-
Foot-And-Mouth Disease
Wikipedia
"The Pathogenesis of Foot-and-Mouth Disease II: Viral Pathways in Swine, Small Ruminants, and Wildlife; Myotropism, Chronic Syndromes, and Molecular Virus-Host Interactions". ... WAHID Interface—OIE World Animal Health Information Database Disease card The European Commission for the Control of Foot-and-Mouth Disease (EuFMD) Species Profile - Foot and Mouth Disease , National Invasive Species Information Center, United States National Agricultural Library . v t e Skin infections , symptoms and signs related to viruses DNA virus Herpesviridae Alpha HSV Herpes simplex Herpetic whitlow Herpes gladiatorum Herpes simplex keratitis Herpetic sycosis Neonatal herpes simplex Herpes genitalis Herpes labialis Eczema herpeticum Herpetiform esophagitis Herpes B virus B virus infection VZV Chickenpox Herpes zoster Herpes zoster oticus Ophthalmic zoster Disseminated herpes zoster Zoster-associated pain Modified varicella-like syndrome Beta Human herpesvirus 6 / Roseolovirus Exanthema subitum Roseola vaccinia Cytomegalic inclusion disease Gamma KSHV Kaposi's sarcoma Poxviridae Ortho Variola Smallpox Alastrim MoxV Monkeypox CPXV Cowpox VV Vaccinia Generalized vaccinia Eczema vaccinatum Progressive vaccinia Buffalopox Para Farmyard pox : Milker's nodule Bovine papular stomatitis Pseudocowpox Orf Sealpox Other Yatapoxvirus : Tanapox Yaba monkey tumor virus MCV Molluscum contagiosum Papillomaviridae HPV Wart / plantar wart Heck's disease Genital wart giant Laryngeal papillomatosis Butcher's wart Bowenoid papulosis Epidermodysplasia verruciformis Verruca plana Pigmented wart Verrucae palmares et plantares BPV Equine sarcoid Parvoviridae Parvovirus B19 Erythema infectiosum Reticulocytopenia Papular purpuric gloves and socks syndrome Polyomaviridae Merkel cell polyomavirus Merkel cell carcinoma RNA virus Paramyxoviridae MeV Measles Togaviridae Rubella virus Rubella Congenital rubella syndrome ("German measles" ) Alphavirus infection Chikungunya fever Picornaviridae CAV Hand, foot, and mouth disease Herpangina FMDV Foot-and-mouth disease Boston exanthem disease Ungrouped Asymmetric periflexural exanthem of childhood Post-vaccination follicular eruption Lipschütz ulcer Eruptive pseudoangiomatosis Viral-associated trichodysplasia Gianotti–Crosti syndromeFLNA, FSHMD1A, SAT1, SAT2, IFNG, ITGAV, JMJD6, IL15, ATG12, DCTN3, ATG5, G3BP2, BECN1, VTN, VIM, G3BP1, SEC62, EIF2AK3, AKT1, CARD8, SEA, IL23A, DDX56, IL21, NOD2, DHX58, NLRP3, STING1, MIR203A, MIR1307, ST3GAL4, RPL13, CCL20, ANXA13, APCS, ATF4, DDX1, DHODH, EEF1G, EGR1, EIF2S1, EIF4G1, ERN1, FANCG, HLA-B, IL2, ITGA5, ITGB6, ITIH4, RPSA, SH2D1A, NME1, PDR, PPIA, EIF2AK2, ERVW-4

-
Death Anxiety (Psychology)
Wikipedia
PMID 24493652 . v t e Death In medicine Cell death Necrosis Avascular necrosis Coagulative necrosis Liquefactive necrosis Gangrenous necrosis Caseous necrosis Fat necrosis Fibrinoid necrosis Temporal lobe necrosis Programmed cell death AICD Anoikis Apoptosis Autophagy Intrinsic apoptosis Necroptosis Paraptosis Parthanatos Phenoptosis Pseudoapoptosis Pyroptosis Autolysis Autoschizis Eschar Immunogenic cell death Ischemic cell death Pyknosis Karyorrhexis Karyolysis Mitotic catastrophe Suicide gene Abortion Accidental death Autopsy Brain death Brainstem death Clinical death DOA Death by natural causes Death rattle Dysthanasia End-of-life care Euthanasia Lazarus sign Lazarus syndrome Medical definition of death Organ donation Terminal illness Unnatural death Lists Causes of death by rate Expressions related to death Natural disasters People by cause of death Premature obituaries Preventable causes of death Notable deaths by year Unusual deaths Mortality Birthday effect Child mortality Gompertz–Makeham law of mortality Infant mortality Karoshi Maternal death Maternal mortality in fiction Memento mori Micromort Mortality displacement Mortality rate RAMR Mortality salience Perinatal mortality After death Body Stages Pallor mortis Algor mortis Rigor mortis Livor mortis Putrefaction Decomposition Skeletonization Fossilization Preservation Cryopreservation Cryonics Neuropreservation Embalming Maceration Mummification Plastination Prosection Taxidermy Disposal Burial Natural burial Cremation Dismemberment Excarnation Promession Resomation Beating heart cadaver Body donation Cadaveric spasm Coffin birth Death erection Dissection Gibbeting Postmortem caloricity Post-mortem interval Other aspects Afterlife Cemetery Consciousness Customs Crematorium Examination Funeral Grief Intermediate state Internet Mourning Online mourning Obituary Vigil Paranormal Ghosts Near-death experience Near-death studies Necromancy Out-of-body experience Reincarnation research Séance Legal Abortion law Administration Capital punishment Cause of death Civil death Coroner Death-qualified jury Death certificate Declared death in absentia Death row Dying declaration Inquest Legal death Murder Necropolitics Prohibition of death Right to die Suspicious death Trust law Will Fields Forensic pathology Funeral director Mortuary science Necrobiology Post-mortem chemistry Post-mortem photography Taphonomy Biostratinomy Thanatology Other Apparent death Dark tourism Darwin Awards Death and culture Death anniversary Death anxiety Death deity Personification of death Dying-and-rising god Psychopomp Death camp Death drive Death education Death from laughter Death hoax Death knell Death march Death messenger Death notification Death panel Death poem Death pose Death-positive movement Death squad Death threat Death trajectory Dignified death Extinction Fan death Festival of the Dead Fascination with death Hierarchy of death Homicide Last rites Martyr Megadeath Museum of Death Necronym Necrophilia Necrophobia The Order of the Good Death Predation Sacrifice human Suicide Assisted suicide Thanatosensitivity The Goodbye Family Category Outline
-
Prosopagnosia
Wikipedia
"First report of prevalence of non-syndromic hereditary prosopagnosia (HPA)" (PDF) . ... Classification D ICD - 10 : R48.8 ICD - 10-CM : R48.3 ICD - 9-CM : 368.16 OMIM : 610382 MeSH : D020238 v t e Symptoms , signs and syndromes associated with lesions of the brain and brainstem Brainstem Medulla (CN 8, 9, 10, 12) Lateral medullary syndrome/Wallenberg PICA Medial medullary syndrome/Dejerine ASA Pons (CN 5, 6, 7, 8) Upper dorsal pontine syndrome/Raymond-Céstan syndrome Lateral pontine syndrome ( AICA ) (lateral) Medial pontine syndrome / Millard–Gubler syndrome / Foville's syndrome ( basilar ) Locked-in syndrome Internuclear ophthalmoplegia One and a half syndrome Midbrain (CN 3, 4) Weber's syndrome ventral peduncle, PCA Benedikt syndrome ventral tegmentum, PCA Parinaud's syndrome dorsal, tumor Claude's syndrome Other Alternating hemiplegia Cerebellum Latearl Dysmetria Dysdiadochokinesia Intention tremor ) Medial Cerebellar ataxia Basal ganglia Chorea Dystonia Parkinson's disease Cortex ACA syndrome MCA syndrome PCA syndrome Frontal lobe Expressive aphasia Abulia Parietal lobe Receptive aphasia Hemispatial neglect Gerstmann syndrome Astereognosis Occipital lobe Bálint's syndrome Cortical blindness Pure alexia Temporal lobe Cortical deafness Prosopagnosia Thalamus Thalamic syndrome Other Upper motor neuron lesion Aphasia
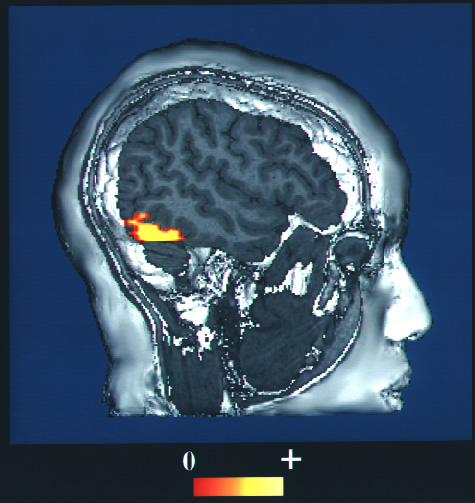
-
Metastasis
Wikipedia
See also [ edit ] Biology portal Medicine portal Abscopal effect Brain metastasis Brown-Séquard syndrome (Sections on cavernous malformation, germinoma, renal cell carcinoma and lung cancer) Collective cell migration Contact normalization Disseminated disease Micrometastasis Mouse models of breast cancer metastasis Positron emission tomography (PET) References [ edit ] ^ "Metastasis", Merriam–Webster online, accessed 20 Aug 2017. ^ "What is Metastasis?" ... Q&A: Metastatic Cancer —from the National Cancer Institute Classification D MeSH : D009362 DiseasesDB : 28954 External resources MedlinePlus : 002260 v t e Overview of tumors , cancer and oncology Conditions Benign tumors Hyperplasia Cyst Pseudocyst Hamartoma Malignant progression Dysplasia Carcinoma in situ Cancer Metastasis Primary tumor Sentinel lymph node Topography Head and neck ( oral , nasopharyngeal ) Digestive system Respiratory system Bone Skin Blood Urogenital Nervous system Endocrine system Histology Carcinoma Sarcoma Blastoma Papilloma Adenoma Other Precancerous condition Paraneoplastic syndrome Staging / grading TNM Ann Arbor Prostate cancer staging Gleason grading system Dukes classification Carcinogenesis Cancer cell Carcinogen Tumor suppressor genes / oncogenes Clonally transmissible cancer Oncovirus Carcinogenic bacteria Misc.

