-
Multiple Endocrine Neoplasia Type 1
Wikipedia
Malignant islet cell tumors due to MEN 1 syndrome often have a more benign course than do sporadically occurring malignant islet cell tumors. [ citation needed ] About 40% of islet cell tumors originate from a β-cell , secrete insulin ( insulinoma ), and can cause fasting hypoglycemia . β-cell tumors are more common in patients < 40 years of age. ... This is known as Knudson's two-hit hypothesis [3] and is a common feature seen with inherited defects in tumor suppressor genes.

-
Cornelia De Lange Syndrome 1
OMIM
Boyle et al. (2015) provided a detailed review of CDLS, including clinical features, diagnosis, and genetic counseling. ... Allanson et al. (1997) evaluated 43 subjects with de Lange syndrome, 30 with classic features and 13 with the mild phenotype. ... Melegh et al. (1996) described a newborn boy with clinical features of de Lange syndrome who manifested dyspnea, hypertonia, and hyperthermia. ... In the mother of a child with typical features, de Die-Smulders et al. (1992) observed mild manifestations. ... Features that proved to be misleading included full or flat brows, a prominent nasal bridge or bulbous tip, and/or a normal or prominent chin.NIPBL, RAD21, HDAC8, SMC3, SMC1A, BRD4, KMT2A, SETD5, PDS5A, ATR, LZTR1, SOS1, RIT1, RAF1, PTPN11, KRAS, CENPJ, CEP63, GPT, CREBBP, ESCO2, NAALADL2, TNKS, MAU2, PHIP, EPS15L1, GALNT14, DYM, GSC, ANKRD11, AFF4, ARSD, PDS5B, STAG2, CTCF, STAG1, CHRD, SHOX2, MYC, HDAC2, GRM1, GH1, FOXF1, EP300, DDX11, CRABP2, CARS1, NIPBL-DT

-
Disease X
Wikipedia
It is also the case that where you have a new disease there is no resistance in the population and that means it can spread fast". [10] Bacterial infection [ edit ] In September 2019, Public Health England (PHE) reported that the increasing antibiotic resistance of bacteria, even to "last-resort" antibiotics such as carbapenems and colistin , could also turn into a potential Disease X, citing the antibiotic resistance in gonorrhea as an example. [36] In popular culture [ edit ] In 2018, the Museum of London ran an exhibition titled "Disease X: London's next epidemic?", hosted for the centenary of the Spanish flu epidemic from 1918. [37] [38] The term features in the title of several fiction books that involve global pandemic diseases, such as Disease (2020), [39] and Disease X: The Outbreak (2019). [40] See also [ edit ] Coalition for Epidemic Preparedness Innovations (CEPI) Global Research Collaboration for Infectious Disease Preparedness (GloPIR-R) Synthetic virology Bioterrorism Planet X References [ edit ] ^ a b c Daszak, Peter (22 February 2020). ... External links [ edit ] Wikiquote has quotations related to: Disease X Blueprint priority diseases World Health Organization (6-7 February 2018) Prioritizing diseases for research and development in emergency contexts World Health Organisation (March 2018) (Video) What is Disease X World Health Organization (16 March 2018) The mystery viruses far worse than flu BBC News (November 2018) v t e Concepts in infectious disease Transmission Basic concepts Asymptomatic carrier Host Incubation period Index case Infectious period Latent period Natural reservoir Subclinical infection Super-spreader Modes Human-to-human transmission Horizontal Vertical Cross-species transmission Spillover infection Vector Zoonosis Reverse zoonosis Routes Airborne disease Blood-borne disease Foodborne illness Waterborne disease Hospital-acquired infection Fomite Fecal-oral route Sexual Modelling Attack rate Basic reproduction number Compartmental models in epidemiology Critical community size Herd immunity Infection rate Serial interval Transmission risks and rates Medication Antimicrobial Antibiotic Antiviral drug Antimicrobial resistance Immunotherapy Phage therapy Vaccination Emerging infections Disease X Emergent virus Other Discovery of disease-causing pathogens Eradication of infectious diseases Pandemic v t e COVID-19 pandemic SARS-CoV-2 (virus) COVID-19 (disease) Timeline Pre-pandemic Crimson Contagion Disease X Event 201 Exercise Cygnus 2019 2020 January February responses March responses April responses May responses June responses July responses August responses September responses October responses November responses December responses 2021 January Locations Africa Northern Algeria Canary Islands Ceuta Egypt Libya Mauritania Melilla Morocco Sudan Tunisia Western Sahara Sahrawi Arab Democratic Republic Eastern Burundi Comoros Djibouti Eritrea Ethiopia Kenya Madagascar Mauritius Mayotte Réunion Rwanda Seychelles Somalia Puntland Somaliland South Sudan Tanzania Uganda Southern Angola Botswana Eswatini Lesotho Malawi Mozambique Namibia South Africa Zambia Zimbabwe Central Cameroon Central African Republic Chad Democratic Republic of the Congo Republic of the Congo Gabon São Tomé and Príncipe Western Benin Burkina Faso Cape Verde Equatorial Guinea Gambia Ghana timeline Guinea Guinea-Bissau Ivory Coast Liberia Mali Niger Nigeria Senegal Sierra Leone Togo Asia Central / North Kazakhstan Kyrgyzstan Russia timeline Tajikistan Turkmenistan Uzbekistan East Hong Kong Japan timeline North Korea South Korea Macau Mongolia Taiwan China Beijing Heilongjiang Henan Hubei lockdown Inner Mongolia Liaoning Shanghai Sichuan Tibet Xinjiang South Afghanistan timeline Bangladesh Bhutan Maldives Nepal timeline Pakistan timeline Sri Lanka India timeline January–May 2020 June–December 2020 economic impact evacuations lockdown migrant workers' crisis union government response Operation Namaste state government responses ) Andaman and Nicobar Islands Andhra Pradesh Arunachal Pradesh Assam Bihar Chandigarh Chhattisgarh Dadra and Nagar Haveli and Daman and Diu Delhi Tablighi Jamaat hotspot Goa Gujarat Haryana Himachal Pradesh Jammu and Kashmir Jharkhand Karnataka Kerala Ladakh Madhya Pradesh Maharashtra Manipur Meghalaya Mizoram Nagaland Odisha Puducherry Punjab Rajasthan Sikkim Tamil Nadu Telangana Tripura Uttar Pradesh Uttarakhand West Bengal Southeast Brunei Cambodia East Timor Indonesia timeline social restrictions Laos Malaysia timeline movement control order Johor Kuala Lumpur Sabah Sarawak Selangor Myanmar Singapore timeline circuit breaker response Thailand timeline Vietnam timeline Philippines ( timeline government response community quarantines Luzon evacuations testing controversy ) Bangsamoro Bicol Region Cagayan Valley Calabarzon Caraga Central Luzon Central Visayas Cordillera Davao Region Eastern Visayas Ilocos Region Metro Manila Mimaropa Northern Mindanao Soccsksargen Western Visayas Zamboanga Peninsula Overseas Filipinos West Armenia Azerbaijan Artsakh Bahrain Georgia Abkhazia South Ossetia Iran Iraq Kurdistan Israel Jordan Kuwait Lebanon Oman Palestine Qatar Saudi Arabia Syria Turkey timeline United Arab Emirates Yemen Europe United Kingdom timeline January–June 2020 July–December 2020 2021 government response social impact economic impact education impact Operation Rescript England 2020 timeline 2021 timeline London Northern Ireland 2020 timeline 2021 timeline Scotland 2020 timeline 2021 timeline Wales 2020 timeline 2021 timeline Crown dependencies Isle of Man Jersey Guernsey Overseas territories Akrotiri and Dhekelia Gibraltar response Eastern Armenia Azerbaijan Belarus Georgia Kazakhstan Moldova Gagauzia Transnistria Russia timeline government responses Crimea Sevastopol Ukraine Crimea Sevastopol Donetsk Luhansk Balkans Albania Bosnia and Herzegovina Kosovo Montenegro North Macedonia Serbia Turkey timeline Microstates Andorra San Marino Vatican City Monaco European Union EFTA countries Austria Belgium Bulgaria Croatia timeline Cyprus Northern Cyprus Czech Republic Denmark Faroe Islands Estonia Finland Åland Islands France Guadeloupe French Guiana Réunion Martinique Mayotte Saint Martin Germany North Rhine-Westphalia Greece Hungary Iceland Ireland timeline January–June 2020 July–December 2020 economic impact social impact Italy lockdown Latvia Liechtenstein Lithuania Luxembourg Malta Netherlands Norway Poland Portugal Romania timeline Piatra Neamț hospital fire Slovakia Slovenia Spain timeline Asturias Canary Islands Ceuta Community of Madrid Melilla Sweden timeline government response Switzerland North America Mexico timeline Central America Belize Costa Rica El Salvador Guatemala Honduras Nicaragua Panama Canada timeline economic impact Alberta British Columbia Manitoba New Brunswick Newfoundland and Labrador Northwest Territories Nova Scotia Nunavut Ontario timeline Peel Region Toronto Prince Edward Island Quebec Montreal Saskatchewan Yukon Caribbean Antigua and Barbuda Bahamas Barbados British Overseas Territories Anguilla British Virgin Islands Cayman Islands Montserrat Turks and Caicos Islands response Cuba Guantanamo Bay Naval Base Dominica Dominican Republic Dutch Caribbean Aruba Curaçao Sint Maarten Caribbean Netherlands Bonaire Saba Sint Eustatius French West Indies Guadeloupe Martinique Saint Barthélemy Saint Martin Grenada Haiti Jamaica Saint Kitts and Nevis Saint Lucia Saint Vincent and the Grenadines Trinidad and Tobago US insular areas Puerto Rico U.S.

-
Basal Cell Nevus Syndrome
OMIM
One of the patients had astrocytoma with severe hydrocephalus. Other features included pits of the palms and soles. ... Schwartz (1978) noted hamartomatous polyps of the stomach and mesenteric cysts as features of the basal cell nevus syndrome. ... Lo Muzio et al. (1999) described the clinical features of Gorlin syndrome in northern Italy. ... Palmar/plantar pits were the most frequent feature seen in affected individuals at all ages. ... Midro et al. (2004) described an 11-year-old girl with an interstitial deletion of 9q22.32-q33.2 associated with a familial translocation t(9;17)(q34.11;p11.2) who had clinical features consistent with basal cell nevus syndrome and some features of nail-patella syndrome (NPS; 161200).
-
Crossbite
Wikipedia
Unilateral crossbites can present with following features in a child Lower midline deviation [14] to the crossbite side Class 2 Subdivision relationships Temporomandibular disorders [15] Treatment [ edit ] A child with posterior crossbite should be treated immediately if the child shifts their mandible on closing, which is often seen in a unilateral crossbite as mentioned above. ... "A multiple logistic regression analysis of the risk and relative odds of temporomandibular disorders as a function of common occlusal features". Journal of Dental Research . 72 (6): 968–979. doi : 10.1177/00220345930720061301 . ... External links [ edit ] Classification D ICD - 10 : K07.2 ICD - 9-CM : 524.27 v t e Orthodontics Diagnosis Bolton analysis Cephalometric analysis Cephalometry Dentition analysis Failure of eruption of teeth Little's Irregularity Index Malocclusion Scissor bite Standard anatomical position Tooth ankylosis Tongue thrust Conditions Overbite Open bite Crossbite Prognathism Retrognathism Maxillary hypoplasia Condylar hyperplasia Overeruption Mouth breathing Appliances ACCO appliance Archwire Activator appliance Braces Damon system Elastics Frankel appliance Invisalign Lingual arch Lip bumper List of orthodontic functional appliances List of palatal expanders Lingual braces Headgear Orthodontic technology Orthodontic spacer Palatal lift prosthesis Palatal expander Quad helix Retainer SureSmile Self-ligating braces Splint activator Twin Block Appliance Procedures Anchorage (orthodontics) Cantilever mechanics Fiberotomy Interproximal reduction Intrusion (orthodontics) Molar distalization SARPE Serial extraction Materials Beta-titanium Nickel titanium Stainless steel TiMolium Elgiloy Ceramic Composite Notable contributors Edward Angle Spencer Atkinson Clifford Ballard Raymond Begg Hans Peter Bimler Samir Bishara Arne Björk Charles B.

