign invaders (such as bacteria). However, excessive inflammation can damage body tissues. Recurring pneumonia often results in the formation of air-filled cysts (pneumatoceles) in the lungs. Frequent skin infections and an inflammatory skin disorder called eczema are also very common in AD-HIES.
Autosomal dominant hyper IgE syndrome (AD-HIES) , formerly known as Job syndrome, affects several body systems including the immune system. AD-HIES is characterized by abnormally high levels of an immune system protein called immunoglobulin E (IgE) in the blood. Signs and symptoms may include recurrent infections (e.g., pneumonia, skin infections), eczema , and occasionally bone and tooth abnormalities. The eczema and skin infections may cause rashes, blisters, collections of pus (abscesses), open sores, and scaling of the skin. Some cases of AD-HIES are caused by mutations in the STAT3 gene. In other cases, the cause is unknown.
They also recommend routinely screening men aged 20 to 35 years and women aged 20 to 45 years if they have other risk factors for coronary heart disease . [36] In 2016 they concluded that testing the general population under the age of 40 without symptoms is of unclear benefit. [37] [38] In Canada, screening is recommended for men 40 and older and women 50 and older. [39] In those with normal cholesterol levels, screening is recommended once every five years. [40] Once people are on a statin further testing provides little benefit except to possibly determine compliance with treatment. [41] Treatment [ edit ] Treatment recommendations have been based on four risk levels for heart disease. [42] For each risk level, LDL cholesterol levels representing goals and thresholds for treatment and other action are made. [42] The higher the risk category, the lower the cholesterol thresholds. [42] LDL cholesterol level thresholds [42] Risk category Criteria for risk category Consider lifestyle modifications Consideration medication No. of risk factors† 10-year risk of myocardial ischemia mmol/litre mg/dL mmol/litre mg/dL High Prior heart disease OR >20% >2.6 [43] >100 >2.6 >100 Moderately high 2 or more AND 10–20% >3.4 >130 >3.4 >130 Moderate 2 or more AND <10% >3.4 >130 >4.1 >160 Low 0 or 1 >4.1 >160 >4.9 >190 †Risk factors include cigarette smoking, hypertension (BP ≥140/90 mm Hg or on antihypertensive medication), low HDL cholesterol (<40 mg/dL), family history of premature heart disease, and age (men ≥45 years; women ≥55 years). ... The American Journal of Clinical Nutrition . 102 (2): 276–94. doi : 10.3945/ajcn.114.100305 . ISSN 1938-3207 . ... "The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials" . Lancet . 380 (9841): 581–90. doi : 10.1016/S0140-6736(12)60367-5 . ... Systematic review and meta-analysis of randomised controlled trials". Arch Dis Child . 95 (9): 673–80. doi : 10.1136/adc.2008.157024 . PMID 20515970 . ... "Gene therapy for familial hypercholesterolemia". Curr Pharm Des . 17 (24): 2575–91. doi : 10.2174/138161211797247550 .
A survey conducted by security firm Avira identified 84% of people fear social networking sites will steal or misuse their personal information, [9] demonstrating the net majority of internet users share, at least partially, in distrusting the digital world.
Molecular Genetic Testing Used in IPEX Syndrome View in own window Gene 1 Test Method Proportion of Pathogenic Variants 2 Detectable by This Method FOXP3 Sequence analysis 3,4 ~99% Gene-targeted deletion/duplication analysis 5 1 reported 6 1.
IPEX syndrome Other names Autoimmune enteropathy type 1 [1] IPEX syndrome is inherited via X-linked recessive Specialty Immunology Symptoms Lymphadenopathy [2] Causes FOXP3 gene mutation [1] Diagnostic method Family history, Genetic test [1] Treatment TPN(nutritional purpose), Cyclosporin A and FK506, Bone marrow transplant [3] [4] Immunodysregulation polyendocrinopathy enteropathy X-linked (or IPEX ) syndrome is a rare disease linked to the dysfunction of the transcription factor FOXP3 , widely considered to be the master regulator of the regulatory T cell lineage. [5] [6] It leads to the dysfunction of regulatory T-cells and the subsequent autoimmunity . [7] The disorder is one of the autoimmune polyendocrine syndromes and manifests with autoimmune enteropathy, psoriasiform or eczematous dermatitis , nail dystrophy , autoimmune endocrinopathies , and autoimmune skin conditions such as alopecia universalis and bullous pemphigoid . [7] [2] Management for IPEX has seen limited success in treating the syndrome by bone marrow transplantation . [8] Contents 1 Symptoms and signs 2 Genetics 3 Mechanism 4 Diagnosis 5 Treatment 6 Research 7 See also 8 References 9 Further reading 10 External links Symptoms and signs [ edit ] Eczema Some of the symptoms and signs of IPEX syndrome are the following: [2] Lymphadenopathy Eczema Hypothyroidism Diarrhea Genetics [ edit ] Mutations in FOXP3 gene causing IPEX syndrome - known in year 2012. IPEX syndrome is inherited in males via an x-linked recessive manner, as the FOXP3 gene, whose cytogenetic location is Xp11.23, is involved in the mechanism of this condition. [5] [6] Mutation of FOXP3 leading to expression of malfunctioning protein is often localised in DNA-binding domain called the forkhead domain. The truncated protein can not bind to its binding-spot on the DNA and thus its function concerning T regulatory lymphocytes development and functioning is impaired. The absence or dysfunction of regulatory T cells is the cause of autoimmune symptoms. [9] Data from 2018 describes over 70 mutations in FOXP3 gene leading to IPEX syndrome. Nonetheless, this number is still changing with new cases and discoveries coming. [10] For example in 2010 there were only 20 mutations of FOXP3 known in the literature. [9] Mechanism [ edit ] This autoimmunity called IPEX is an attack from the body's own immune system against the body's own tissues and organs. [4] Early age onset of this disease in males causes severe enlargement of the secondary lymphoid organs , and insulin dependent diabetes [ medical citation needed ] This condition indicates the loss of CD4+ CD25+ T regulatory cells, and express the transcription factor Foxp3.