-
Urinary Tract Infection
Wikipedia
In post-menopausal women, sexual activity does not affect the risk of developing a UTI. [4] Spermicide use, independent of sexual frequency, increases the risk of UTIs. [4] Diaphragm use is also associated. [29] Condom use without spermicide or use of birth control pills does not increase the risk of uncomplicated urinary tract infection. [4] [30] Sex Women are more prone to UTIs than men because, in females, the urethra is much shorter and closer to the anus . [31] As a woman's estrogen levels decrease with menopause , her risk of urinary tract infections increases due to the loss of protective vaginal flora . [31] Additionally, vaginal atrophy that can sometimes occur after menopause is associated with recurrent urinary tract infections. [32] Chronic prostatitis in the forms of chronic prostatitis/chronic pelvic pain syndrome and chronic bacterial prostatitis (not acute bacterial prostatitis or asymptomatic inflammatory prostatitis ) may cause recurrent urinary tract infections in males. ... External links Classification D ICD - 10 : N39.0 ICD - 9-CM : 599.0 MeSH : D014552 DiseasesDB : 13657 External resources MedlinePlus : 000521 eMedicine : emerg/625 emerg/626 Patient UK : Urinary tract infection medicine portal v t e Diseases of the urinary tract Ureter Ureteritis Ureterocele Megaureter Bladder Cystitis Interstitial cystitis Hunner's ulcer Trigonitis Hemorrhagic cystitis Neurogenic bladder dysfunction Bladder sphincter dyssynergia Vesicointestinal fistula Vesicoureteral reflux Urethra Urethritis Non-gonococcal urethritis Urethral syndrome Urethral stricture Meatal stenosis Urethral caruncle Any/all Obstructive uropathy Urinary tract infection Retroperitoneal fibrosis Urolithiasis Bladder stone Kidney stone Renal colic Malakoplakia Urinary incontinence Stress Urge Overflow Authority control GND : 4023498-8 LCCN : sh85141416 NDL : 00946940

- Exhibitionism Wikipedia
-
Hairy Cell Leukemia
Wikipedia
The presence of additional lymphoproliferative diseases is easily checked during a flow cytometry test, where they characteristically show different results. [16] The differential diagnoses include: several kinds of anemia , including myelophthisis and aplastic anemia , [22] and most kinds of blood neoplasms, including hypoplastic myelodysplastic syndrome , atypical chronic lymphocytic leukemia, B-cell prolymphocytic leukemia , or idiopathic myelofibrosis . [16] Classification [ edit ] When not further specified, the "classic" form is often implied. ... External links [ edit ] Classification D ICD - 10 : C91.4 ICD - 9-CM : 202.4 ICD-O : M9940/3 MeSH : D007943 DiseasesDB : 5589 SNOMED CT : 118613001 External resources MedlinePlus : 000592 eMedicine : med/937 About HCL at the US National Cancer Institute History of HCL and the Godmother of HCL v t e Leukaemias , lymphomas and related disease B cell ( lymphoma , leukemia ) (most CD19 CD20 ) By development/ marker TdT+ ALL ( Precursor B acute lymphoblastic leukemia/lymphoma ) CD5 + naive B cell ( CLL/SLL ) mantle zone ( Mantle cell ) CD22 + Prolymphocytic CD11c+ ( Hairy cell leukemia ) CD79a + germinal center / follicular B cell ( Follicular Burkitt's GCB DLBCL Primary cutaneous follicle center lymphoma ) marginal zone / marginal zone B-cell ( Splenic marginal zone MALT Nodal marginal zone Primary cutaneous marginal zone lymphoma ) RS ( CD15 +, CD30 +) Classic Hodgkin lymphoma ( Nodular sclerosis ) CD20+ ( Nodular lymphocyte predominant Hodgkin lymphoma ) PCDs / PP ( CD38 +/ CD138 +) see immunoproliferative immunoglobulin disorders By infection KSHV ( Primary effusion ) EBV Lymphomatoid granulomatosis Post-transplant lymphoproliferative disorder Classic Hodgkin lymphoma Burkitt's lymphoma HCV Splenic marginal zone lymphoma HIV ( AIDS-related lymphoma ) Helicobacter pylori ( MALT lymphoma ) Cutaneous Diffuse large B-cell lymphoma Intravascular large B-cell lymphoma Primary cutaneous marginal zone lymphoma Primary cutaneous immunocytoma Plasmacytoma Plasmacytosis Primary cutaneous follicle center lymphoma T/NK T cell ( lymphoma , leukemia ) (most CD3 CD4 CD8 ) By development/ marker TdT+ : ALL ( Precursor T acute lymphoblastic leukemia/lymphoma ) prolymphocyte ( Prolymphocytic ) CD30+ ( Anaplastic large-cell lymphoma Lymphomatoid papulosis type A ) Cutaneous MF+variants indolent: Mycosis fungoides Pagetoid reticulosis Granulomatous slack skin aggressive: Sézary disease Adult T-cell leukemia/lymphoma Non-MF CD30 -: Non-mycosis fungoides CD30− cutaneous large T-cell lymphoma Pleomorphic T-cell lymphoma Lymphomatoid papulosis type B CD30 +: CD30+ cutaneous T-cell lymphoma Secondary cutaneous CD30+ large-cell lymphoma Lymphomatoid papulosis type A Other peripheral Hepatosplenic Angioimmunoblastic Enteropathy-associated T-cell lymphoma Peripheral T-cell lymphoma not otherwise specified ( Lennert lymphoma ) Subcutaneous T-cell lymphoma By infection HTLV-1 ( Adult T-cell leukemia/lymphoma ) NK cell / (most CD56 ) Aggressive NK-cell leukemia Blastic NK cell lymphoma T or NK EBV ( Extranodal NK-T-cell lymphoma / Angiocentric lymphoma ) Large granular lymphocytic leukemia Lymphoid+ myeloid Acute biphenotypic leukaemia Lymphocytosis Lymphoproliferative disorders ( X-linked lymphoproliferative disease Autoimmune lymphoproliferative syndrome ) Leukemoid reaction Diffuse infiltrative lymphocytosis syndrome Cutaneous lymphoid hyperplasia Cutaneous lymphoid hyperplasia with bandlike and perivascular patterns with nodular pattern Jessner lymphocytic infiltrate of the skin General Hematological malignancy leukemia Lymphoproliferative disorders Lymphoid leukemiasBRAF, MAP2K1, CD22, CCND1, TNF, ITGAX, MAP2K7, SOX11, BCL2, CDKN1B, KRT20, CD200, IL2RA, IGH, IGHV4-34, MS4A1, IL6, BTK, TP53, IL2, KLF2, MAPK1, EPHB2, ISG20, CD19, IFNG, IGHV3-69-1, NCAM1, IGHV3OR16-7, ITGAE, ZHX2, MME, IFNA1, NOTCH2, ADA, RAF1, IFNA13, IL1B, CDKN2A, LOC102724971, BCR, RHOH, LOC102723407, FCRL5, CD79B, TBX21, ANXA1, ZNRD2, ABCB6, DCTN6, CIB1, TMED7-TICAM2, NSA2, PCAT1, IGHV3-21, MYMX, BCL2L11, PPP1R13L, GRAP2, DOK2, SYNJ2, SKAP1, ENC1, CLLS2, TICAM2, CD160, IFNL3, RAPH1, IGLJ3, TMED7, IGHV3-30, TGFB1, DBR1, SHC3, IGHV3-33, AICDA, CPAT1, MARCKSL1, PPARGC1A, B3GAT1, WLS, TNFRSF13C, SMUG1, PRAME, ACSBG1, TBC1D9, ALKBH3, IGHV1-2, ABL1, ABCB1, TERC, IL3, IFI27, HLA-DRB1, HEXB, GSTP1, CXCR3, GDF1, FLT3, F2R, ERBB2, CTTN, EGF, CYP19A1, CSF3, CR2, CDKN2B, CD81, CD44, CD40, CD38, CD1D, ATM, APOH, ANXA5, IL1A, IL3RA, TRBV20OR9-2, IL4, SYN2, SPN, ROCK1, RNH1, PTH, PSMD9, PIK3CG, PIK3CD, PIK3CB, PIK3CA, ENPP1, SERPINA5, PAX5, NDUFAB1, MYD88, ABCC1, KMT2A, MCL1, LTA, ITK, ITGB2, IL10, CXCL8, H3P23