-
Paragangliomas 4
OMIM
For a general phenotypic description and a discussion of genetic heterogeneity of familial paragangliomas, see PGL1 (168000). Clinical Features Bogdasarian and Lotz (1979) reported a family in which affected individuals had multiple catecholamine-secreting head and neck paragangliomas and retroperitoneal pheochromocytomas. ... She was initially found to have a paravertebral extraadrenal pheochromocytoma with features of a paraganglioma on histologic examination of the resected tumor, but further analysis showed an area with morphology of a differentiating neuroblastoma. ... SDHB mutation carriers were more likely than SDHD mutation carriers to develop extraadrenal pheochromocytomas and malignant disease, whereas SDHD mutation carriers had a greater propensity to develop head and neck paragangliomas and multiple tumors. For the index cases, there was no difference between 43 SDHB and 19 SDHD mutation carriers in the time to first diagnosis (34 vs 28 years, respectively; p = 0.3).
-
Coffin-Lowry Syndrome
GeneReviews
Typically SIDAs begin between mid-childhood and the teens. Characteristic facial features may be more apparent with age. ... The authors are aware of an individual with a proven RPS6KA3 pathogenic missense variant who is able to work in a fast-food restaurant [C Skinner, personal communication]. ... Craniofacial. Characteristic craniofacial features (see Figure 1, Figure 2, and Figure 3) may be more apparent with age. ... Individuals with a variety of chromosome disorders may have features of Coffin-Lowry syndrome. Two examples: McCandless et al [2000] reported a family with del(10)(q25.1q25.3) in which affected members had findings suggestive of CLS. Concannon et al [2002] reported an individual with features of CLS and a complex chromosome rearrangement (involving chromosomes 2,3,7, and 11).
-
Ataxia–telangiectasia
Wikipedia
Current consensus is that special screening tests are not helpful, but all women should have routine cancer surveillance. Skin [ edit ] A–T can cause features of early aging such as premature graying of the hair. ... Vision (ability to see objects in focus) is normal. [25] Control of eye movement is often impaired, affecting visual functions that require fast, accurate eye movements from point to point (e.g. reading). ... A variety of laboratory abnormalities occur in most people with A–T, allowing for a tentative diagnosis to be made in the presence of typical clinical features. Not all abnormalities are seen in all patients. ... There are other rare disorders that can be confused with A–T, either because of similar clinical features, a similarity of some laboratory features, or both. ... Differentiation of these disorders is often possible with clinical features and selected laboratory tests.
-
Leri-Weill Dyschondrosteosis
OMIM
See also Langer mesomelic dysplasia (LMD; 249700), a more severe phenotype that results from homozygous defect in the SHOX or SHOXY genes. Clinical Features The disorder was first described by Leri and Weill (1929). ... Diagnosis Ogata et al. (2001) reviewed the clinical features and diagnostic and therapeutic implications of SHOX haploinsufficiency and overdosage. They suggested that identification of Madelung deformity is important in the clinical diagnosis of SHOX haploinsufficiency and that gonadal suppression therapy may mitigate the clinical features, including mesomelic short stature. ... Gatta et al. (2007) pointed out that the MLPA analysis can be carried out on a buccal swab, and that this technique represents a fast, simple, and high throughput approach in the screening of SHOX deletions. ... The proposita had short forearms with a short, bowed radius, cubitus valgus with limited motion at the elbows, fusion of C1 and C2 vertebrae, and other skeletal anomalies. Many of the features suggested dyschondrosteosis. Whatever the precise diagnosis, the findings implicated one of the breakpoints as causative (Hecht and Hecht, 1984).
-
Epidermolysis Bullosa
Wikipedia
Rather, it is speculated that cross-correction from tissue-resident graft-derived immune cells contributes to the observed clinical benefit. [19] A pilot study performed in 2015 suggests that systemic granulocyte-colony stimulating factor (G-CSF) may promote increased wound healing in people with dystrophic epidermolysis bullosa. [20] Transplanting skin derived from genetically modified stem cells onto the wound surfaces has been studied with a report of improvements in one person. [21] Monitoring [ edit ] The Epidermolysis Bullosa Disease Activity and Scarring index (EBDASI) is a scoring system that objectively quantifies the severity of epidermolysis bullosa. ... "Epidermolysis bullosa pruriginosa: dystrophic epidermolysis bullosa with distinctive clinicopathological features". British Journal of Dermatology . 130 (5): 617–625. doi : 10.1111/j.1365-2133.1994.tb13109.x . ... PMID 29144448 . ^ Development, reliability, and validity of a novel Epidermolysis Bullosa Disease Activity and Scarring index (EBDASI). 2014. Loh CH, Kim J, Su JC, Daniel BS, Venugopal SS, Rhodes LM, Intong LR, Law MG, Murrell DF.ITGB4, KRT14, KRT5, EXPH5, COL7A1, COL17A1, PLEC, LAMC2, LAMB3, DST, ITGA6, ITGA3, ACTB, LAMA3, CCL21, SDC2, TMSB4X, CXCL12, TGM3, TP53, BEST1, PLOD3, TGM5, CD160, B3GAT1, SLCO1B3, KLHL24, FERMT1, RPS27A, LAD1, MMP1, FLII, CEACAM5, DMD, DSC2, DSC3, DSP, EBM, GPT, LDLR, HMGB1, HSPG2, IL1A, IL1B, IL6, KRT18, ERBIN

-
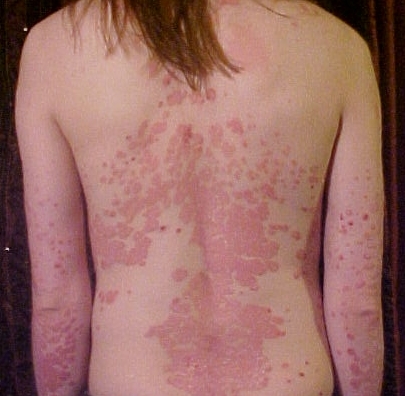
Psoriasis
Wikipedia
Mild psoriasis has been defined as a percentage of body surface area (BSA)≤10, a psoriasis area severity index (PASI) score ≤10, and a Dermatology Life Quality Index (DLQI) score ≤10. [64] Moderate to severe psoriasis was defined by the same group as BSA >10 or PASI score >10 and a DLQI score >10. [64] The DLQI is a 10-question tool used to measure the impact of several dermatologic diseases on daily functioning. ... It assesses the severity of lesions and the area affected and combines these two factors into a single score from 0 (no disease) to 72 (maximal disease). [66] Nevertheless, the PASI can be too unwieldy to use outside of research settings, which has led to attempts to simplify the index for clinical use. [67] Management [ edit ] Schematic of psoriasis treatment ladder While no cure is available for psoriasis, [46] many treatment options exist.CARD14, STAT3, VEGFA, ERAP1, TYK2, IL12B, IL23R, TRAF3IP2, TNFAIP3, TNIP1, IL13, RNF114, NOS2, IFIH1, GJB2, DDX58, FBXL19, CSMD1, SERPINB8, HLA-C, LCE3B, LCE3C, IL23A, TNF, IL1B, IL4, IL6, TP53, CRP, S100A7, IL36RN, TGFA, APOE, REL, CSF2, PPARG, ZNF816, CAT, IFNLR1, PCNA, NFKBIA, PTTG1, RUNX3, MKI67, REN, VNN3, LCE3D, VNN1, SOD2, HCAR2, TAGAP, VNN2, CYP2S1, CARM1, AP1S3, ZC3H12C, CP, ITGB2, TTC7A, IL19, SPTLC2, HLA-B, IL10, ARPC4, CCHCR1, PDE4A, PTPN22, NFKB1, NOD2, TLR4, RUNX1, TNFSF15, IL2RA, COG6, IL27, HCP5, ITGAL, CDKAL1, GATD3A, AHR, NOTCH1, TNFRSF1A, CD40, NFATC1, FUT2, ERAP2, AIM2, ZMIZ1, CD226, SPATA2, ANKRD55, SMAD3, CD6, JAZF1, IL1R1, STAT2, FNDC3A, BTD, BANK1, ZGLP1, PRKRA, PTPN2, TNFRSF6B, ZNF365, GLYAT, F9, KLRC1, FNBP1, SBNO2, SYNE2, PLCL2, FLG, ESR2, RAD50, PSORS4, ERN1, PKIG, ADGRL2, MYT1L, CLEC16A, PARK7, SLC9A8, ATXN2L, CRB1, UBBP4, SP140, DNAJB4, FGFR1OP, POLI, USP49, IL17RA, BPTF, ETS1, ELOA, SH2B3, PTPRN2, SUOX, STK11, SON, SLIT3, ATXN2, RPS26, RPS6KB1, REV3L, REG1A, IFNG, IFNGR2, RDX, IL1A, HLA-DRB1, IL6R, MAPK1, CXCL8, PRKCQ, PRKCB, PLAU, IL15, IL17A, IL18, NPPA, MST1, MGMT, ICAM5, HLA-DQA1, ELMO