A number sign (#) is used with this entry because X-linked immunodysregulation, polyendocrinopathy, and enteropathy (IPEX) is caused by mutation in the FOXP3 gene (300292) on chromosome Xp11. Description IPEX is an X-linked recessive immunologic disorder characterized by onset in infancy of severe diarrhea due to enteropathy, type 1 diabetes mellitus, and dermatitis. Other features may include hypothyroidism, autoimmune hemolytic anemia, thrombocytopenia, lymphadenopathy, hepatitis, and nephritis. The disorder may be fatal before age 2 years if not aggressively treated. Long-term therapeutic options include immunosuppression and hematopoietic stem cell transplantation (review by d'Hennezel et al., 2012).
Immunodysregulation polyendocrinopathy enteropathy x-linked (IPEX) syndrome is a rare autoimmune disease. it affects only males and starts in the first six months of life. The symptoms of IPEX syndrome include severe diarrhea , diabetes , skin conditions (such as eczema , erythroderma , or psoriasis ), and thyroid disease ( thyroiditis ). IPEX syndrome is caused by changes (mutations) of the FOXP3 gene, which is located on the X chromosome. There are several other diseases that are very similar to the IPEX syndrome, caused by mutations in other genes and that affect both males and females. Treatment of IPEX syndrome consists of medications that limit immune system function; a bone marrow transplantation is the only treatment that can cure the disease, but it may have several complications.
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome primarily affects males and is caused by problems with the immune system. The immune system normally protects the body from foreign invaders, such as bacteria and viruses, by recognizing and attacking these invaders and clearing them from the body. However, the immune system can malfunction and attack the body's own tissues and organs instead, which is known as autoimmunity. IPEX syndrome is characterized by the development of multiple autoimmune disorders in affected individuals. Although IPEX syndrome can affect many different areas of the body, autoimmune disorders involving the intestines, skin, and hormone-producing (endocrine) glands occur most often.
A rare immunodysregulatory disease characterized by refractory diarrhea, endocrinopathies, cutaneous involvement, and infections. Epidemiology Immune dysregulation-polyendocrinopathy-enteropathy-X-linked (IPEX) syndrome prevalence is unknown. The disease has probably been underestimated, and milder clinical phenotypes surviving to adult life are being described. Clinical description IPEX syndrome most commonly develops during the first few days or weeks of life and affects exclusively boys. It classically manifests with the sequential appearance of the triad of enteropathy, autoimmune disease (particularly Type I diabetes mellitus), and cutaneous involvement, but the clinical features and severity of the disease can vary considerably between individuals.
Use of psychotropic medications such as benzodiazepines in people with intellectual disability requires monitoring and vigilance as side effects occur commonly and are often misdiagnosed as behavioral and psychiatric problems. [40] Epidemiology Intellectual disability affects about 2–3% of the general population. 75–90% of the affected people have mild intellectual disability. ... Levine and Marks 1928 IQ classification [65] [66] IQ Range ("ratio IQ") IQ Classification 175 and over Precocious 150–174 Very superior 125–149 Superior 115–124 Very bright 105–114 Bright 95–104 Average 85–94 Dull 75–84 Borderline 50–74 Morons 25–49 Imbeciles 0–24 Idiots United States Special Olympics USA team in July 2019 In North America , intellectual disability is subsumed into the broader term developmental disability , which also includes epilepsy , autism , cerebral palsy , and other disorders that develop during the developmental period (birth to age 18). ... In several U.S. states , and several European Union states, persons with intellectual disabilities are disenfranchised. [80] [81] The European Court of Human Rights ruled in Alajos Kiss v.
Additional cases of acute phosphate nephropathy have been reported to FDA and described in the literature since these were issued." [2] Contents 1 Natural history 2 Signs and symptoms 3 Diagnosis 3.1 Pathophysiology 3.2 Types of assessment 4 Risk factors 5 Management 5.1 Prevention 5.2 Treatment 6 References 7 Further reading Natural history [ edit ] Mannitol and large volume of saline were first used as bowel preparation agents prior to colonoscopy. [6] As the use of Mannitol causes the production of methane , hydrogen , and other flammable gases, it was reported to be associated with colonic explosion. [6] Large volume of saline was also reported to significantly impact the electrolyte balance and net fluid within the body. [6] Later in the 1990, the polyethylene-glycol electrolyte lavage solution or PEG-ELS, was formulated with more effectiveness and safety to use. [7] PEG-ELS was not widely adopted due to its requirement of consuming an enormous volume. [6] Then, OSP (C.B.