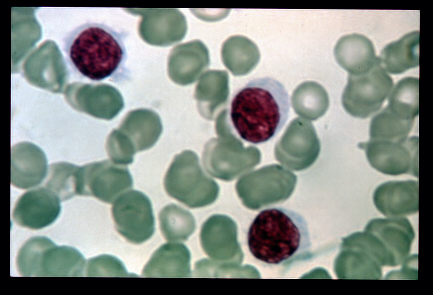
-
Tinnitus
Wikipedia
Children with hearing loss have a high incidence of tinnitus, even though they do not express the condition or its effect on their lives. [113] [114] Children do not generally report tinnitus spontaneously and their complaints may not be taken seriously. [115] Among those children who do complain of tinnitus, there is an increased likelihood of associated otological or neurological pathology such as migraine, juvenile Meniere's disease or chronic suppurative otitis media. [116] Its reported prevalence varies from 12% to 36% in children with normal hearing thresholds and up to 66% in children with a hearing loss and approximately 3–10% of children have been reported to be troubled by tinnitus. [117] See also Medicine portal Auditory hallucination Health effects from noise List of people with tinnitus List of unexplained sounds Phantom vibration syndrome Zwicker tone References ^ a b c d Levine, RA; Oron, Y (2015).IFNA2, RNR1, BDNF, ARC, ABAT, COCH, CLCNKB, AIFM1, KCNJ5, MPL, MPV17, TMC1, NAGA, PDGFB, SDHB, SDHC, SDHD, SLC12A3, SMARCB1, VHL, DNAH11, FKRP, SLC44A4, KCNQ4, JAK2, MFN2, AFG3L2, DKK1, P2RX2, TUBB6, SLC39A14, STRN4, SUFU, TET2, DIABLO, OSBPL2, NF2, CACNA1A, DSPP, CYP11B2, CYP11B1, CCND1, CACNA1D, TNF, CSF2, LAMC2, SLC17A6, PART1, NT5E, PTPN4, SLC17A7, GDNF, IL2, KCNE3, DCX, SPIN1, CYP2E1, DIAPH1, CRYGD, IL1B, PYCARD, COX8A, COMT, TYMS, CNR1, CNC2, AQP2, ACTG1, ACR, PTCRA, HCCAT5, THEMIS, EPS8, THAS, TRI-AAT9-1, OTC, IL6, IL10, IL1A, KCNE1, HSPA4, KCNQ2, GTF2I, GSTP1, GRM7, GRIN2B, ABR, NHS, GJB2, IL1RN, PEPD, CFP, PRKAR1A, GDF10, GAP43, SLC6A4, SLC8A1, GAD2, SLC18A3, SLC25A1, F11, TCEA1, ERVW-4

-
Postpartum Depression
Wikipedia
Sleep deprivation can lead to physical discomfort and exhaustion, which can contribute to the symptoms of postpartum depression. [39] Risk factors [ edit ] While the causes of PPD are not understood, a number of factors have been suggested to increase the risk: Prenatal depression or anxiety [40] A personal or family history of depression [41] Moderate to severe premenstrual symptoms [42] Stressful life events experienced during pregnancy [43] [44] Postpartum blues [40] Birth-related psychological trauma Birth-related physical trauma History of sexual abuse [45] [46] Childhood trauma [45] [46] [47] Previous stillbirth or miscarriage [42] Formula-feeding rather than breast-feeding [41] Cigarette smoking [41] Low self-esteem [40] Childcare or life stress [40] Low social support [40] Poor marital relationship or single marital status [40] Low socioeconomic status [40] [48] A lack of strong emotional support from spouse, partner, family, or friends [39] Infant temperament problems/ colic [40] Unplanned/unwanted pregnancy [40] Low vitamin D levels [49] [50] Breastfeeding difficulties [51] Administration of labor-inducing medication synthetic oxytocin [34] Of these risk factors a history of depression, and cigarette smoking have been shown to have additive effects. [41] Some studies have found a link with low levels of DHA in the mother. [52] Chronic illnesses caused by neuroendocrine irregularities including irritable bowl syndrome and fibromyalgia typically put individuals at risk for further health complications.SLC6A4, TTC9B, HP1BP3, HTR1A, OPRM1, PER2, MTHFR, GABRD, IL10, OXTR, TPO, BDNF, SAGE1, NR3C1, CCN6, ADIPOQ, FHL5, ACOT7, PART1, MARCHF11, AGO2, POTEKP, TAL1, SCLY, ARID4B, KRT88P, CREB3L1, POTEM, COPD, ACTBL2, VSX1, SERPINA3, PRL, ABO, ACTG1, ACTG2, ATP5F1A, OPN1SW, TSPO, SERPINA6, CRH, CRP, EGR3, ESR1, GALR1, GATA3, HPD, HSD11B1, IL6, KRT7, NR3C2, MPI, NPPA, STIN2-VNTR
-
Hangover
Wikipedia
Potentially dangerous daily activities such as driving a car or operating heavy machinery are also negatively influenced. [5] In mid-2017, it was reported that one company in the UK allows sick days when hung over. [62] Research [ edit ] Psychological research of alcohol hangover is growing rapidly. [ citation needed ] The Alcohol Hangover Research Group had its inaugural meeting in June 2010 as part of the Research Society on Alcoholism (RSA) 33rd Annual Scientific Meeting in San Antonio , Texas , USA. [ citation needed ] In 2012, Éduc'alcool, a Quebec -based non-profit organization that aims to educate the public on the responsible use of alcohol, published a report noting hangovers have long-lasting effects that inhibit the drinker's capabilities a full 24 hours after heavy drinking. [63] See also [ edit ] Auto-brewery syndrome Comedown (drugs) Food drunk References [ edit ] ^ a b c d Stephens R, Ling J, Heffernan TM, Heather N, Jones K (23 January 2008). ... A colorful article on hangovers, their cause and treatment along with reference to famous Soho residents, such as Jeffrey Bernard , Dylan Thomas , and Francis Bacon . v t e Psychoactive substance-related disorder General SID Substance intoxication / Drug overdose Substance-induced psychosis Withdrawal : Craving Neonatal withdrawal Post-acute-withdrawal syndrome (PAWS) SUD Substance abuse / Substance-related disorders Physical dependence / Psychological dependence / Substance dependence Combined substance use SUD Polysubstance dependence SID Combined drug intoxication (CDI) Alcohol SID Cardiovascular diseases Alcoholic cardiomyopathy Alcohol flush reaction (AFR) Gastrointestinal diseases Alcoholic liver disease (ALD): Alcoholic hepatitis Auto-brewery syndrome (ABS) Endocrine diseases Alcoholic ketoacidosis (AKA) Nervous system diseases Alcohol-related dementia (ARD) Alcohol intoxication Hangover Neurological disorders Alcoholic hallucinosis Alcoholic polyneuropathy Alcohol-related brain damage Alcohol withdrawal syndrome (AWS): Alcoholic hallucinosis Delirium tremens (DTs) Fetal alcohol spectrum disorder (FASD) Fetal alcohol syndrome (FAS) Korsakoff syndrome Positional alcohol nystagmus (PAN) Wernicke–Korsakoff syndrome (WKS, Korsakoff psychosis) Wernicke encephalopathy (WE) Respiratory tract diseases Alcohol-induced respiratory reactions Alcoholic lung disease SUD Alcoholism (alcohol use disorder (AUD)) Binge drinking Caffeine SID Caffeine-induced anxiety disorder Caffeine-induced sleep disorder Caffeinism SUD Caffeine dependence Cannabis SID Cannabis arteritis Cannabinoid hyperemesis syndrome (CHS) SUD Amotivational syndrome Cannabis use disorder (CUD) Synthetic cannabinoid use disorder Cocaine SID Cocaine intoxication Prenatal cocaine