1, ATG5, NR5A2, KEAP1, SCRN1, ZFYVE16, SETD1A, PRORP, GART, IKBKE, GBAP1, RGS6, NPEPPS, GCKR, GNA12, HLA-A, OSMR, FIBP, SKAP2, GPR35, BSN, ACAD8, ASAP2, KSR1, IL18R1, TNFRSF14, VDR, UBE2L3, GPR183, GPR160, DENND1B, LRRC43, SLC39A11, TMEM258, MYRF, TRIM65, HIPK1, ITPRID1, BLOC1S2, CAMK2G, FAM177A1, CAVIN1, LURAP1L, PUDPP2, PRDM1, UBAC2, IL37, CAMP, FLG-AS1, ZNF831, ANTXR2, NXPE1, LRRK2, NRAD1, ZPBP2, RAVER1, RBM45, RNF145, LINC01620, CASR, PUS10, CMC1, LINC02085, CAST, C17orf67, IRGM, STX1B, ADCY7, TH2LCRR, LINC00824, LINC01147, LINC00993, LINC02863, CCR5AS, ADCY3, CHROMR, LINC01714, LINC02571, ACTA2, LINC01932, ACO1, H3P13, LINC01250, IFNG-AS1, LINC02213, TRAF3IP2-AS1, C1orf141, LINC01185, TEX41, MIR146A, IRF1-AS1, KPRP, LINC00598, RTEL1-TNFRSF6B, INS-IGF2, C1orf68, SPATA48, ELOA-AS1, ALDH2, ZNF816-ZNF321P, RMI2, KRT17, IL17F, UBASH3A, ELOVL6, CHST8, DAP, CARD9, DAG1, FOXP3, IGF2-AS, TLR7, HDAC7, KLF13, GAL3ST2, THADA, CPEB4, BACH2, IL21, CREM, GIPC2, RASIP1, FAM118A, PPP2R3C, ATG16L1, RIC8B, INAVA, EXOC2, CS, CLCN6, IL22, DELEC1, MFSD4B, IL17B, ANKRD30A, AP5B1, DNMT3A, KCNH7, UQCR10, NKD1, MYPN, CFL1, DONSON, PAK5, IL20, KIAA1109, QTRT1, CDKN2A, TSPAN14, CDKN2D, CDSN, NDFIP1, ACE, PIK3CG, MTHFR, IL9, PIK3CD, IL17C, LEP, EGF, EIF2AK1, CCL2, CCRL2, S100A9, TGFB1, PIK3CA, IL2, PIK3CB, CCR6, DEFB4A, HT, TLR2, ADIPOQ, TRBV20OR9-2, TP63, CCL27, PRL, IL1RL2, MIR21, MIR31, JAK1, TEK, AREG, PI3, DEFB4B, TAP1, EGFR, MICA, PSORS1C1, IFNA13, ADAM33, TSLP, IFNA1, IL33, S100B, IL1RN, S100A8, CCL20, IGF1, NLRP3, IL24, SIRT1, RPSA, SLCO6A1, LCN2, NGF, IL1F10, H3P28, EBNA1BP2, RABEPK, TAC1, ARHGEF2, GSTK1, ARMH1, RETN, MIR155, MIR203A, PSMD7, MAPK3, CCL17, SLC2A1, TLR9, PSORS6, STAT4, PON1, PSORS9, LANCL1, H3P10, GOT2, CHI3L1, AKT1, EDN1, GSTM1, CXCL10, MAPK14, TIMP3, NXF1, PLAAT4, CSTA, HMOX1, FLNB, HLA-DPB1, TNFRSF1B, POLDIP2, AHSA1, ADAM17, HMGB1, S100A1, MTX1, MAPK8, SOAT1, FCGR3A, SLC22A4, MYDGF, FGF7, SELE, CRK, CCL22, CCL5, CCND1, S100A7A, ADAMTSL5, SERPINB3, CD79A, GRAP2, ACR, CXCL1, GRN, KIR2DS1, USO1, KRT16, NFKBIZ, CCR5, GSTT1, MIR223, MIR99A, LOC102724971, PPARD, JUN, AIMP2, KYNU, RNF19A, PSORS5, WNT5A, SEC14L2, LOC102723407, DEFB103A, DEFB104B, ANXA6, SERPINA1, CXCR2, IFI27, IFN1@, CSF3, DEFB103B, APOB, POU5F1, RARRES2, CCL4L1, APOA1, TREX2, KLK7, HP, IL36G, CD1D, ZNF750, SOCS3, SLC9A3R1, GATA3, RIPK1, MBTPS1, GSTM2, CDR3, ACKR2, FOSL1, FZD5, TRPV1, FN1, HIF1A, SLURP1, CD1A, DEFB104A, TGM1, CASP1, CALCA, TAP2, PSORS1C2, CA2, HES1, CRH, C4B, C4A, HSP90AA1, PECAM1, CAV1, DUSP1, LTA, JUNB, TLR8, LGALS3, IL17D, LOXL2, OPRM1, CCR2, HPGDS, KDR, MMP2, AGER, IVL, GATD3B, PSORS1C3, CXCL9, MIF, ITGAX, LORICRIN, KIR3DL1, ITGAE, ISG20, NFE2L2, NF1, PIPOX, FLT4, SLC22A5, HLA-DOA, ACE2, HRH4, NLN, OR2AG1, CLDN3, SEMA3A, CPQ, S100A14, TIMP1, HAX1, TGM3, TGM2, HBEGF, MMP9, PDPN, TFPI, SYT1, HNRNPA1, ITGAM, MUC1, RPTOR, ALB, C5AR1, ABCC1, MRC1, STAT1, TRIM21, TSC22D3, SPP1, SOX9, MIB1, GNLY, FOXO1, AGT, DUT, TNFRSF4, ISYNA1, JAK2, CD14, GABPA, JUND, TNFSF10, GCG, SLC12A8, GORASP1, DEFB1, IL25, CCL4L2, ACD, GEM, CYP24A1, NR1I2, WNK1, ABCG2, KRT14, BCL10, CYP27A1, TGM5, IL32, CYP27B1, MYOM2, CCR10, CCR4, PCLAF, CD69, NR1H3, UBE2N, UVRAG, CD27, SHBG, FOS, VIP, CD274, MC1R, IL22RA2, TNFSF8, FOSB, LPA, CD34, MBL2, AKT3, CMKLR1, DPP4, OPN4, CCR7, ACTB, CD40LG, ROBO3, SLC6A4, S100A4, COPD, IFI16, ETFA, TRIM32, TNFRSF11B, KLK6, DKK1, CTNNB1, F2RL1, SLC52A1, PTEN, MIR126, CTLA4, FABP4, CCN1, OLR1, ANGPT2, RBP1, WDHD1, IL26, NT5E, RELA, ANGPT1, FCGR2A, RPE65, IL4R, IL7, ESR1, SLCO1C1, PDE3A, PDCD1, MIR424, CYP1A1, ABCB1, ELANE, REG3A, CX3CR1, IL12RB2, CTSS, PLAT, CXCR1, MIR17HG, QPCT, PLG, POMC, KLK5, EPAS1, IL20RB, MIR210, ORM1, CTSG, NOTCH4, OAS2, LCE3A, BDNF, SELP, SAA1, FGF2, SAA2, CX3CL1, CKAP4, FCGR3B, S1PR1, SELPLG, CXCL11, SERPINB4, HTR2A, IRF5, CCL4, C4B_2, MALT1, NAT9, CCL19, IRF2, NOTCH2, NOS3, BSG, BTNL2, BCL2, IFNL1, IRF1, CTSL, MUL1, CRHR2, CLEC7A, TREM1, MAVS, NAA16, NBAS, DUOX1, SLC12A9, DGCR8, CCR1, AKR1B10, CKLF, FYCO1, MED15, CLU, CYBB, RIPK4, IL17RD, PSORS7, TET2, KIR2DL5A, LYNX1, MARCKSL1, PGLYRP4, PIAS4, CYLD, CLC, CTSV, CRHR1, TNFRSF12A, IL20RA, CYP2E1, CYP3A4, CYP2D6, COX8A, CST6, TRMO, CYP2B6, PROK2, SUGP1, SCLY, ACOT13, CRABP1, IL22RA1, TNFAIP8L2, JAM2, ZC4H2, SULF2, CXCL16, WDR11, TRIB3, SPHK2, ITLN1, CTSB, AZI2, CD244, CYP2C19, GRHL3, CALML5, POMP, SUCO, MOCOS, CNR1, ATOD1, COMT, SEMA6A, NCOA5, CCN2, PRO2268, MSX2P1, NANS, KIR3DL2, CHRNA4, MIR193B, APP, MIR17, MIR187, MIR197, MIR19A, MIR20A, MIR221, BIRC2, TNFSF12-TNFSF13, SLURP2, MXRA7, MIR330, MIR340, MIR369, ANXA2, PLF, WG, PRR9, MIR146B, APRT, MIR145, MIR143, C10orf99, ATP5F1A, ASIP, TICAM2, STS, ARG1, IL31, SUMO4, NPSR1, RTL1, MIR130A, AQP3, AQP1, FASLG, FAS, MIRLET7B, MIR122, MIR125A, MIR125B1, MIR492, MIR486-1, CLMN, POU5F1P3, MIR4516, AHCY, AGTR1, JAG1, ADRA1B, PARP1, MIR6731, ADH1B, ADCYAP1, ADA, LINC02210-CRHR1, UPK3B, LOC110806262, LYNX1-SLURP2, LINC02605, H3P9, ACACA, H3P23, AOC1, COMMD3-BMI1, ALOX12, PWAR6, MIR744, ANXA1, POU5F1P4, SLC25A5, NME1-NME2, HNRNPA1P10, ANPEP, POTEF, MIR876, C20orf181, ALOX15, AMH, CD24, KLLN, PRINS, ALOX15B, MIR320B1, TMED7-TICAM2, AAA1, HCP5B, LINC01193, BCL3, LINC02693, IL17RC, CDC6, CD68, CD59, CD47, ENTPD1, DMKN, MUC16, CD38, SCARB1, CDCA5, PIK3IP1, TXNRD3, PGLYRP3, TNFRSF13C, TNFRSF8, CD86, CD28, CD19, CDH13, RNASE7, SPINK7, CEBPD, VASH2, EHMT1, WLS, GRHL2, NAA25, ZC3H12A, SCUBE1, CETN1, SLC38A1, CDK2, CDKN2B, SHARPIN, CDKN1A, TLCD3B, ANGPTL6, STK40, CDK5, CDK4, CD1C, OSCP1, CD1B, BMX, KIAA1324L, VAMAS6, C3, IPMK, CADM2, BST2, PCSK9, TAB3, BMP6, CACNA1A, CXCL17, SLC9C2, TCAIM, HCG22, BMP4, BMI1, CFB, TNFRSF17, VPS51, OLIG3, CCNB1, YDJC, CCKBR, PSORS8, CCKAR, CCK, TEPSIN, B3GNTL1, GLIS1, SDF4, CASP5, SLC25A20, PLB1, CASP3, FGD5, CAPN1, AMOT, CBLL2, SPRED1, C16orf82, HSPA14, NES, TMED7, SEA, RPA1, RYR1, S100A2, ID4, ID1, ICAM1, S100A12, IARS1, TSPAN31, MSMO1, HTR3A, SCN7A, CCL3, CCL7, HTR1A, HTC2, CCL21, CXCL5, SDC1, TRIM27, RENBP, IFNAR1, PTPN11, PSMB9, IL1RAP, PSMD9, PTH, PTGS1, PTPN1, IGH, PTPN6, PTPRC, IFNB1, PTX3, PZP, MOK, RARA, RARRES1, RBP4, IGF2, IGF1R, CXCL12, SETMAR, SOST, SFRP4, TCF3, TCF7L2, TCF19, TRGV5, HLA-G, HLA-E, THBS1, TIMP4, HLA-DRB5, TLE1, TLR1, HLA-DQB1, HLA-DQA2, HLA-DMB, TNNT1, TNXB, HLA-DMA, TPM1, TPM2, NAT2, TBCC, TAZ, SOD3, SFTPD, ST6GAL1, HSPD1, SLC22A1, SLPI, SNAI1, SNRNP70, HSPB2, HSPB1, TAT, SRF, HSPA1B, HSPA1A, STAT6, AURKA, HOXA5, TAL1, FOXA1, PSMB8, PSAP, IL3, MAPK9, MMP10, MMP12, MMP19, MNAT1, CD200, COX2, MYD88, NAGLU, NAP1L1, NCAM1, NEDD8, NFATC2, NFATC4, NFKBIL1, IRF3, NHS, NM, NME1, NME2, MMP8, MME, MGST2, LRP6, KLK1, KLRC2, KRT7, KRT10, LAG3, LAMP1, LGALS1, LNPEP, LRP5, MGAT5, LYN, LYZ, SMAD2, SMAD7, MCAM, SMCP, MDM2, MFAP1, IRAK1, NRTN, NTRK1, PLCD1, IL16, IL15RA, IL13RA2, IL13RA1, PKD1, PLA2G1B, PLA2G2A, IL12RB1, POLR2G, PHB, IL12A, PPARA, IL9R, PPBP, PPP3R1, PPP6C, PRELP, PRKCZ, TNFRSF9, PGR, NTS, SERPINB2, NTSR1, NR4A2, OPRK1, OSM, OXA1L, P2RX7, FURIN, SERPINE1, CNTN3, PGF, PRKN, INSRR, PDC, PDE4B, PDE4D, PDE7A, SERPINF1, PF4, TPO, TRAF6, TRPC6, DDX39A, LYVE1, SUB1, RAB40B, COPS5, SPINK5, KLK11, LILRA3, EFEMP1, SLC7A9, PWP1, FANCE, FABP7, KLK8, FABP1, TREX1, F13A1, COPE, IKZF3, NLRP1, FCN2, HPSE, NCKAP1, TACC2, SPRY2, TRAIP, FLT1, TLR6, FOXO3, SEMA4D, ZNRD2, MRPL28, SLCO1B1, KIR3DS1, POSTN, KHDRBS3, FOXC1, DCTN6, TNFSF13B, B3GNT2, PTTG2, MASP2, F3, EZH2, CD93, NXT1, EHF, SLC17A5, EDA, ECM1, IL36B, ATN1, R3HCC1L, TRAJ23, TBK1, OPTC, DNASE1, DLAT, DHFR, CRNN, AKR1C1, IL21R, DBH, TAS2R13, FGF21, TES, STAB1, TNFRSF13B, EPHB2, ENG, EMP1, DMXL2, SATB2, ELN, SERPINB1, BRD4, SMUG1, PCDHGA12, EIF4E, PLXNB2, EGR1, SH3BP4, TFIP11, PRDX5, LPAR1, PNISR, FLT3, LHFPL6, TXK, PDZK1IP1, NR0B2, GALR3, HAT1, LMO4, GSTP1, TNFSF11, AOC3, AKR1C3, SOCS1, TNFSF12, GSK3B, TNFRSF11A, TNFRSF10B, CXCL2, NRP1, CD84, GGH, GRIN1, CDK5R1, PLA2G10, FZD8, NRIP1, WT1, TYR, CFH, UGT8, VCAM1, GZMB, GZMA, WNT7B, WNT10B, XBP1, FGF23, YWHAZ, ZFP36, ZNF148, CXCR4, PSORS3, DDX39B, PLA2G7, NR4A3, APLN, GPX4, GPX1, MTOR, RNF7, AATK, ISG15, GAPDH, IFI6, BMS1, FZD2, ACKR1, FPR2, NAPSA, FAM20B, FGF19, FOSL2, ACOT8, TANK, EDIL3, BCAP31, EBI3, GCHFR, GH1, SPHK1, USP2, FFAR2, MCHR1, BAIAP3, BTRC, HSPB3, PGLYRP1, GPR15, GPRC5A, SYNGR1, CBLIF, CXCR3, LRRFIP1, GLP1R, RPS6KA5, MAPKAPK2, CD163, PPIG, GJA1, NAT1