exposure (PCE) SUD Cocaine dependence Hallucinogen SID Acute intoxication from hallucinogens (bad trip) Hallucinogen persisting perception disorder (HPPD) Nicotine SID Nicotine poisoning Nicotine withdrawal SUD Nicotine dependence Opioids SID Opioid overdose SUD Opioid use disorder (OUD) Sedative / hypnotic SID Kindling (sedative–hypnotic withdrawal) benzodiazepine : SID Benzodiazepine overdose Benzodiazepine withdrawal SUD Benzodiazepine use disorder (BUD) Benzodiazepine dependence barbiturate : SID Barbiturate overdose SUD Barbiturate dependence Stimulants SID Stimulant psychosis amphetamine : SUD Amphetamine dependence Volatile solvent SID Sudden sniffing death syndrome (SSDS) Toluene toxicity SUD Inhalant abuse v t e Headache Primary ICHD 1 Migraine Familial hemiplegic Retinal migraine ICHD 2 Tension Mixed tension migraine ICHD 3 Cluster Chronic paroxysmal hemicrania SUNCT ICHD 4 Hemicrania continua Thunderclap headache Sexual headache New daily persistent headache Hypnic headache Secondary ICHD 5 Migralepsy ICHD 7 Ictal headache Post-dural-puncture headache ICHD 8 Hangover Medication overuse headache ICHD 13 Trigeminal neuralgia Occipital neuralgia External compression headache Cold-stimulus headache Optic neuritis Postherpetic neuralgia Tolosa–Hunt syndrome Other Vascular Authority control GND : 4339907-1

-
Arsenic Poisoning
Wikipedia
An experiment of Hu et al. (2002) demonstrated increased binding activity of AP-1 and NF-κB after acute (24 h) exposure to +3 sodium arsenite, whereas long-term exposure (10–12 weeks) yielded the opposite result. [109] The authors conclude that the former may be interpreted as a defense response while the latter could lead to carcinogenesis. [109] As the contradicting findings and connected mechanistic hypotheses indicate, there is a difference in acute and chronic effects of arsenic on signal transduction which is not clearly understood yet. [ citation needed ] Oxidative stress [ edit ] Studies have demonstrated that the oxidative stress generated by arsenic may disrupt the signal transduction pathways of the nuclear transcriptional factors PPARs, AP-1, and NF-κB, [94] [109] [110] as well as the pro-inflammatory cytokines IL-8 and TNF-α. [94] [109] [110] [111] [112] [113] [114] [115] The interference of oxidative stress with signal transduction pathways may affect physiological processes associated with cell growth, metabolic syndrome X, glucose homeostasis, lipid metabolism, obesity, insulin resistance , inflammation, and diabetes-2. [116] [117] [118] Recent scientific evidence has elucidated the physiological roles of the PPARs in the ω- hydroxylation of fatty acids and the inhibition of pro-inflammatory transcription factors (NF-κB and AP-1), pro-inflammatory cytokines (IL-1, -6, -8, -12, and TNF-α), cell4 adhesion molecules (ICAM-1 and VCAM-1), inducible nitric oxide synthase, proinflammatory nitric oxide (NO), and anti-apoptotic factors. [94] [111] [116] [118] [119] Epidemiological studies have suggested a correlation between chronic consumption of drinking water contaminated with arsenic and the incidence of Type 2-diabetes. [94] The human liver after exposure to therapeutic drugs may exhibit hepatic non-cirrhotic portal hypertension, fibrosis, and cirrhosis. [94] However, the literature provides insufficient scientific evidence to show cause and effect between arsenic and the onset of diabetes mellitus Type 2. [94] Diagnosis [ edit ] Arsenic may be measured in blood or urine to monitor excessive environmental or occupational exposure, confirm a diagnosis of poisoning in hospitalized victims or to assist in the forensic investigation in a case of fatal over dosage. ... PMID 21750349 . ^ Joca, L; Sacks, JD; Moore, D; Lee, JS; Sams R, 2nd; Cowden, J (2016). "Systematic review of differential inorganic arsenic exposure in minority, low-income, and indigenous populations in the United States".CCL20, AQP9, SKIL, SOD2, SRP68, SSBP1, TRAPPC10, TNF, TNFAIP6, TP53, UBE2E1, PIAS1, USO1, IER3, USP13, CCRL2, CD83, MINPP1, AKAP9, ZNF267, N4BP2L2, RUFY3, TNIK, TRA2A, SOX18, ZFAND6, ZNF331, PELI1, AS3MT, TAF1D, MIR145, SFPQ, CCL3L3, CCL4, ATXN7, CD44, CRP, ERCC1, ERCC3, ERCC4, GOLGA4, CXCL2, CXCL3, HSPA1B, ID2, IL1A, IL1B, IL1RN, INPP5A, KCNJ2, KRT10, MT1A, MTHFR, GADD45B, NDUFB8, PNP, NR4A2, PDE4B, PFKFB3, PTX3, RFX3, RGS1, GSR, ALAD
-
Community-Acquired Pneumonia
Wikipedia
Other immune problems that increase the risk of developing pneumonia range from severe childhood immune deficiencies, such as Wiskott–Aldrich syndrome , to the less severe common variable immunodeficiency . [10] Pathophysiology [ edit ] The symptoms of CAP are the result of lung infection by microorganisms and the response of the immune system to the infection. ... External links [ edit ] Infectious Diseases Society of America/American Thoracic Society Consensus Guidelines on the Management of Community-Acquired Pneumonia in Adults PDF v t e Diseases of the respiratory system Upper RT (including URTIs , common cold ) Head sinuses Sinusitis nose Rhinitis Vasomotor rhinitis Atrophic rhinitis Hay fever Nasal polyp Rhinorrhea nasal septum Nasal septum deviation Nasal septum perforation Nasal septal hematoma tonsil Tonsillitis Adenoid hypertrophy Peritonsillar abscess Neck pharynx Pharyngitis Strep throat Laryngopharyngeal reflux (LPR) Retropharyngeal abscess larynx Croup Laryngomalacia Laryngeal cyst Laryngitis Laryngopharyngeal reflux (LPR) Laryngospasm vocal cords Laryngopharyngeal reflux (LPR) Vocal fold nodule Vocal fold paresis Vocal cord dysfunction epiglottis Epiglottitis trachea Tracheitis Laryngotracheal stenosis Lower RT / lung disease (including LRTIs ) Bronchial / obstructive acute Acute bronchitis chronic COPD Chronic bronchitis Acute exacerbation of COPD ) Asthma ( Status asthmaticus Aspirin-induced Exercise-induced Bronchiectasis Cystic fibrosis unspecified Bronchitis Bronchiolitis Bronchiolitis obliterans Diffuse panbronchiolitis Interstitial / restrictive ( fibrosis ) External agents/ occupational lung disease Pneumoconiosis Aluminosis Asbestosis Baritosis Bauxite fibrosis Berylliosis Caplan's syndrome Chalicosis Coalworker's pneumoconiosis Siderosis Silicosis Talcosis Byssinosis Hypersensitivity pneumonitis Bagassosis Bird fancier's lung Farmer's lung Lycoperdonosis Other ARDS Combined pulmonary fibrosis and emphysema Pulmonary edema Löffler's syndrome / Eosinophilic pneumonia Respiratory hypersensitivity Allergic bronchopulmonary aspergillosis Hamman-Rich syndrome Idiopathic pulmonary fibrosis Sarcoidosis Vaping-associated pulmonary injury Obstructive / Restrictive Pneumonia / pneumonitis By pathogen Viral Bacterial Pneumococcal Klebsiella Atypical bacterial Mycoplasma Legionnaires' disease Chlamydiae Fungal Pneumocystis Parasitic noninfectious Chemical / Mendelson's syndrome Aspiration / Lipid By vector/route Community-acquired Healthcare-associated Hospital-acquired By distribution Broncho- Lobar IIP UIP DIP BOOP-COP NSIP RB Other Atelectasis circulatory Pulmonary hypertension Pulmonary embolism Lung abscess Pleural cavity / mediastinum Pleural disease Pleuritis/pleurisy Pneumothorax / Hemopneumothorax Pleural effusion Hemothorax Hydrothorax Chylothorax Empyema/pyothorax Malignant Fibrothorax Mediastinal disease Mediastinitis Mediastinal emphysema Other/general Respiratory failure Influenza Common cold SARS Coronavirus disease 2019 Idiopathic pulmonary haemosiderosis Pulmonary alveolar proteinosis v t e Pneumonia Infectious pneumonias Bacterial pneumonia Viral pneumonia Fungal pneumonia Parasitic pneumonia Atypical pneumonia Community-acquired pneumonia Healthcare-associated pneumonia Hospital-acquired pneumonia Ventilator-associated pneumonia Severe acute respiratory syndrome Pneumonias caused by infectious or noninfectious agents Aspiration pneumonia Lipid pneumonia Eosinophilic pneumonia Bronchiolitis obliterans organizing pneumonia Noninfectious pneumonia Chemical pneumonitis Idiopathic pneumonia syndromeCRP, COPD, TNF, IL6, ACE, IL10, HMGB1, MBL2, MMP9, ALB, TLR4, ADM, LNPEP, IRF5, SFTPB, SERPINB6, IL1RN, SLC9A6, HACD1, GRN, SLC2A10, SORBS1, BRD4, CTAA1, ANGPT2, COL4A5, CAP1, AVP, DELEC1, SDC4, NLRP3, SFTPD, SMPD1, TIMP1, TLR9, FGF21, HAVCR1, VDR, ARHGEF5, BHLHE40, BECN1, TIMELESS, SAFB, RGS6, RTN3, CCL18, A2M, RASA1, RARB, AGTR1, AMBP, BAG1, CAMP, CFTR, CHI3L1, DDX3X, FCGR2A, FCN2, HSPA1A, HSPA1B, HSPA2, TNC, IL1B, IL4, CXCL8, IL13, LTA, MCL1, MEFV, MRC1, MUC1, MYLK, SERPINE1, PTX3, MIF
-
Gonorrhea
Wikipedia
Other complications include inflammation of the tissue surrounding the liver , [60] a rare complication associated with Fitz-Hugh–Curtis syndrome ; septic arthritis in the fingers, wrists, toes, and ankles; septic abortion ; chorioamnionitis during pregnancy; neonatal or adult blindness from conjunctivitis ; and infertility . ... Gonorrhea at Curlie "Gonorrhea – CDC Fact Sheet" Classification D ICD - 10 : A54 ICD - 9-CM : 098 MeSH : D006069 DiseasesDB : 8834 External resources MedlinePlus : 007267 eMedicine : article/782913 Patient UK : Gonorrhea v t e Sexually transmitted infections (STI) Bacterial Chancroid ( Haemophilus ducreyi ) Chlamydia , lymphogranuloma venereum ( Chlamydia trachomatis ) Donovanosis ( Klebsiella granulomatis ) Gonorrhea ( Neisseria gonorrhoeae ) Mycoplasma hominis infection ( Mycoplasma hominis ) Syphilis ( Treponema pallidum ) Ureaplasma infection ( Ureaplasma urealyticum ) Protozoal Trichomoniasis ( Trichomonas vaginalis ) Parasitic Crab louse Scabies Viral AIDS ( HIV-1/HIV-2 ) Cancer cervical vulvar penile anal Human papillomavirus (HPV) Genital warts ( condyloma ) Hepatitis B ( Hepatitis B virus ) Herpes simplex HSV-1 & HSV-2 Molluscum contagiosum ( MCV ) General inflammation female Cervicitis Pelvic inflammatory disease (PID) male Epididymitis Prostatitis either Proctitis Urethritis / Non-gonococcal urethritis (NGU) v t e Proteobacteria -associated Gram-negative bacterial infections α Rickettsiales Rickettsiaceae / ( Rickettsioses ) Typhus Rickettsia typhi Murine typhus Rickettsia prowazekii Epidemic typhus , Brill–Zinsser disease , Flying squirrel typhus Spotted fever Tick-borne Rickettsia rickettsii Rocky Mountain spotted fever Rickettsia conorii Boutonneuse fever Rickettsia japonica Japanese spotted fever Rickettsia sibirica North Asian tick typhus Rickettsia australis Queensland tick typhus Rickettsia honei Flinders Island spotted fever Rickettsia africae African tick bite fever Rickettsia parkeri American tick bite fever Rickettsia aeschlimannii Rickettsia aeschlimannii infection Mite-borne Rickettsia akari Rickettsialpox Orientia tsutsugamushi Scrub typhus Flea-borne Rickettsia felis Flea-borne spotted fever Anaplasmataceae Ehrlichiosis : Anaplasma phagocytophilum Human granulocytic anaplasmosis , Anaplasmosis Ehrlichia chaffeensis Human monocytotropic ehrlichiosis Ehrlichia ewingii Ehrlichiosis ewingii infection Rhizobiales Brucellaceae Brucella abortus Brucellosis Bartonellaceae Bartonellosis : Bartonella henselae Cat-scratch disease Bartonella quintana Trench fever Either B. henselae or B. quintana Bacillary angiomatosis Bartonella bacilliformis Carrion's disease , Verruga peruana β Neisseriales M+ Neisseria meningitidis/meningococcus Meningococcal disease , Waterhouse–Friderichsen syndrome , Meningococcal septicaemia M− Neisseria gonorrhoeae/gonococcus Gonorrhea ungrouped: Eikenella corrodens / Kingella kingae HACEK Chromobacterium violaceum Chromobacteriosis infection Burkholderiales Burkholderia pseudomallei Melioidosis Burkholderia mallei Glanders Burkholderia cepacia complex Bordetella pertussis / Bordetella parapertussis Pertussis γ Enterobacteriales ( OX− ) Lac+ Klebsiella pneumoniae Rhinoscleroma , Pneumonia Klebsiella granulomatis Granuloma inguinale Klebsiella oxytoca Escherichia coli : Enterotoxigenic Enteroinvasive Enterohemorrhagic O157:H7 O104:H4 Hemolytic-uremic syndrome Enterobacter aerogenes / Enterobacter cloacae Slow/weak Serratia marcescens Serratia infection Citrobacter koseri / Citrobacter freundii Lac− H2S+ Salmonella enterica Typhoid fever , Paratyphoid fever , Salmonellosis H2S− Shigella dysenteriae / sonnei / flexneri / boydii Shigellosis , Bacillary dysentery Proteus mirabilis / Proteus vulgaris Yersinia pestis Plague / Bubonic plague Yersinia enterocolitica Yersiniosis Yersinia pseudotuberculosis Far East scarlet-like fever Pasteurellales Haemophilus : H. influenzae Haemophilus meningitis Brazilian purpuric fever H. ducreyi Chancroid H. parainfluenzae HACEK Pasteurella multocida Pasteurellosis Actinobacillus Actinobacillosis Aggregatibacter actinomycetemcomitans HACEK Legionellales Legionella pneumophila / Legionella longbeachae Legionnaires' disease Coxiella burnetii Q fever Thiotrichales Francisella tularensis Tularemia Vibrionaceae Vibrio cholerae Cholera Vibrio vulnificus Vibrio parahaemolyticus Vibrio alginolyticus Plesiomonas shigelloides Pseudomonadales Pseudomonas aeruginosa Pseudomonas infection Moraxella catarrhalis Acinetobacter baumannii Xanthomonadaceae Stenotrophomonas maltophilia Cardiobacteriaceae