-
Cardiovascular Disease
Wikipedia
They include family history, coronary artery calcification score, high sensitivity C-reactive protein (hs-CRP), ankle–brachial pressure index , lipoprotein subclasses and particle concentration, lipoprotein(a), apolipoproteins A-I and B, fibrinogen , white blood cell count, homocysteine , N-terminal pro B-type natriuretic peptide (NT-proBNP), and markers of kidney function. [56] [57] High blood phosphorus is also linked to an increased risk. [58] Depression and traumatic stress There is evidence that mental health problems, in particular depression and traumatic stress, is linked to cardiovascular diseases. ... Obesity and diabetes mellitus are often linked to cardiovascular disease, [70] as are a history of chronic kidney disease and hypercholesterolaemia . [71] In fact, cardiovascular disease is the most life-threatening of the diabetic complications and diabetics are two- to four-fold more likely to die of cardiovascular-related causes than nondiabetics. [72] [73] [74] Screening Screening ECGs (either at rest or with exercise) are not recommended in those without symptoms who are at low risk. [75] This includes those who are young without risk factors. [76] In those at higher risk the evidence for screening with ECGs is inconclusive. [77] Additionally echocardiography , myocardial perfusion imaging , and cardiac stress testing is not recommended in those at low risk who do not have symptoms. [78] Some biomarkers may add to conventional cardiovascular risk factors in predicting the risk of future cardiovascular disease; however, the value of some biomarkers is questionable. [79] [80] Ankle-brachial index (ABI), high-sensitivity C-reactive protein (hsCRP), and coronary artery calcium , are also of unclear benefit in those without symptoms as of 2018. [81] The NIH recommends lipid testing in children beginning at the age of 2 if there is a family history of heart disease or lipid problems. [82] It is hoped that early testing will improve lifestyle factors in those at risk such as diet and exercise. [83] Screening and selection for primary prevention interventions has traditionally been done through absolute risk using a variety of scores (ex. ... A 2015 Cochrane Review found some evidence that interventions aiming to reduce more than one cardiovascular risk factor may have beneficial effects on blood pressure, body mass index and waist circumference; however, evidence was limited and the authors were unable to draw firm conclusions on the effects on cardiovascular events and mortality. [111] For adults without a known diagnosis of hypertension, diabetes, hyperlipidemia, or cardiovascular disease, routine counseling to advise them to improve their diet and increase their physical activity has not been found to significantly alter behavior, and thus is not recommended. [112] Another Cochrane review suggested that simply providing people with a cardiovascular disease risk score may reduce cardiovascular disease risk factors by a small amount compared to usual care. [113] However, there was some uncertainty as to whether providing these scores had any effect on cardiovascular disease events.NOS3, LPA, APOB, APOE, ADRB1, ICAM1, ALB, LPL, GH1, APOC3, MPO, MTHFR, NPPB, VWF, EDN1, VCAM1, PON1, ACE, CETP, PTGS2, CST3, CCL2, AGT, CRP, HP, CYP2C19, NPY, SELE, CBS, SORT1, PON2, GPX1, GRK2, GRK5, SLC12A2, TNNT2, AGER, HMGCR, CHDH, LDLR, FTO, PCSK9, VDR, ABCG8, FBN2, LIPC, SH2B3, CASZ1, CYP11B2, DNMT1, TNC, DBH, INSR, PRKAG2, ADH1B, MEPE, BACE1, SCAF1, GCKR, MYBPC3, FUT2, CDKAL1, WRN, NPC1, PLPP3, RORA, MRAS, NPR1, SLC7A1, MCF2L, GUCY1A1, COL4A1, SEPTIN9, TNFRSF11B, OPRL1, PLG, TANGO2, EBF1, GOLGA6A, PLA2G2A, PKHD1, AAGAB, IL1B, PIK3CG, ZNF831, SERPINE1, UPF3A, CELSR2, EPHX2, COPD, HNF1A-AS1, PAX2, NDUFAF6, ENPEP, OR10A4, EFCAB13, PRDM16, DPYSL2, HOXB-AS3, BAHCC1, LINC01122, PREX1, CYBA, LINC02245, RBP4, CASC15, REG1A, PTX3, REN, PLEKHG1, REXO1, LIN54, JPH2, RGL3, SAA1, LINC01344, IGF1, DPP4, DECR1, DPEP1, PPARA, PPARG, EBF2, DLG2, HS1BP3, PRKCE, TMBIM1, PTH, CCDC148, OLR1, DBP, THADA, ACE2, CPEB2, ABHD17C, THAP9, LCORL, ESR1, TIMD4, CADPS2, FGD5, PRDM6, GML, HMOX1, GLP1R, LINC01312, FREM1, ZFAND2A, CDCA5, IL6, LSP1, NAV1, DOCK7, UBASH3B, HOXB3, MBL2, ADO, LGALS3, LEP, HOXC4, APOA5, IL18, ARHGAP42, OR10AD1, HIVEP2, INPP5A, HGFAC, IL10, AGAP1, C1QTNF4, LCN2, PHF13, APCDD1L-DT, NLRP3, GTF2I, KNG1, KPNA4, SLC9B1, NR3C1, HOXB5, GCG, ESR2, HPR, FES, NFATC2, NFE2L2, CCDC170, BICC1, NMT1, DBH-AS1, SVEP1, FABP4, FGB, TTC41P, NPNT, F3, RNF214, F2, MECOM, EHMT1, ALG9, MYO9B, MXI1, KDM2B, ANO5, NAA38, NR3C2, MMP2, GABPA, TCHP, UNC13D, MMP9, DCUN1D5, ZCCHC7, TMEM72-AS1, NKAIN4, PDILT, NCALD, MXD3, CABCOCO1, APOLD1, DLEU7, CMIP, NFKBIA, ZDHHC18, ATXN2, SELENBP1, PKD2L1, PDYN-AS1, ATP2B1, RERE, S1PR2, SLC9A3R2, KL, ADIPOQ, DNAJC27-AS1, DDX23, MYEOV, FGF21, GDF15, ZGLP1, GOSR2, USP34, KANK2, PNKD, APOA1, AVP, ZPR1, CACNA1D, MACROD1, VPS51, PLCE1, HSD17B12, AXIN1, MAD1L1, ATL1, APOL1, RUVBL1, UNC5C, DELEC1, UBN1, BDNF, UHRF1, ALDH1A2, LINC00452, HNRNPFP1, HRCT1, APLN, HIPK2, GIT2, C22orf31, HOTTIP, CERT1, CCND2-AS1, CRTC1, ZNF268, PDE10A, ADM, ADK, PLCB1, UTS2, ADCY9, SWAP70, CLUAP1, SORCS3, NT5C2, HSF2BP, LINC02646, ABO, HIBADH, IL1RAPL1, ABCA1, SORBS1, AGTR1, ATG7, C5orf67, ANGPT2, LHFPL2, TSPOAP1-AS1, GLYAT, TARID, DLEU1, LINC02227, LINC01478, LIPC-AS1, AHSG, ALOX5, CFDP1, ZFPM2, C1QTNF7-AS1, ALDH2, LINC01169, LINC02576, SIRT1, FGF23, RPS6, CACNB2, CHI3L1, CD36, TNF, TLR4, CCHCR1, TGFB1, ARL15, TERF2, PLA2G7, PAQR5, PGPEP1, RNF43, TXNL4B, BANK1, MIR21, ZC3HC1, ARMC4, RCBTB1, SPP1, CLCN6, SOS2, TBC1D19, ABCF3, SOX6, SLC5A2, SHBG, SELP, RETN, C1GALT1, CD14, BCAS3, CCND2, SPTBN5, WNT2B, YES1, IRF1-AS1, PALM2AKAP2, WT1-AS, LIMA1, VEGFA, CAD, ANGPTL3, ADAMTS13, MGP, PGR-AS1, IL1A, HGF, ELN, APLNR, DSPP, COX2, FLNA, SMUG1, BMS1, IL6R, TCF7L2, ACACA, BGLAP, IL33, ADRB2, NOX4, PIK3CA, ACTB, CXCL12, SOST, MTCO2P12, SIRT3, CDKN2A, PIK3CB, NHS, TM6SF2, TNNI3, PIK3CD, CDKN2B-AS1, EDNRA, COMT, CX3CR1, RAPGEF5, ROS1, RARRES2, MOK, GPT, SIRT6, HSD11B1, PNPLA3, MIR155, S100A9, S100A12, ANPEP, AGTR2, ANGPTL4, LIPG, GIP, CD40LG, CTSS, F5, HFE, IMPACT, ATM, HIF1A, CXCL8, SCARB1, MIR146A, GNB3, CYP19A1, ADA, PDE5A, THBD, TXN, ANGPTL2, LOX, CHGA, PPIA, ATP6AP2, CP, PLA2G1B, SAA2, UCP2, UMOD, S100A8, SCN5A, SLC33A1, CXCR4, SLC6A4, MTTP, WASF1, UGT1A1, SELENOS, CXCR6, ABCG1, CLOCK, NOS1, MMP1, RMC1, PROS1, THBS1, MMP8, PTGS1, LEPR, NPPC, PTPN1, MMP12, TNFSF11, NTS, KDR, VKORC1, KCNQ1, PPIG, SOD1, PLA2G6, KLF2, STAT3, ANGPT1, ADRA2B, CIMT, FABP3, CD34, MIR126, G6PD, MIR210, AHR, CAV1, NQO1, FBN1, GSTK1, C20orf181, KIF6, CCR5, CFH, SLCO6A1, BTF3P11, MIR499A, CDKN2B, ELANE, ENG, EPO, KLK3, ARSA, HLA-DRB1, HSPD1, FSHMD1A, SOD3, SOD2, COL4A2, SSTR4, TM7SF2, MIR221, TH, MIR34A, MFAP1, FSD1L, FSD1, ACCS, USF1, CAT, CASP1, MAS1, CA2, PLA2G10, BRS3, BRCA2, MIR145, ACKR3, FOXO3, FABP2, PAPPA, SERPINF1, C1QTNF9, DHX40, PLAG1, AGXT2, P2RY12, PPARD, F7, NPR3, F11, MAPK1, NPPA, MAPK3, SHMT1, CYP7A1, CXCL16, NOS2, TRIB3, PTGIS, RAPSN, CTH, CRYGD, GJA1, CCL5, APOM, SELENOP, ACSS2, BRCA1, TYRP1, PSAT1, GPR42, TMEM54, LPAR2, PARP1, POLDIP2, ADRA1A, APRT, NPEPPS, LBP, IRS1, TBPL1, WASF2, PADI4, IL17A, ALOX5AP, KCNH2, CACUL1, ABCC6, PPARGC1A, PLA2G15, HPGDS, F2RL3, TNFRSF11A, DNER, ITGB3, HSPB2, NR1I2, HSPB1, CAPN10, ABCB6, HSPB3, GPER1, B2M, CYP2J2, RFC1, ROCK1, PART1, NAMPT, RENBP, CYBB, MIR122, CTSK, GDF11, AMPD1, TRIB1, SLC35A1, MALAT1, DDAH2, PDE4A, FSTL1, PGF, ABCB1, MMRN1, CST12P, ADD1, PLAT, DUSP1, PLTP, ADH1C, POMC, ADRB3, AHSA1, PRKAA1, CBSL, PEAR1, ABCG5, AKR1B1, NOD1, BRD4, S100A1, CPB2, MAPK14, TP53, MIR22, NOX1, TFPI, CCR2, MIR33A, IL32, IL1RL1, TLR2, TNFRSF1A, TNFRSF1B, IL23A, MIR208A, VCL, CALR, MGAM, CALCR, SLC25A20, AIMP2, GP6, C4B, TSPO, KLF15, GRAP2, FADS2, RNF19A, SI, MIR132, CRK, CRH, KEAP1, PDGFC, APOA4, TNFSF10, COX8A, MIR143, HTR2A, SLC2A1, ST2, SLC2A2, SLC2A4, GAL3ST1, ROCK2, CNR1, SLC17A5, HAVCR1, CLU, NTN1, ARG1, SERPINA5, ANGPTL8, FABP1, INSRR, MSRA, GSTT1, CD180, FN1, EPRS1, FOXO1, FLAD1, BPIFB4, ADM2, PHACTR1, GLUL, FCGR3A, IGFBP3, HSPA5, ADIPOR2, IGFBP1, MMP7, LTA, SPX, IL4, FGG, MUSK, MME, HSPA1A, LCAT, FCGR3B, GOT2, MTR, HABP2, MEG3, WDR1, CNR2, NR1H4, DIANPH, SOAT1, GPR35, MRGPRX3, CUBN, LYPD4, APOA2, TRIM21, MTOR, APOC1, APOC2, LTC4S, ABCC9, SLC22A3, C3, CYP4F3, SLC2A9, HLA-B, INS, ANK2, CXCL5, DDAH1, ANXA1, SELPLG, NRG1, TUBB1, SFRP5, SFTPD, FZD4, COX1, FLNC, FLT1, SLC6A2, CHRNA5, CISD1, SLC6A8, MIR150, MRGPRX4, FMO3, HLP, MIR208B, APP, GHS, IL37, CD47, XPR1, CD44, MAP3K5, GDF2, CD38, GSTM1, CRISP2, TRAF1, HSP90B2P, RHOA, TTR, RNU1-1, MEFV, MEF2A, PDE3A, MDM2, SIRT7, GCH1, VASP, CD5L, ANKRD1, TNFSF12-TNFSF13, VN1R17P, GPR166P, ASIC3, BEST1, MET, CD69, MAOA, HDL3, MIR214, JAK3, TAC1, IL15, MIR222, MIR223, MIR28, TAT, PCSK7, HNF1A, MIR29A, OXER1, GPBAR1, TET2, GAS6, PTPN22, KMT2A, BAG3, CALCA, CDH13, GPX3, PXDN, KCNJ5, GATA4, HOPX, TREM1, HFM, CR1, LTA4H, IL11, IGF2BP2, HSPA4, PON3, IGF2, MRGPRX1, IRS2, PPBP, C1QL1, ETV3, PTPA, MLC1, PRKAA2, PRKAB1, NPR2, PRKCD, MIA3, BID, NOD2, NOTCH3, AOC3, DAPK3, LGR6, PROC, NEDD4L, CYP11B1, PSEN1, NOTCH1, FBLN1, ARSI, PER2, ADAM10, WNK1, LOC110806262, ENPP1, HTR2C, PDZK1, PECAM1, PAH, PF4, P2RX7, RIPK2, SERPINA1, EGR1, ACLY, PITX2, OGN, TNFSF12, RAPGEF4, PLA2G4A, EPOR, PLAU, DKK1, MYOCD, PINK1, IFNG, PLN, ERG, KCNIP3, HAMP, ARMS2, TRPV1, S100B, KLF14, KLF5, RLN2, CCN2, CTF1, MBL3P, DEGS1, POSTN, ALOX12, UCMA, NBN, FBLN5, CRYZ, BSG, IL6ST, MIRLET7G, MIR106A, RPS6KA1, RAPGEF3, PNPLA2, AKR1A1, FADS1, FGA, RGS2, GIPR, IL1RN, LPAR3, PTGIR, GRK3, HMGB1, GPR151, CYP3A4, GPRC6A, CYP2D6, LRP5, ZC3H12A, OGT, CYP2C9, SCD, CYP1B1, CYP1A2, SAT1, FGFR4, NGF, AKT1, ZNF385B, SETD2, NAAA, AGO2, MYLIP, BRD7, PPM1K, SLC27A6, MIR486-1, BTLA, PDCD4, PDLIM3, GGTLC5P, GGT2, SFTPA1, GGTLC3, NCF1, MAT2B, TOR2A, MIR652, CYTOR, SERP1, COA3, UCA1, REM1, MIR642A, MUC16, POTEF, TMEM43, PLB1, TARDBP, CAMTA1, SZT2, PPARGC1B, TP53COR1, PROKR2, TPSD1, TRS-AGA2-3, SFTPA2, FUNDC1, ACTRT1, HIF1A-AS1, MIR499B, TSPAN15, ARL2BP, APOA1-AS, PHLPP1, RBM45, ARL6IP1, LPIN1, ERICD, CNOT1, RN7SL263P, SIRT1-AS, SIRT2, ATF6, LOC107987479, GNPDA2, LOC110599569, CARD8, ADGRL1, TDGF1P6, TTC7B, NRG4, DAPK2, ATRNL1, TUBA3D, AKR1B1P3, TXNRD3, COMMD7, TIRAP, GGTLC4P, CYGB, FBXO8, PSS, OSBPL11, C1QTNF3, TES, CILP2, C1QTNF6, LRIG1, LINC01672, OMA1, SLC22A12, BEAN1, POU2F3, TXN2, PRDX5, SAMM50, GCA, CERNA1, ZNF318, MTCH2, SENP1, BRD7P3, AAA1, MIR483, MIR98, NPS, SLC44A2, LPAL2, SMYD2, MIR130B, PPP1R2C, PDGFD, MIR125A, PDXP, TBX20, MIRLET7C, C1QTNF12, ASRGL1, ARFGEF3, JPH3, AS3MT, AHRR, ZBTB12, SEMA6A, LINC01194, MIR139, SPIRE1, SPHK2, MIR142, ENAH, KDM3A, MIR17, CENPJ, MIR148A, ARID5B, USE1, SESN2, GPRC5C, MIR144, STOX1, APOL6, TBATA, ZEB1-AS1, MYDGF, COL18A1, CLDND1, JCAD, CHD8, CHD7, BTBD8, TAS2R50, BCL11B, IS1, ACD, TNFAIP8L2, HCAR2, GPIHBP1, BPIFA4P, KCTD1, SERPINA13P, UBE2Z, PAGR1, NOX5, KCTD15, SLC22A24, MAPKAP1, MBOAT7, NEAT1, NIBAN2, GORASP1, ECHDC3, NANOS3, STN1, TNRC6C, SPTBN4, IL27, PRM3, FNDC5, TRPV4, HEATR6, SLC27A1, ENHO, FRMD3, FBRS, AGBL2, HHIP, TICAM2, HCP5B, HSD17B13, ATF7IP, PLXNA3, UBIAD1, ANGPT4, MIR378A, UCN2, CRIM1, KLF3, CD320, TLR8, TNFRSF12A, KRT90P, MIR342, MIR99B, WNT16, MIR331, TRPV2, SYNPO2, PCYOX1, DCDC2, ADTRP, MIR17HG, HSPA14, INSIG2, ABHD5, ADIPOR1, DAB2IP, DSE, CERS2, CALML6, RHOD, VSX1, MIR451A, MIR429, BDNF-AS, IL22, MIR383, PAQR7, IRX4, FOXP3, MLXIPL, MIR382, TNNI3K, CNDP1, MIR99A, SLC35E3, PPP6R3, RLS1, MIR216A, RMST, ATG16L1, PRMT6, NADSYN1, SPRED2, NAT10, MIR200B, SCLY, MIR197, ZBTB7C, PRSS55, MGLL, MIR188, MIR185, KIAA1109, GPRC5D, TRPM7, DYM, TRPM4, MARCHF5, MED15, LSR, GHRL, PTCRA, MIR96, SPZ1, ARID4B, ZGPAT, INPP5K, IL17D, TPCN1, FAM3B, HHIPL1, MIR30C2, UGT1A6, MIR30C1, MIR29C, COG2, A2M, NRM, GCLC, GRIA1, FFAR2, GPR15, UTS2R, CXCR3, GP5, GJA5, CXCL2, GJA4, GGT1, GDF10, MSTN, GCK, GC, GRN, GSN, EDNRB, HLA-DQA1, HSD11B2, HRH1, HPD, HOXD13, HNRNPA1, HNF4A, HLA-DMA, HBA1, HLA-C, HDLBP, HDAC2, HTT, HCRTR2, HCRT, GAP43, GALR1, GAD1, ETV2, FAAH, F13A1, F12, F9, F8, F2RL1, FBL, ACKR1, ERCC3, EPHX1, EPHA1, SLC29A1, EGFR, EFNB2, FANCD2, FAT1, FCGR2A, FDFT1, FDXR, FER, FGF2, FHL1, FHL2, VEGFD, FOXC2, FOXM1, FLNB, FLT3LG, FOLH1, FOS, FOSB, HSF1, HSF4, HSPA1B, MC3R, MFAP4, MEN1, MAP3K3, MEIS1, DNAJB9, ADAM11, MBP, LRP1, MB, MAT1A, MAPT, MAP4, EPCAM, LRPAP1, MIF, FOXO4, MMP3, MMP11, MMP13, MMP14, MPI, MPZ, MRC1, MSR1, MST1, MT1A, MT1B, CYTB, ND5, RNR2, MTRR, LRP6, LOXL1, HSPA2, IL5, ITGB4, ITGB2, IRF6, IRF5, CXCL10, TNFRSF9, IL4R, LNPEP, IL1R1, IGFBP4, IGFBP2, IGFALS, IDH2, IDE, ITPA, JAK2, JUN, JUNB, JUND, KCNE1, KCNJ11, KCNMA1, KCNN4, KLKB1, KRT7, LGALS1, LGALS2, LGALS3BP, LIPA, LIPE, LMNA, EEF1A1, EDN2, TRNL2, ATP4A, CCND1, BCHE, BBS4, BBS1, ATP5PF, ALDH7A1, ATP12A, BCL2L1, ATHS, ATF3, ARSD, ARNTL, ARG2, AR, BCL2A1, BCL3, LPAR1, BMPR1A, CAV3, CASR, CAPN1, CAMP, CAPN5, SERPING1, BMP2, TNFRSF17, BHMT, CEACAM1, BGN, CFB, BDKRB2, BCR, AQP7, AQP4, AQP1, ACYP2, AKT2, NR0B1, AGRP, ACAN, ADRA2A, ADAM8, ACVRL1, APOH, ACP1, ACHE, ACAT1, ABL1, ABCA2, AAVS1, ALCAM, ALDH9A1, ALOX15, ALPL, ALPP, AMBP, AMCN, AMD1, AMD1P2, AMH, ANG, ANXA5, ANXA7, AOC2, APCS, APEX1, BIRC3, RUNX1, RUNX3, SERPINH1, NKX2-5, CYP2B6, CYP1A1, CTSL, CTSD, CTRL, CTNNB1, CSTB, CREM, VCAN, CSN3, CSN2, CSF3, CSF1, CRY1, CYP2C18, CYP3A5, CYP27A1, DACH1, DCN, DDT, DES, DHCR7, DMBT1, DMD, TRDMT1, DNMT3A, DRD4, TSC22D3, HBEGF, ECE1, TYMP, CRMP1, CPOX, CD6, CDK8, CENPC, CEBPD, CEBPB, CEBPA, CEACAM5, CDK9, CDC42, CORT, CDC25C, CD74, CD40, CD86, CD28, CD19, CES1, CETN1, CGA, CHGB, CHM, CISH, CLCN3, CLCN7, LTB4R, CMM, COL5A1, COL5A2, COL9A1, COL9A2, COL9A3, COMP, KLF6, TRNL1, MUC1, PACSIN2, MBTPS1, TMEM11, IQGAP1, IL18R1, IL18RAP, CREG1, TNFRSF10B, TNFRSF25, GGH, GALNT4, BECN1, VAMP8, SOCS1, TP63, DENR, SOCS2, CCN6, TNFAIP6, ARTN, DCLK1, OSMR, HGS, ARHGEF1, ANGPTL1, AIP, CCRL2, HDAC3, KALRN, TIMELESS, AP1G2, SPHK1, VNN1, ALKBH1, CYP4F2, RGS5, SOAT2, VDAC1, YWHAZ, XDH, WNT5A, NSD2, VTN, VHL, UTRN, NR0B2, UCP1, UCN, TYMS, TXNRD1, TNFSF4, TRPC1, LRP8, BSND, LEPQTL1, LAG5, TFEB, TFPI2, ARHGEF5, FOSL1, AAAS, HMGA2, AKAP1, TAM, ESS2, KDM5D, DYSF, ACOX2, BRAP, MSC, GPR55, CD163, PROCR, GJB6, NES, TRAF3IP2, OGA, CORIN, TXNIP, CIB1, FSTL3, STK25, FST, PPIH, IFI30, SPON2, APBB3, CCL27, HPSE, PROKR1, GADD45G, COPS8, PRDX3, GPR75, STIP1, KCNQ1OT1, KLK11, ESM1, WDR5, ADAMTS5, WDHD1, SLC2A6, NISCH, WIF1, EFS, SPRY2, SLIT2, H6PD, ELMO1, PCLAF, RIPOR2, NOS1AP, PPP6R2, NUP155, GMFG, SPRY1, ADAMTS1, CHST3, MAP4K4, AIM2, MED23, COX5A, MAGI2, RNF10, MFN2, SLC23A2, SCO2, NAALAD2, SRA1, PDCD6IP, SLC17A4, DNM1L, PTPRU, GPC6, PPIF, LRPPRC, G3BP1, ABI2, PRG4, TRAF6, TLR1, MYH7, PRKD1, EIF2AK2, MAPK10, MAPK9, MAPK8, MAPK7, PRKG1, PRKCA, MASP1, PRKAR2A, PRKAR1A, PREP, PPID, PODXL, SEPTIN4, PRL, RELN, TIMP4, RAC1, RNASE3, RNASE1, RIEG2, RELA, RASGRF1, RARA, PTPN11, HTRA1, PTGER3, PTGER2, PTGDS, TAS2R38, PTAFR, PSMA6, PMP22, PMM2, PLK1, NOTCH4, OPA1, OGG1, NR4A2, NUCB2, NT5E, NPY1R, NNMT, PLIN1, NME1, NM, NFKB1, NF1, MYOD1, MYLK, OPRK1, SLC22A18, OSM, OXA1L, PAEP, SERPINB2, PC, PCBP1, PDE4D, PDGFB, ENPP3, PDR, PEPD, SLC25A3, PIN1, PIP, PLAUR, BRD2, RNU1-4, ROBO1, STAT4, ADAM17, SYT1, ABCC8, SULT1A1, STK11, SULT1E1, SERPINA3, SMS, STAT2, ST13, SSB, SRM, SOS1, SORL1, TACR1, TAGLN, TAL1, TBXAS1, TRA, TERC, TERF1, TERT, TFR2, TFRC, TGFBR1, TGFBR2, THBS2, THBS4, THPO, TIAM1, TIMP1, SNAP25, SMPD1, RPE, SCN2A, CCL20, CCL7, CCL3, SCT, SCN10A, SCN8A, ACSM3, SMO, SAG, RYR3, RYR2, RYR1, RPS20, RPS6KB1, CCL21, CCL22, CCL23, SFRP1, SHC1, SLC1A5, SLC5A1, SLC6A7, SLC8A1, SLC9A1, SLC10A1, SLC12A3, SLC19A1, SLC20A2, SLC22A2, SLPI, SMARCA4, LOC113664106

-
Succinic Semialdehyde Dehydrogenase Deficiency
GeneReviews
Diagnosis Suggestive Findings Succinic semialdehyde dehydrogenase (SSADH) deficiency should be suspected in individuals with the following clinical, imaging, EEG, and supportive laboratory findings: Clinical features. Late-infantile to early-childhood onset, slowly progressive or static encephalopathy characterized by: Cognitive deficiency Prominent expressive language deficit Hypotonia Epilepsy Hyporeflexia Ataxia Neuroimaging features Cranial MRI that demonstrates: A pallidodentatoluysian pattern [Pearl et al 2009c], showing increased T 2 -weighted signal involving the globus pallidi bilaterally and symmetrically, in addition to the cerebellar dentate nuclei and subthalamic nuclei T 2 -hyperintensities of subcortical white matter and brain stem Cerebral atrophy Cerebellar atrophy Delayed myelination Magnetic resonance spectroscopy that demonstrates elevated levels of GABA and related compounds in the Glx peak (e.g., GHB [gammahydroxybutyrate, also known as 4-hydroxybutyric acid], glutamate, and homocarnosine) EEG findings. ... Clinical Characteristics Clinical Description SSADH deficiency is characterized by a relatively non-progressive encephalopathy presenting with hypotonia and delayed acquisition of motor and language developmental milestones in the first two years of life. Common clinical features include intellectual disability, behavior problems, and motor dysfunction. ... GABA transaminase deficiency was first reported in an index sibship and an additional unrelated proband [Medina-Kauwe et al 1999], and then in an infant identified using MR spectroscopy [Tsuji et al 2010].

-
Alkaptonuria
GeneReviews
Alkaptonuria is caused by deficiency of homogentisate 1,2-dioxygenase, an enzyme that converts homogentisic acid (HGA) to maleylacetoacetic acid in the tyrosine degradation pathway. The three major features of alkaptonuria are the presence of HGA in the urine, ochronosis (bluish-black pigmentation in connective tissue), and arthritis of the spine and larger joints. ... Diagnosis Suggestive Findings Alkaptonuria should be suspected in individuals with any of the following major features: Dark urine or urine that turns dark on standing. ... A deep purple or black discoloration may be seen on the skin of the hands, corresponding to the underlying tendons, or in the web between the thumb and index finger. Arthritis, often beginning in the spine and resembling ankylosing spondylitis in its large-joint distribution. ... Comprehensive genome sequencing (when available) including exome sequencing, genome sequencing, and mitochondrial sequencing may be considered if serial single-gene testing (and/or use of a multigene panel) fails to confirm a diagnosis in an individual with features of alkaptonuria. For an introduction to comprehensive genomic testing click here.
-
Palmoplantar Keratoderma, Mutilating, With Periorificial Keratotic Plaques
OMIM
The digital constriction ('pseudoainhum') may progress to autoamputation of fingers and toes (Olmsted, 1927). Clinical Features Olmsted (1927) reported an Italian American boy who had onset of disease at age 18 months and, at 5 years, had symmetric massive yellow-gray horny plaques on his palms, divided into a mosaic pattern by several deep fissures. ... Several mitoses were seen within the basal and occasional suprabasal keratinocytes, and the mitotic index (2.5%) was greater than that of a control specimen. ... Because of extreme foot pain, the patient walked on hands and knees, resulting in mild palmar keratoderma, and used a wheelchair from age 3 years. Other features included increased sweating, thin brittle fingernails, and fine, dry, curly, and unmanageable hair. ... Examination of skin biopsies from 1 of the boys showed features suggesting that the disorder is related to epidermal hyperproliferation, including numerous centrioles by electron microscopy, increased mitotic count by histology, and increased numbers of silver-staining nucleolar organizer regions (AgNORs) on histochemical analysis.
-
Von Willebrand Disease, Type 2
OMIM
For a general discussion and a classification of the types of von Willebrand disease, see VWD type 1 (193400). Clinical Features Von Willebrand disease type 2, like VWD type 1, is characterized by excessive mucocutaneous bleeding, such as epistaxis and menorrhagia, and prolonged bleeding after surgery (Mannucci, 2004). ... Riddell et al. (2009) concluded that the defect was distinct from VWF type 2M, in that type 2M is also characterized by impaired binding to platelet GP1BA and can show a full range of associated VWF multimers. Other Features An association between aortic stenosis and hemorrhage from gastrointestinal angiodysplasia has long been recognized. ... Type IIE is associated with a reduction of high molecular weight (HMW) VWF multimers and a lack of outer proteolytic bands on gel electrophoresis, indicating reduced proteolysis. Genetic analysis of 38 such index cases identified 22 different mutations in the VWF gene, most of them affecting cysteine residues clustered in the D3 domain. ... She had very low levels of VWF and F8, and absent binding of VWF to F8. Clinical features included epistaxis, hematomas, and hematemesis throughout childhood.
-
Congenital Disorder Of Glycosylation, Type Ia
OMIM
In a review, Hagberg et al. (1993) stated that CDG I had been diagnosed in 45 Scandinavian patients and presented different clinical phenotypic features of the syndrome according to period of life. ... Hagberg et al. (1993) summarized the features of CDG IIa and compared them with those of CDG I. ... Barone et al. (2008) reported 2 adult Sicilian brothers with CDGIa confirmed by genetic analysis (601785.0001; 601785.0003). Clinical features in both patients included early-onset cerebellar atrophy, mental impairment, pigmentary retinopathy, and dysmorphic features. The younger brother, patient 2, was more severely affected and had additional features, including abnormal subcutaneous fat distribution, inverted nipples, genu valgum and flat and inverted feet. ... Patients from 12 families had a typical type I transferrin profile, but one had a variant profile and another, who had many clinical features of CDG type I, had a normal profile.