Cardiobacterium hominis HACEK Aeromonadales Aeromonas hydrophila / Aeromonas veronii Aeromonas infection ε Campylobacterales Campylobacter jejuni Campylobacteriosis , Guillain–Barré syndrome Helicobacter pylori Peptic ulcer , MALT lymphoma , Gastric cancer Helicobacter cinaedi Helicobacter cellulitis Authority control GND : 4138035-6 LCCN : sh85055863 NDL : 00569533ERBB2, BCL6, SULT2A1, ZAP70, NR3C1, BCL2, SPG21, IRF4, SPACA9, IL6, TLR4, IL10, CD274, MME, PWWP3A, TNF, CXCL8, CFH, NOD2, GKN1, ESR1, VOPP1, LEP, CTNNB1, MET, ABCB1, GPRASP1, CD163, MTA2, SDC1, TP53, MUC16, HOXA10, CDH1, CEACAM3, C4BPA, C4BPB, THEMIS, RD3, CIB2, CKAP5, ZEB2, MIR503, SCO2, HDAC6, MPZL2, NOD1, YAP1, MCRS1, POSTN, CIB1, ATG5, AHCYL1, ARPP21, HSPH1, PPARGC1A, MALT1, MIR489, MIR451A, CKAP4, MIR377, MIR33B, MIR589, MIR371A, VPS9D1-AS1, LOC107987479, TNFAIP3, CCAT2, TP73, TTR, TXN, TYMS, NNT-AS1, UMPS, UVRAG, VDAC1, VIP, XRCC1, MIR1298, MFT2, CUL4B, PPFIA3, TP63, BECN1, URI1, APLN, SQSTM1, SOCS3, PRDM4, SIRT1, GPR182, HAVCR2, IFIH1, FRTS1, FTO, FSD1, MIR195, MIR139, ARHGAP24, FSD1L, BRSK1, HDGFL2, MALAT1, NPVF, WDR20, SOX2-OT, CDCA5, CPXM2, GSC, RASSF6, MLKL, TIGIT, IL27, ADGRF1, MIR200C, TNMD, NID2, MIR224, TFAP2E, RAI14, MIR93, IL22, FOXP3, SOST, TMEM8B, IL17D, LZTFL1, MIR23B, MIR215, CPAT1, SARS2, KIF26B, FBXW7, TLR2, INTS13, MYDGF, MIR214, MIR21, SLC12A9, IL21, ABCA4, SPP1, TGFB2, ERBB4, CYP2D6, DPYD, ATN1, TSC22D3, E2F1, TYMP, EGF, EGFR, EGR1, EMP1, DMTN, ERBB3, ERCC2, CSF3R, FDPS, FES, FKBP5, FOXF1, FOXO1, FLT4, MSTN, GLI2, GNA12, GRN, HINT1, HLA-F, CYLD, CSF3, IFNG, FOXL2, ABO, ADM, ADRB2, AFP, AGER, AHR, AMH, KLK3, ATM, CCND1, CEACAM1, PRDM1, BRCA1, CCR4, CALCR, CAT, CCND2, CD40LG, CD44, CDKN1A, CDKN1B, CDKN1C, CDKN2A, CDX2, CEACAM5, CLU, HMGB1, IGF2, TGFB1, SARS1, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PTCH1, PTH, PTGS2, MOK, RAP1B, RGS3, RPE65, RRM1, CXCL12, SLC26A4, SKP2, SLPI, SNAI2, ABL1, STAT3, TAZ, ZEB1, TRBV20OR9-2, TEAD4, TFF3, TFPI, TFRC, SERPINA1, FURIN, IL2, MATN1, IL15, IL16, IL17A, ILF2, INPPL1, INSRR, IRF1, IRF2, ITGAV, JUN, KIF2A, L1CAM, MAX, NPR2, CD46, MMP8, MMP9, ABCC1, MTHFR, MTRR, MUC4, MUTYH, MYC, NDUFAB1, NFE2L2, NME1, H3P10

-
Lymphoma
Wikipedia
CD5, CD10 , surface Ig Frequently occurs outside lymph nodes, very indolent, may be cured by local excision Nodal marginal zone B cell lymphoma Follicular lymphoma About 40% of lymphomas in adults Small "cleaved" [cleft] cells ( centrocytes ) mixed with large activated cells ( centroblasts ), usually nodular ("follicular") growth pattern CD10 , surface Ig About 72–77% [33] Occurs in older adults, usually involves lymph nodes, bone marrow and spleen, associated with t(14;18) translocation overexpressing Bcl-2 , indolent Primary cutaneous follicle center lymphoma Mantle cell lymphoma About 3 to 4% of lymphomas in adults Lymphocytes of small to intermediate size growing in diffuse pattern CD5 About 50 [34] to 70% [34] Occurs mainly in adult males, usually involves lymph nodes, bone marrow, spleen and GI tract , associated with t(11;14) translocation overexpressing cyclin D1 , moderately aggressive Diffuse large B cell lymphoma , not otherwise specified About 40 to 50% of lymphomas in adults Variable, most resemble B cells of large germinal centers, diffuse growth pattern Variable expression of CD10 and surface Ig Five-year survival rate 60% [35] Occurs in all ages, but most commonly in older adults, may occur outside lymph nodes, aggressive Diffuse large B-cell lymphoma associated with chronic inflammation Epstein–Barr virus-positive DLBCL of the elderly Lymphomatoid granulomatosis Primary mediastinal (thymic) large B-cell lymphoma Intravascular large B-cell lymphoma ALK+ large B-cell lymphoma Plasmablastic lymphoma Primary effusion lymphoma Large B-cell lymphoma arising in HHV8-associated multicentric Castleman's disease Burkitt lymphoma/leukemia < 1% of lymphomas in the United States Round lymphoid cells of intermediate size with several nucleoli, starry-sky appearance by diffuse spread with interspersed apoptosis CD10, surface Ig Five-year survival rate 50% [36] Endemic in Africa, sporadic elsewhere, more common in immunocompromised and children, often visceral involvement, highly aggressive Mature T cell and natural killer (NK) cell neoplasms T-cell prolymphocytic leukemia T-cell large granular lymphocyte leukemia Aggressive NK cell leukemia Adult T-cell leukemia/lymphoma Extranodal NK/T-cell lymphoma, nasal type Enteropathy-associated T-cell lymphoma Hepatosplenic T-cell lymphoma Blastic NK cell lymphoma Mycosis fungoides / Sezary syndrome Most common cutaneous lymphoid malignancy Usually small lymphoid cells with convoluted nuclei that often infiltrate the epidermis, creating Pautrier microabscesseses CD4 5-year survival 75% [37] Localized or more generalized skin symptoms, generally indolent, in a more aggressive variant, Sézary's disease , skin erythema and peripheral blood involvement Primary cutaneous CD30-positive T cell lymphoproliferative disorders Primary cutaneous anaplastic large cell lymphoma Lymphomatoid papulosis Peripheral T-cell lymphoma not otherwise specified Most common T cell lymphoma Variable, usually a mix small to large lymphoid cells with irregular nuclear contours CD3 Probably consists of several rare tumor types, often disseminated and generally aggressive Angioimmunoblastic T cell lymphoma Anaplastic large cell lymphoma Precursor lymphoid neoplasms B-lymphoblastic leukemia/lymphoma not otherwise specified B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities T-lymphoblastic leukemia/lymphoma 15% of childhood acute lymphoblastic leukemia and 90% of lymphoblastic lymphoma . [29] : 635 Lymphoblasts with irregular nuclear contours, condensed chromatin, small nucleoli and scant cytoplasm without granules TdT , CD2 , CD7 It often presents as a mediastinal mass because of involvement of the thymus .ATM, PRF1, BLM, WAS, BCL2, EZH2, PTEN, CDKN2A, MYD88, MLH1, SH2D1A, KRAS, LIG4, NRAS, CD274, CDKN2B, TP53, FAS, NOTCH1, MTHFR, CDK4, BRAF, TNFAIP3, SOCS1, IKZF1, RHOA, NOTCH2, JAK3, MTR, BCL11B, PON1, EPHX1, MIR143, OGG1, CDK6, MAD1L1, DOCK8, RAC2, TWF1, PTGER4, CSF3R, HMGA1, EPM2A, NFKB1, IGH, BCL10, STAT3, BCL6, MS4A1, CD19, RTEL1, MSH2, MDM2, MAGT1, NBN, CR2, DCLRE1C, RAG2, TNFRSF13B, MSH6, RUNX1, PMS2, NFKB2, ZAP70, XIAP, TNFRSF13C, ADA, RAG1, TP63, CHEK2, ITK, XRCC4, PTPN11, HLA-DRB1, HLA-DQA1, RB1, RECQL4, WIPF1, CASP10, HLA-DQB1, PRKCD, TERT, SPINK1, APC, RAD54B, PNP, DNASE1L3, ICOS, TINF2, IL2RG, NCAM1, IL6, NPM1, MCL1, IL7R, CD81, CXADR, CFTR, PARN, IL10, CD44, CD40LG, CD40, KIF11, MYC, RMRP, TNFRSF8, CSF3, PAX5, PIK3CD, PDCD1, PIK3CG, PIM1, PIK3CB, IL2, DKC1, PIK3CA, PRKAR1A, IFNG, LYST, MAPK1, MME, JAK2, HLA-DRB9, MGMT, CDKN1B, ABCB1, PGM3, PRSS1, IL4, IRF4, H3P10, TNFSF12, NR1I3, USB1, PRMT5, BMI1, TRIM13, CTC1, ZFP91, OR10Q2P, AICDA, BTK, ARHGAP24, TCL1B, PDLIM7, KRT20, RAD54L, BCR, TNFSF13B, TCL1A, WRAP53, NSUN2, ARR3, IGHV3-69-1, IGHV3OR16-7, FOXP1, SMUG1, TBC1D9, NHP2, CTRC, CCND1, NOP10, CHD7, BIRC3, MALT1, ALK, DDX41, LOC102723407, CASR, COMMD3-BMI1, TRBV20OR9-2, TNF, TP73, SPG7, MIR155, TERC, LOC102724971, CXADRP1, VEGFA, ZFP91-CNTF, AKT1, PTGS2, CARD11, CD38, CD79B, STAT5B, PTPRC, CD22, HIF1A, GZMB, TMED7, STAT5A, CD27, H3P23, TICAM2, TMED7-TICAM2, IFI27, IL15, PRL, CASP3, PSMD9, KIT, MIR17HG, PRDM1, ZNRD2, CXCR4, DCTN6, IL2RA, LGALS1, FGFR1, CTNNB1, MIR21, SOAT1, MMP9, NOS2, BRCA2, CDKN1A, REL, TGFB1, CD28, SKP2, SOX11, CXCL12, CD34, GBA, HDAC9, TIAM1, CD79A, IGF1, EPHB2, CBLL2, KMT2A, IL18, TNFSF10, ISG20, PWWP3A, MUL1, PRKN, CREBBP, NOS1, SUB1, NPR2, LYN, BCL2L11, BAX, CIB1, JUNB, RASGRP1, JUN, FCER2, CD99, NME1, LOC105379528, H2AX, MET, NR0B2, HPSE, LAIR1, XRCC1, TIA1, HSP90AA1, LMO2, CDR3, MAPK3, PTPN6, TRAF1, TRAF3, LOC390714, IL3RA, PLK1, SLC16A1, CRK, CCND3, CD70, BRCA1, ABL1, CSF2, AHI1, GCSAM, KMT2D, KLRK1, MAP3K20, ITGAM, DDIT3, JUND, POU2AF1, B2M, TNFSF13, FASLG, MTOR, FBXW7, FOS, CD47, PER2, AIMP2, TNFRSF9, CD80, UCHL1, TIMP1, NR4A1, CD1D, LINC01194, TYK2, POLDIP2, RNF19A, HLA-A, CDK2, AKT2, CASP8, SYK, SIRT1, STAT6, CASP2, FOXP3, AHSA1, TET2, GRAP2, ASRGL1, MAPK14, MUC1, LONP1, ACTB, ETS1, ESR1, CSF1R, ABCB6, HDAC6, PPARG, CTLA4, MYCN, CXCL13, KLRC4-KLRK1, EPHA7, EP300, ENO2, LAMTOR2, PDCD2, IL21R, MYB, CREB1, IL5, DNMT3A, THBS1, PDCD1LG2, STAT1, IL9, ATN1, ERVW-1, IL1B, DLL1, IDH2, IL1A, FCRL4, TCF3, ANPEP, TRG, ATRAID, NXT1, AHR, CCR4, CCR5, MCTS1, TLR7, PTPN2, CD14, BIRC5, MIR150, CFL1, LAMTOR1, CDKN2C, MAP2K1, MDM4, RPSA, CD48, S100A9, PSMB6, RAC1, TPPP2, CD74, MYDGF, CYP1A1, CDC25A, DAPK1, KIR3DL1, MTX1, SF3B6, PDGFRA, PSMB9, SLPI, SLC19A1, NAT2, PDGFRB, DHX9, TNFSF8, CHEK1, APAF1, BCL11A, CD33, ITGB2, ABCC1, CCL5, DCC, CCND2, NCR3LG1, FCGR3A, EGFR, BMP6, EIF4EBP1, CDK2AP2, WT1, XPA, EGR1, YY1, GSTT1, GSTP1, GSTM1, FLT4, GRN, MAFK, TRRAP, GLB1, PHB2, H3P12, BAK1, RPP14, ACVRL1, MRPL28, SERPING1, BCL2L1, FLI1, FLII, TNFRSF14, FOSB, ZNF197, TNFRSF10A, TLX1, HLA-G, ESR2, H3P9, MIR17, EEF1E1, H3P8, UBE2I, TYMS, MIR18A, ERBB2, CARTPT, LXN, PTHLH, CDC42, CDH13, ODC1, GNLY, LIMD1, PTPN1, HERPUD1, PLSCR4, PRLR, CDK9, CDC25B, ANXA5, ROR1, CCR7, RASGRF1, RARA, NLRP2, ALLC, BCL3, DLEC1, CCR6, CTCF, PAG1, NTRK1, NRP1, MVP, CFLAR, CDK1, TNFRSF17, BSG, PRC1, PIAS2, MARCKSL1, CLTA, SERPINA5, PPIG, TBL1XR1, RAPH1, DLC1, CXCR5, CLEC10A, CKS1B, CLTC, SERPINA1, SLC16A3, SLC16A4, ERVK-6, CEL, APOBEC3G, PML, CMA1, BACH2, IL21, CEBPA, CREBZF, MTA1, PRKCB, CDKN2D, SCYL1, WLS, RHOJ, BBC3, BCOR, TNFRSF10B, PART1, BLNK, TAL1, KDM6A, VCAM1, WEE1, MAP3K7, BRD4, CD5, ZFP36, IL22, STAT4, IGK, SST, ZBTB17, ZBTB7A, PHLPP1, ACSBG1, ATR, UCP1, REG1A, SETD2, MCAT, POLM, TPM3, HSP90B1, TLR3, TLR1, TLE1, RUNX2, TIMP2, THY1, TCHH, TRAF6, GNL3, TWIST1, IGKV3-20, TFRC, UCN, SOS1, SOD2, STAB1, SNCA, CCL3, CCL2, SATB1, SAI1, TNFSF11, COPS5, DYNLL1, S100A4, BCL2A1, HPGDS, CASZ1, HRK, RNF2, TNFRSF6B, APOE, BATF3, RET, IL24, PIM2, CCL4, CHP1, SNAP25, IL23A, NR4A3, MLLT10, NINL, CALM3, SLC2A1, IL17D, CALCA, TLR9, SELE, CALM2, SDC1, CALM1, CX3CL1, CCL17, CNR1, NAT1, GSTK1, PARP1, AFP, LBR, STMN1, LAMC2, LAG3, BTLA, DAP, KIR3DL2, KIR2DS1, TMTC3, WG, TXLNA, MIR92A1, ITGA4, IL27, SCFV, CKS1BP7, IRF7, LGALS7, HOTAIR, DNER, FN1, MUC16, CD46, MCM2, GAPDH, NLRP3, LGALS7B, RBM45, CYP2B6, LMNA, GEM, CYP2E1, SLCO6A1, RICTOR, GUCY2D, CXCL9, IL1R1, EGF, MIR19B1, IGKV@, MIR19A, E2F1, IGF2, IFNA13, IFNA1, RCAN1, HOXC5, IAPP, HRAS, HTC2, HSPA5, HSPA4, IGL, IL1RN, IRF5, HLA-DPB1, INPP5D, CXCL10, NQO1, IL13, SERPINA9, CLEC4D, HGF, DLX5, STING1, MIR203A, IL7, DNMT1, DNMT3B, MIR20A, HLA-C, FOXO1, MIR142, BMF, PARP9, ERCC2, ERCC5, NFATC1, CSNK2A2, NEDD9, NEDD8, FH, LINC02605, FGFR3, ERVK-32, EZH1, FGF2, EPAS1, GLIS2, ABO, MYCL, ERVK-20, FCGR3B, SYVN1, MRC1, FCGR2A, F9, MTAP, COX2, H3P13, MTCO2P12, NGF, COX8A, MEF2B, AMH, ADRA2B, EML4, MIR152, BMP4, SUZ12, ADRA1A, ACAP2, HS3ST2, AXL, PRAME, SEC14L2, TMEM97, ATRX, DLEU1, MIR210, ATIC, PATZ1, CBX7, MIR221, ACOX1, ISCU, TFG, MIR222, MIR223, NOMO1, MLYCD, MIR29A, MIR30A, KLF2, MIR7977, H3P19, TP53COR1, PPP1R15A, CD2AP, LOC110806263, SLC23A1, MIR187, PTPN22, MYCBP, IBTK, HSPB8, BRS3, MIR182, GREM1, PIGK, SMC4, LAT, SND1, MIR16-1, MIR200B, MIR197, NAMPT, DICER1, NOCT, SERPINC1, MIR15B, MIR15A, PRPF6, SPRY2, MIR205, CADM1, SLC23A2, SH3BP4, H3P11, EIF3K, ROR1-AS1, MIR320A, CFDP1, ATF7, IGF2BP1, SSX2B, RAB40B, CKAP4, MIR633, FAM72B, SLC27A5, GGTLC5P, IGLL5, MIR31, SOX30, NCF1, INAFM2, SMC2, RBM14-RBM4, ANXA8, UBE2C, TPPP, TMED2, IGF2BP3, ANXA8L1, GGTLC3, GNA13, GGTLC4P, FAM72A, PLK4, CD226, PLK2, HSPH1, ADM, CCR2, CXCR6, FRS2, IGF2BP2, MIR711, BCL2L2, ADAM28, GGT2, HCST, BCL2L2-PABPN1, ANP32B, P3H3, H4C15, TPX2, MIR376A1, TARP, SETX, PIM3, MSE, PARP4, KDM4C, SPART, MIR93, RBM14, RCOR1, MIR34B, MIR34A, KIR2DS2, ATMIN, SMCHD1, ERVK-18, ATF6, CORO1A, MIR494, RASSF1, BGN, PTGDR2, DCTN3, KAT5, BIK, TREX1, STK38, BAD, POU5F1P4, POU5F1P3, RRAS2, IKZF3, IKZF2, MIR193B, NXF1, SEC31A, LINC00273, VOPP1, IGKV1-5, ASS1, FOXP2, NAPRT, TIGAR, RTN4, SCGB3A1, SALL4, MTG1, APEX1, DERL3, CCDC34, DPP9, AHRR, ZNF608, HACE1, PCDH10, WDR48, CXCL16, DIXDC1, TP53INP2, HSH2D, KIF14, PCBP4, UTP3, ACKR3, DIABLO, BIRC2, ATF7IP, CCAR1, DHX32, KRT222, USE1, PBK, H4-16, C10orf90, ALOX15, CHPT1, SLAMF6, CARD16, TIRAP, SPHK2, DBA2, DUSP22, SMYD2, CMTM1, PPP1R14A, ALPI, PLEKHA2, ADGRG7, MAP3K19, CDCA7, SUV39H2, AMELX, PALB2, E2F8, MYH14, CAMKMT, NANOG, DHDDS, DOHH, CCDC51, ANXA1, FCRL5, ST6GALNAC5, RNF34, ELL3, ULBP1, FIP1L1, TRIM11, AMT, MIXL1, RTL10, HAVCR2, AIRE, RIOX2, INSM2, CPAT1, GAS5, PRDM15, KDM2B, TSPYL2, SMOC2, RNASE7, IL25, VTCN1, ALPP, HVCN1, RTN4R, SETD3, CENPM, ASCC2, ABRAXAS1, AMD1, ANXA2, PASD1, ENOSF1, RBM38, RAB7B, ARRB2, LRP12, SLC35B2, SMARCAL1, TSPAN33, ABCB5, GEMIN4, ZC3H12D, F11R, ARRB1, SGPP1, NDUFA13, RDH11, LEF1, PLEKHO1, UBR5, CYP4V2, TRPV2, ARNTL, ISYNA1, SENP1, UHRF1, KCNRG, IGHJ5, IGHV4-59, IGHV4-34, ASCL1, MIR141, IGHV3-52, MIR139, IGHV3-41, MIR127, MIR125A, MIR124-1, STS, IGLV6-57, MIR10B, UOX, JAG1, IGKV3-15, IGKV2-29, MALAT1, ASPG, PYCARD, PGP, ETV7, CTCFL, RC3H1, CLEC4C, DDX53, DDX4, OTUD4, LY6K, NKX2