-
Heart Failure
Mayo Clinic
Irregular heart rhythms may cause the heart to beat too fast, creating extra work for the heart. ... Irregular heartbeats, especially if they are very frequent and fast, can weaken the heart muscle and cause heart failure. ... This quick and painless test records the electrical signals in the heart. It can show how fast or how slowly the heart is beating.NPPB, GRK2, TNF, ADRB1, NOS3, ACE, ATP2A2, NPPA, EDN1, HIF1A, AGT, AGTR1, IL6, PPARGC1A, CCL2, HMOX1, SOD2, MSTN, ADRB3, RAC1, HTR2B, GDF15, NRG1, PTH, ADIPOQ, IL1B, CRP, CCN2, AVP, REN, VEGFA, NR3C2, ALB, PIK3CG, EDNRA, GCG, PPP1R1A, PPARG, SIRT1, NFE2L2, CYBB, ADRA2C, PTGS2, TNFRSF1A, UCN2, SLC9A1, NOX4, SERPINE1, PRL, PON1, NOS2, VWF, EDNRB, RETN, PEBP1, TLR2, AVPR2, OLR1, POMC, EPHX2, GSK3B, CSF2, CS, XDH, CXCL8, NPR1, RBP4, IFNG, SOD3, TNNT2, ACACA, UCP1, CFD, CSF3, ACADS, ROCK2, GATM, HAND2, NRIP1, CAT, PDGFRA, TBX20, BAMBI, CSRP3, NOX1, PCK1, CIDEA, CASP3, PTGS1, MMP9, PDPK1, FIP1L1, INS, ITGB1, IGF1, MAP2K7, CALR, HSPB1, GHRL, KAT8, RYR2, PRKAR2B, SCN5A, TP53, LGALS3, CXCL2, GPX4, PLN, SOD1, ELOVL6, SOX4, MME, PLAT, ACE2, APLN, FASN, ADM, DSTN, FBLN5, ATP2A1, ACLY, ADRB2, APOC1, APCS, ATP1A3, PRKCA, SLC8A1, IL10, IL17A, ERBB2, MTPN, MMP2, CASQ2, POSTN, SIRT3, NR3C1, MYH7, MYH6, MMP1, HSF1, TIMP1, PRKCB, BNIP3, NOS1, GNAQ, AGTR2, ADAM17, SCD, NPY, TNFRSF11B, ALOX15, P2RX4, NCAM1, OPA1, TH, MAS1, HMGB1, PARP1, UCP3, TIMP2, BCL2, ENG, FXYD1, HP, SRI, BAX, PGF, KCNK2, CX3CR1, CORIN, KCNE1, HTR2A, IL6R, ERBB4, ECE1, TIMP4, DDR1, ARRB1, PPP1CC, DYNLL1, UCP2, RAMP2, FASLG, PLCD1, RNLS, MDH2, ENDOG, TUBB, FKBP1B, MMP13, DNMT1, MED1, LIF, CYP11B1, PLD2, MMP14, DIO3, MMP8, NOS1AP, FOS, TOMM70, OGT, CHRM4, CHRNA4, BECN1, SELP, BIRC5, NPTXR, FOSL1, SCN8A, CASP12, MYOD1, ALOX12, NGFR, GRK1, FTH1, ACAN, FAS, CDKN2B-AS1, TCF7L2, SLC39A8, FTO, RFC1, POLK, CELSR2, ADAMTSL1, SEMA5B, IL1A, ANGPT1, CMTM7, TM7SF2, TLR4, AMPD1, CERT1, ADRA1A, S100A1, EHMT1, REG1A, APLNR, GRK5, GPR42, FSD1L, C1orf21, ADRA2B, ANGPT2, SLC6A8, SSTR4, COPD, ST2, SLC5A2, LIPC, FADS1, LMNA, GRHL1, GCKR, GH1, ITPK1, GJA1, GLP1R, BRS3, CHDH, NUBPL, LCN2, TGFB1, OTUD7A, CYTH3, MAML3, ZGLP1, CST3, BEST1, MAPK14, FADS2, MIR21, LINC01782, SLC30A3, PIK3CD, LIPC-AS1, PIK3CB, PIK3CA, FGF23, CXCR6, SUGP1, CPNE5, ACTB, SIK3, ALDH1A2, MIR423, ZPR1, CALCR, BAZ1B, CA5A, VPS51, ANKS1B, ACKR3, LPAR2, MYBPC3, CELSR1, DSPP, TTN, PLCG1, BAG3, DPP4, DNAH8, TNNI3, ZCCHC8, DMD, FSD1, TTR, ATP2B1, C8orf37-AS1, ZFHX3, PPARA, IL18, CAD, DECR1, DES, LEP, HLA-C, KNG1, ATM, CYP11B2, GOLGA6A, APRT, RCBTB1, MFAP1, DNM1L, MIR25, MUC2, MARCKSL1, MRGPRX3, AIMP2, AGER, MAPK1, AKT1, VN1R17P, FZD4, BDNF, GPR166P, MRGPRX4, ALDH2, RNF19A, POLDIP2, LOX, STAT3, MRGPRX1, GPRC6A, SPP1, GPR151, LPAR3, AHSA1, CRK, LGR6, OXER1, GRAP2, HGF, UTS2, SLC33A1, PDE5A, HSPB7, GABPA, UTRN, GATA4, MEF2A, PGR-AS1, SGCD, KLF15, HAVCR1, JPH2, MTOR, PRKAA2, CAV3, BIN1, HSPA4, IL6ST, CSH1, CSH2, CYP2D6, MTCO2P12, MIR499A, CCHCR1, SELENBP1, CTF1, PCSK9, ERCC8, COX2, CRMP1, PPARGC1B, MPO, PKP2, DENR, CHGA, SHBG, EGFR, DAPK2, LRPPRC, MFN2, NM, HSPA9, TRPC3, MIR214, SRF, PPP1R2C, SMAD3, CENPJ, IL4, PRKAA1, PITX2, IL33, NPR2, NPPC, PRKD1, PTEN, INSRR, NFKB1, LPA, SERPINA3, DSP, TNFRSF1B, ATXN1, MMRN1, TIMELESS, MIR150, TRPC6, SDC4, MIR34A, BRD4, APOE, ANXA5, MIR665, EPO, TFAM, CD36, TAZ, CD34, TAC1, FSTL1, BRCA1, XBP1, ANKRD1, PRRT2, ARHGEF5, MUC16, HCN4, MIR221, SERPINA1, GINGF2, PENK, THBS2, PPIA, TLX1NB, MB, MAOA, SETD2, AXL, MIR126, TRPV1, PTPA, MIR22, PIN1, PTK2B, MIR19A, MIR223, MAP3K5, ABCB6, ABCB1, CCR2, FSTL3, ACVRL1, TXN, MIR199A1, RBM20, MIR146A, IL37, MIR199A2, TPM1, PINK1, MIF, CDK9, TPI1, DBP, CHAMP1, MS4A1, S100B, PNPLA2, RND3, TET2, FST, PSEN1, SCARA3, RBFOX1, SEMA4D, OSM, P2RX1, HCRTR2, NLRP3, PTPN1, IL34, P2RX7, NR4A1, TLX2, KRT20, IGFBP7, MOV10L1, P2RY2, MORF4, DNER, ROS1, MOK, RLN2, TNFRSF12A, AAVS1, NPR3, MAPK8, GRK3, SMYD1, SLC2A1, PRKAB1, HDAC4, FOXO3, TNNI3K, SLPI, CCK, CCN5, MLC1, ERBIN, PDE9A, PDK1, PIM1, CFLAR, P2RX6, MPRIP, NOD1, RACK1, SERPINA5, PDE4D, SLCO1B1, PDE4A, ARID1A, PDE3A, TNFRSF10B, LONP1, ANGPTL2, P2RX2, NNT, SPON1, PPIG, CD163, CXCR4, DUOX1, WNT5A, PCSK6, RIPK3, SMARCA4, SLN, GAL, SLC9A3, SLC6A2, SLC5A1, GP6, MAPK3, CASZ1, SGK1, RNF111, CCL21, CCL19, ISYNA1, SIRT6, NR1H4, RYR1, HDAC5, PTX3, RPGR, LUC7L3, ROCK1, DUOX2, STIM1, HAND1, QRSL1, VIP, VDR, PKD2, UMOD, ADCY10, GAL3ST1, UCN, SUMO1, TWIST1, TRPC1, TRAF6, CTCF, MFN1, GDF11, TNNC1, CHEK2, TLR3, HDAC9, TIMP3, TGM2, PYCARD, ZEB1, TBK1, UBR5, SLC35A1, MIR155, HSPD1, AVPR1A, F2RL1, NEXN, F8, IGHG3, IGFBP4, F10, IGF2, HCN2, ICAM1, BDH1, TNC, HTR4, BMI1, CPB1, ATP2B4, CXCR2, MIR212, ITPR1, ARRB2, HOPX, C20orf181, CRHR2, STS, G6PD, MIR208B, MIR210, ITGA2B, IRF1, INSR, CXCL10, ILK, CREM, CP, BMPR2, ERN1, BNIP3L, CASP1, MIR24-1, MIR27B, TRIM72, MIR342, MIR340, MIR29B1, GNB3, CRYGEP, GHSR, MIR29B2, MIR30A, FXN, GAS6, TMTC3, CAPN1, GSN, GSR, FGF2, HSF2, MIR216A, HRC, DBH, BSG, UCN3, FGFR4, RBM24, HFE, CFH, VEGFD, CAMK2D, CNP, MIR425, FBL, CRH, KCNH2, TMBIM1, ACTC1, CREBZF, ADCY6, MIR195, MYLK, PRDM16, MIR132, ACR, DSG2, COX1, DUSP1, S1PR1, MMP12, P2RX5-TAX1BP3, CYP1B1, HAMP, KMT2A, NQO1, P2RY1, P2RX5, P2RX3, ABCA4, CST12P, MIR18A, NT5E, ACHE, SLC24A3, CCN3, BIRC6, CYP2J2, CNOT3, ACADM, MMP3, MIR130B, ANXA2, MECP2, CRX, LAMP2, APP, APOA1, CRYGC, ENPEP, COL18A1, LTBP2, LUM, CTSD, CTNNA1, ANG, CTNNB1, ELAVL2, CHI3L1, MGP, ATG7, MIR19B1, ALAS2, AKR1B1, GNPTAB, TXN2, SLC17A5, MIR197, AMACR, TOR1AIP1, SMUG1, BACE1, DDAH1, MIR296, MIR30C1, MIR30C2, MIR30E, OPTC, MIR17, RBFOX2, MIR199B, MIR208A, OSBP2, MIR20A, FOXP1, OPLAH, IL17RA, PHLDA3, DIANPH, MIR185, PART1, BRD1, MIR183, ITGB1BP2, MIR200B, MIR23A, LRIT1, MIR182, MTO1, FAM20C, MIR33A, LIAS, KLRC4-KLRK1, PRDX3, PPR1, TMX2-CTNND1, CKAP4, FERMT2, LILRB4, BACE1-AS, SUGT1, NPPA-AS1, MIR1306, BVES, HLP, TRAP, WDHD1, INSL6, FASTK, FAME3, NISCH, AK6, PDXDC2P, CXCL13, PERCC1, SLU7, GNLY, SCGN, LINC02210-CRHR1, OPN1MW3, LILRB1, NPY4R2, LINC-ROR, MIR4491, OGA, YME1L1, TCFL5, PDE10A, MAP4K5, MIR744, SLC39A14, TRS-AGA2-3, PDS5A, NEDD4L, UFL1, MIR375, MIR328, MIR324, MIR148B, SF3B1, MIR409, LMOD2, POTEKP, PLIN5, TNFSF12-TNFSF13, MIR99A, MIR93, POFUT1, ARC, NRON, USP18, MIR652, MIR147B, MIR675, ECD, ACOT7, KLRK1, RNA18SN5, OPN1MW2, MIR650, POTEM, MIR92B, TRIM32, MON2, FRMD4B, MIR539, NCF1, GPD1L, PDS5B, COL6A4P1, MIR148A, ATP5MD, DDIT4, EGLN1, MCU, MLIP, UGT1A1, ADO, TRPM4, ORAI1, GTPBP3, MYLK3, SPZ1, MARCHF1, MOCOS, WIPI1, TCHP, EVA1A, BCL2L12, UNC93B1, DPP9, MTG1, UFM1, CGB8, NBAS, KLF13, TXNRD3, AZIN2, NLK, CMPK1, TGS1, OPN4, RTEL1, TIMM50, CGB5, MAP3K20, TPCN1, MYOCD, CNTN5, TLR9, ORAI3, CYTL1, SLC52A1, AGGF1, RMDN3, ZC4H2, USE1, CACNG6, RBM25, MYL7, BEX1, RHOU, CXCL16, ZKSCAN7, DPP10, TXNDC5, KCNT1, YLPM1, NLN, PDSS2, METTL3, KCNK13, PLSCR4, MAP3K7CL, GIGYF1, GORASP1, DCAF6, WNK1, ADI1, CDK5RAP3, ZC3H12A, DGLUCY, IMPACT, MEG3, ADM2, SLC30A10, CAAP1, SHCBP1, MYH14, SYBU, FKRP, ENAH, TMEM109, VKORC1, UBE2Z, GDE1, FHAD1, MIR145, BTBD8, SLC27A6, HIPK2, CHPT1, NRBP2, UBE2T, C1QTNF9, FFAR4, SLC9C1, LGALS13, ACTBL2, SYPL2, GNL2, RMC1, EMC10, MDFIC, KCNIP3, KCNIP2, EHD3, MCIDAS, SLCO1B3, C1QTNF1, MIR127, MIR144, MIR143, VPS4A, OXGR1, MIR142, MIR137, MIR134, BMP10, EIF3K, TOR2A, MIR106B, MIR100, NPSR1, MAT2B, MALAT1, ENHO, KCP, GSTK1, NEAT1, EHD2, RICTOR, SLCO6A1, LGALS16, UBAP1, BFAR, ASB14, FUNDC1, KLF14, TRPV2, PRKAG2, EMB, TPCN2, RBM45, CMPK2, SUCO, MYOM3, DNAAF1, JDP2, JAML, OMA1, GPBAR1, RMDN2, PPM1K, AMZ1, NOX3, PRSS55, SYCP3, DHRS7C, NLRP6, ASXL1, IL22, AK3, NTM, APIP, PPP1R18, ADAMTS16, HFM, RMDN1, SLC30A8, WBP2NL, ANGPTL4, MMP28, SELE, CIB1, GHRH, FRZB, XRCC6, GAB1, GAD1, GALNT1, GAPDH, GATA3, GC, GCH1, OPN1MW, GDF2, GFAP, CBLIF, FMOD, GNA12, GOLGB1, GP1BA, CXCR3, UTS2R, GPR17, GPR35, GPT, GRIN1, CXCL1, GSK3A, GSTP1, FN1, FLT1, EMD, FKTN, ENO2, EPHA3, EYA4, ESR1, ESR2, ESRRA, ETS1, ETV3, F2, F9, FABP4, FBLN1, FDPS, FLNA, FDXR, FGF4, FGF13, FGFR1, FKBP1A, FKBP1AP1, FKBP1AP2, FKBP1AP3, FKBP1AP4, FOXC1, FOXM1, FOXO1, GUCA2B, GUSB, H2AX, LPL, JAK2, JARID2, KCNA2, KCND3, KCNK3, KCNQ1, LAD1, LAMC2, LCAT, LDLR, LEPR, LGALS1, LTBP3, HADHA, LY75, MAP6, MATN1, MC4R, CD46, SMCP, MDM2, ME1, MEF2C, MEOX1, MGAT1, MICE, ITPR3, ITPR2, ITGB3, ITGB2, HADH, HDAC2, HDC, HK2, HLA-DQA1, HLA-DRB1, HMBS, HMGB2, HNRNPD, HRH2, PRMT1, HSD11B1, HSPA5, HSPG2, ID2, IGF2R, IGFBP1, IGFBP2, IGFBP5, IKBKB, IL1RN, IL4R, IL11, IL13RA1, ILF3, CTTN, ELAVL1, PRMT5, C3AR1, ATP5F1A, ALDH7A1, AVPR1B, BAD, BCL2A1, BGLAP, BGN, BMP4, BMPR1A, BRCA2, BTD, TSPO, C5AR1, ATF3, CA3, CALCA, CAPG, CAST, CASR, CBR3, CD14, TNFRSF8, SCARB2, CD40, CD44, CD68, ATP2A3, ARSL, ELANE, ADORA2B, ABCA1, ABCB7, ABL1, ABO, ACP5, ACTA2, ACTG1, ACTG2, ADCYAP1, ADCYAP1R1, ADD1, ADORA2A, JAG1, ARSD, AHSG, ALCAM, AMBP, ANK2, ANPEP, ANXA6, APOF, XIAP, AQP2, ARG1, RHOA, ARSB, CD69, CDH15, CDK8, DCN, CTSB, CTSG, CTSK, CTSS, CYP1A1, CYP2B6, CYP2C19, CYP2E1, CYP3A4, CYP3A5, DAG1, DAO, GADD45A, CDKN1A, DDT, TIMM8A, DLD, DMPK, DNASE1, DNM2, DNMT3A, DRD2, DUSP2, E2F6, EGR1, EIF4EBP1, CTRL, CTNND1, CTH, CSF1, CDS1, CETP, CGB3, CHGB, CHRM2, LYST, CIRBP, CISH, CLCN3, CLCNKA, CLU, CMA1, CCR4, CCR5, CCR7, CNN1, CNR2, COL3A1, COL11A2, KLF6, CPT1A, CPT2, CRABP1, CRHR1, CSE1L, CXCL9, MITF, MAP3K11, MAP3K12, VDI, VEGFB, VSNL1, NSD2, WNT1, WNT2, XIST, XRCC1, YY1, YWHAE, PCGF2, ZBTB17, SCG2, VCAM1, SLMAP, MANF, REEP5, LAP, TFEB, TCL1A, AKAP1, RBM10, DYSF, IKBKG, PLPP3, STC2, VCP, UVRAG, AFDN, TERF2, SPTBN1, SSB, STAT5A, STAT5B, STC1, SULT1E1, STK11, STRN, SYK, SYT1, TAGLN, TEAD1, TFPI, UGT2B7, TFR2, TFRC, TGFB2, THBS1, THRA, TJP1, TMSB4X, TOP2B, TPT1, CRISP2, TRAF2, TRAF3, PSMG1, TP63, DGAT1, HIPK3, BMS1, MED12, CCS, KCNE2, NR2E3, HDAC6, ABCC9, NR1H3, COX17, IL18BP, DPP3, PPIF, FRY, URI1, NAMPT, RASA4, ATP6AP2, ALYREF, CALCOCO2, STUB1, SPEG, MARCHF6, CNPY2, KLF2, RAPGEF3, YAP1, PIEZO1, MLEC, KMT2B, FAM53B, TNFSF12, TNFSF10, CDS2, SUCLA2, PROM1, NR1I2, SPHK1, NAE1, WASF1, CLDN10, HGS, IL1RL1, MAP3K13, XPR1, IL32, MSC, KL, TSIX, ABCG2, FHL5, PICK1, TBPL1, BCAR1, AKAP12, ISG15, SPTAN1, SPRR2B, SPARC, PF4, OPRD1, OPRM1, ORM1, P4HB, FURIN, PAFAH1B1, PAM, PAPPA, PCMT1, PDC, PDE1C, PDK4, PFN1, PPP1CB, PGK1, PIGF, PITX3, PKD1, PLA2G2A, PLAU, PMP22, POLR2A, PON2, PPA1, PPARD, PPBP, OGN, OGDH, NRF1, NOTCH3, MMP7, MPI, MRC1, MST1, MTTP, TRNT, MMUT, MYC, MYF6, MYL2, MYL3, MYL4, MYOC, PPP1R12A, NAGLU, NDUFAB1, NFATC2, NFATC4, NFE2L1, NFIL3, NGF, NHS, NME3, NNMT, NOTCH1, PPID, PPP1R3A, SOAT1, SFTPB, RPE65, RPL32, RRAD, RYR3, S100A8, S100A12, SAT1, SCN7A, CCL5, CX3CL1, SDC1, CXCL12, SFTPD, PPP1R7, SGCA, SGTA, SIX1, SLC4A3, SLC4A1, SLC6A4, SLC12A2, SLC18A3, SLC22A5, SMPD2, SUMO3, SUMO2, RORC, RNASE3, RHD, RGS4, PPP2R1A, PPP3CA, PPP5C, PPT1, NPY4R, SRGN, PRKCD, PKN1, PKN2, MAPK9, MAP2K3, PROC, PRTN3, PSEN2, PTGER4, PTHLH, PTN, PTPN11, PURB, PVALB, RAB1A, RARRES2, RELA, RENBP, RGS2, HEAT2
-
Travelers' Diarrhea
Wikipedia
"Isolates from Colonic Spirochetosis in Humans Show High Genomic Divergence and Potential Pathogenic Features but Are Not Detected Using Standard Primers for the Human Microbiota" . ... Zucherman, Ed., Principles and Practice of Travel Medicine , John Wiley and Sons, 2001. p.153 Google books preview Archived 2017-09-08 at the Wayback Machine ^ Shlim, DR (1 December 2005).SERPINA3, LTF, POTEKP, ACTBL2, ACOT7, FHL5, TBCA, SULT1E1, SLC9A3, TNFRSF11B, IL10, ACTG1, GUCY2C, GUCA2B, GUCA2A, GCY, EMD, MAP3K8, CD14, ACTG2, POTEM

-
Rubinstein-Taybi Syndrome 1
OMIM
Hennekam and Van Doorne (1990) commented on short upper lip and pouting lower lip, a feature documented in many photographs by Hennekam et al. (1990). ... Bloch-Zupan et al. (2007) reported detailed orodental features of 40 patients with RSTS ranging in age from 4 to 30 years. ... Many had behavioral problems, such as poor attention span and autistic features, and worsening of behavior over time was reported in about 37%. ... They commented that the typical features of the syndrome increasingly developed in early infancy toward the total 'Gestalt' by the age of 2 years. ... A 2-month-old girl with typical features showed a de novo pericentric inversion of one chromosome 16; her karyotype was 46,XX,inv(16)(p13.3;q13).CREBBP, EP300, SMOC1, EIF4E, PCBP4, PAG1, OPN1LW, MECP2, HTC2, RECQL4, ENOSF1, KAT6B, TWIST1, TNFRSF13B, SRCAP, ZEB2, TMPRSS11D, NIPBL, BMP2, SMARCA4, SMARCA2, SLC20A1, CREB1, POU1F1, OGG1, IGFALS, TNC, HBA2, HBA1, FOXG1, SHH