-3, ALOX12, PGPEP1, AQP3, C1orf56, ACSF3, PINX1, IL34, AKIRIN2, NRG4, TMEM176A, TRPV6, CACUL1, IL17RB, NRSN1, MLKL, RHOF, SIRT7, RHOH, NEAT1, NT5DC3, HDAC7, GDE1, NCR3, EEF1AKNMT, NUTM1, TTC41P, WWOX, ADA2, PIMREG, CTAG1A, RIPK4, LNX2, UNC13D, POLE3, TRIM65, KLHDC8B, CRTC2, AR, MAFB, ELANE, TELO2, MKI67, MAOA, MBL2, CX3CR1, MDK, CUX1, MECP2, MEFV, MEN1, CTPS1, MGST1, CIITA, CCN2, MIP, CTAG1B, ITGAL, FOXO4, CSNK2A1, MMP1, MMP3, MMP7, MNDA, MOS, MPL, MPO, MSH3, MST1R, CSF1, MVD, SMAD5, SMAD2, SMAD1, MAD2L1, ITGB1, EIF6, JAK1, GADD45A, ACE, DCK, KCNA3, KDR, KIR2DL4, KLRC1, KLRD1, CD55, KRT8, CYP17A1, LAMP1, LCK, LDHA, LEP, CYP3A4, LGALS3, LGALS9, LRP1, LTA, LTB, LUM, LYL1, CYP1B1, MXI1, MYBL1, CRYZ, PCM1, PCSK1, PCSK2, PDE4A, PDE4B, PDGFA, SERPINF1, PFDN5, PGF, CLU, CHGA, CHD1, CETP, PKNOX1, PLCG2, PLD2, PLG, PLXNA2, PMS1, CEBPB, POMC, POU2F2, POU5F1, PPIA, PPID, PPP1R3A, PREP, CD52, PCNA, PBX1, CRP, PAX3, ATF2, NDUFAB1, NELL1, NF1, NFATC2, CR1, NFKBIA, CPOX, COMT, CNOT2, COL11A2, COL4A3, NOTCH3, NPC1, PLK3, NSF, NT5E, OAS3, ODF1, OPCML, OPRK1, OPRM1, P2RX7, PAEP, PRDX1, PAK1, PAK2, ITGAX, ISL1, PRKDC, GGT1, FLT3, FMOD, FTH1, FYN, G6PD, XRCC6, GAS6, GATA3, EIF4A1, GCG, GCSH, GDNF, GFI1, GH1, DDX6, GH2, GJB2, GNA12, GOT2, GPI, CXCR3, GPR34, GPR42, GPX1, GPX4, GRB2, GRIA3, NR3C1, FLNB, FOXO3, FKBP1AP4, FKBP1AP3, ELF4, ENG, EPHA4, EIF5A, EIF4G2, ERBB4, ERG, ETV5, ETV6, EWSR1, F3, F8, F10, PTK2B, FAU, FBN1, EIF4E, FCGR2B, FCGRT, FES, FGF6, EIF4A2, GPC5, VEGFD, FKBP1A, FKBP1AP1, FKBP1AP2, GRM3, GSK3A, GSK3B, IRF8, ID3, ID4, IDH1, ARID3A, IFNB1, SLC26A3, DPYSL3, DPYD, IGHG3, IKBKB, DPP4, DNTT, DYNC1H1, DNASE1, CXCL8, IL9R, IL10RA, IL12A, IL12B, IL12RB1, IL17A, DES, INSM1, IRAK1, IRF1, IRF3, IRS1, ID1, ICAM3, EIF1AX, ICAM1, EGR3, HBB, HBZ, HCCS, HDAC2, HEXA, HFE, HIC1, UBE2K, HK2, HLA-B, EDNRA, S1PR1, HMBS, HMGB1, HMGB2, HMMR, HNRNPK, TLX2, HPR, HPRT1, HSF1, HSPA1A, HSPA1B, HSPA1L, HSPA2, HTR1A, CDO1, CDKN1C, SNAP91, PICALM, PAX8, CAPN1, RAB7A, CAMLG, KAT6A, RASSF7, PTP4A2, CALR, SLC7A5, COIL, TKTL1, ARID1A, H4C9, GFI1B, TTR, H4C1, H4C4, H4C6, H4C12, H4C11, H4C3, H4C8, H4C2, H4C5, H4C13, H4C14, GPR65, RAE1, BTG2, LAPTM5, ZMYM2, ZNF32, TYRO3, UBC, UBE2B, SUMO1, SLC35A2, UGT1A, UCK2, UNG, UVRAG, VAV1, VCP, CASP9, VEGFC, VHL, VIM, VIP, VWF, CASP7, WNT5A, WRN, XBP1, XBP1P1, ELF3, CASP5, XRCC3, CASP1, ZFX, SLC43A1, CNTNAP1, DGKZ, SLC7A7, FCGR2C, SMC3, LPAR2, IL1RL1, SLC16A7, AURKB, C1QBP, KLF4, COX5A, BUB1B, LPXN, NCR1, ITM2B, MAP4K4, NAPSA, TBPL1, PTGES, BCAR1, CDC42BPB, APOBEC3B, NFE2L3, BUB1, SART3, BTG1, HDAC4, MTSS1, SPATA2, SLC7A6, AIP, USO1, PKD2L1, DDR1, CA12, BECN1, HYAL2, ADAM19, TNFSF14, CA9, FMNL1, TNFSF9, TNFRSF18, DLK1, TNFRSF10C, INPP4B, CES2, CCN4, HDAC3, ALKBH1, DLEU2, NR1I2, SQSTM1, MCM3AP, SGCE, MBD2, PLOD3, NOL3, MAP3K14, SOCS3, TNFRSF4, TTK, MAPK8, SCT, RIT1, CD37, RNASE3, BRD2, ABCE1, RORB, ROS1, RPE65, RPL22, RPS27A, RRBP1, RYR1, S100A8, CCL1, TSG101, CCL19, CCL20, CXCL11, SEL1L, SET, SHBG, SHH, SHMT1, PMEL, SKI, SLAMF1, SLC2A3, SLC7A4, RHEB, RGS13, RGS1, RFC1, MAP2K7, CDK7, RELN, PRTN3, PSMD2, PTGDR, PTGDS, CDH1, PTK2, PTK6, PTK7, PTN, PTPN13, PTPRA, PTPRJ, PTPRO, PVT1, RAD23B, RAD51, RAD51B, RAF1, CD68, CD59, RBL2, RBP1, OPN1LW, RELB, SLC22A2, SMARCA1, SMARCA4, CD247, CD3G, TERF1, TERF2, CD3E, TFAM, TGFA, TGFBR1, TGM2, TH, THOP1, CCNT1, TJP1, TK1, TLE2, TLE3, TLE4, KRIT1, TNFRSF1B, TOP1, TOP2A, RUNX3, TP53BP2, CBFA2T3, NR2C2, TRAF2, CAT, TSN, TRGC1, TRB, SUMO3, TCP1, SUMO2, FSCN1, SNRPN, CD9, SPIB, CD8B, SPINK2, SPN, SPP1, SPTA1, SRC, SRI, TRIM21, SSB, SSTR4, SSX2, ST14, STAT2, CD8A, CD6, SULT1E1, STK4, AURKA, STXBP2, TAT, CNTN2, ZEB1, XPO1

-
Nonketotic Hyperglycinemia
GeneReviews
Agents/circumstances to avoid: Valproate, which raises blood and CSF glycine concentrations and may increase seizure frequency; vigabatrin, which has resulted in rapid loss of function when used to treat seizures, particularly in those with attenuated NKH who have West syndrome. Genetic counseling. NKH is inherited in an autosomal recessive manner.
-
Friedreich Ataxia
GeneReviews
Pes cavus is common (55%) but generally causes little problem for affected individuals. Restless leg syndrome is common in individuals with Friedreich ataxia, affecting 32%-50% of individuals in two studies [Frauscher et al 2011]. ... Other early-onset ataxias may be distinguishable by virtue of their characteristic clinical features (see also Ataxia Overview): Ataxia-telangiectasia Ataxias associated with pathogenic variants in mitochondrial DNA (see Mitochondrial Disorders Overview) Behr syndrome (spasticity, ataxia, optic atrophy, and intellectual disability) (OMIM 210000) X-linked sideroblastic anemia and ataxia (OMIM 301310) Marinesco-Sjögren syndrome (cerebellar ataxia, cataracts, intellectual disability, short stature, and delayed sexual development) Deafness-dystonia-optic neuronopathy syndrome Late-onset hexosaminidase A deficiency (ataxia, upper and lower motor neuron disorders, dementia, and psychotic episodes) [Perlman 2002] Two autosomal dominant ataxias with sensory neuropathy – spinocerebellar ataxia type 4 (SCA4) [Flanigan et al 1996] and SCA25 [Stevanin et al 2004] – may present with FRDA-like phenotypes (see Ataxia Overview).FXN, NFE2L2, PIP5K1B, ATXN1, GABPA, DLD, EPO, CTCF, PPARGC1A, FTMT, GPAA1, ISCU, APTX, SETX, KIF1B, AHSA1, COG5, AGTR1, MFN2, PCLAF, CRTC1, TJP2, GRAP2, PDLIM1, HDAC3, AIMP2, CIR1, RNF19A, SIRT3, CHMP1B, VTRNA2-1, MIR323A, MIR155, C16orf82, CTCFL, HAMP, JPH3, SLC17A7, ATXN10, TWNK, RNF126, GLRX5, PYCARD, MLH3, POLDIP2, VIM, USP7, TP53, TYMS, TTPA, LY6E, LPA, KCNJ5, KCNC3, IFNG, IFNB1, HSPA9, HFE, GAA, ATN1, TIMM8A, CSF3, MAPK14, CRK, CD34, CASP3, CACNA1A, ATXN3, MLH1, NCF2, SPG7, APOA1, TNNT2, TNNT1, TFRC, TFPI, TEAD1, SRF, ATXN2, PDYN, S100A4, PVALB, PTGS2, MAPK1, PPP2R2B, PPARG, POLG, FTX
-
Neuronal Ceroid Lipofuscinosis
Wikipedia
Retrospectively, these papers disclose that the authors grouped together different types of the syndrome. Furthermore, Batten, at least for some time, insisted that the condition that he described was distinctly different from Tay–Sachs disease , the prototype of a neuronal lysosomal disorder now identified as GM2 gangliosidosis type A.TPP1, CLN6, CLN3, CLN8, PPT1, MFSD8, CLN5, ATP13A2, KCTD7, ARSG, CTSD, PPT2, CLCN3, CLCN6, DNAJC5, GRN, FBXL3, NCL, CTSF, ATP5MC1, TARDBP, CAPN3, CLN9, PSAP, CPQ, AGER, TMEM106B, TBCK, SLC39A7, ZDHHC15, CAPN8, TFEB, PPP5C, TAC1, SOD2, CALD1, DPYSL2, GAD1, GAD2, KCNMA1, LGALS1, MAS1, MECP2, TRPM1, PKD1, POLG, POU4F1, APP, MAPK13, SNCA, PRCD
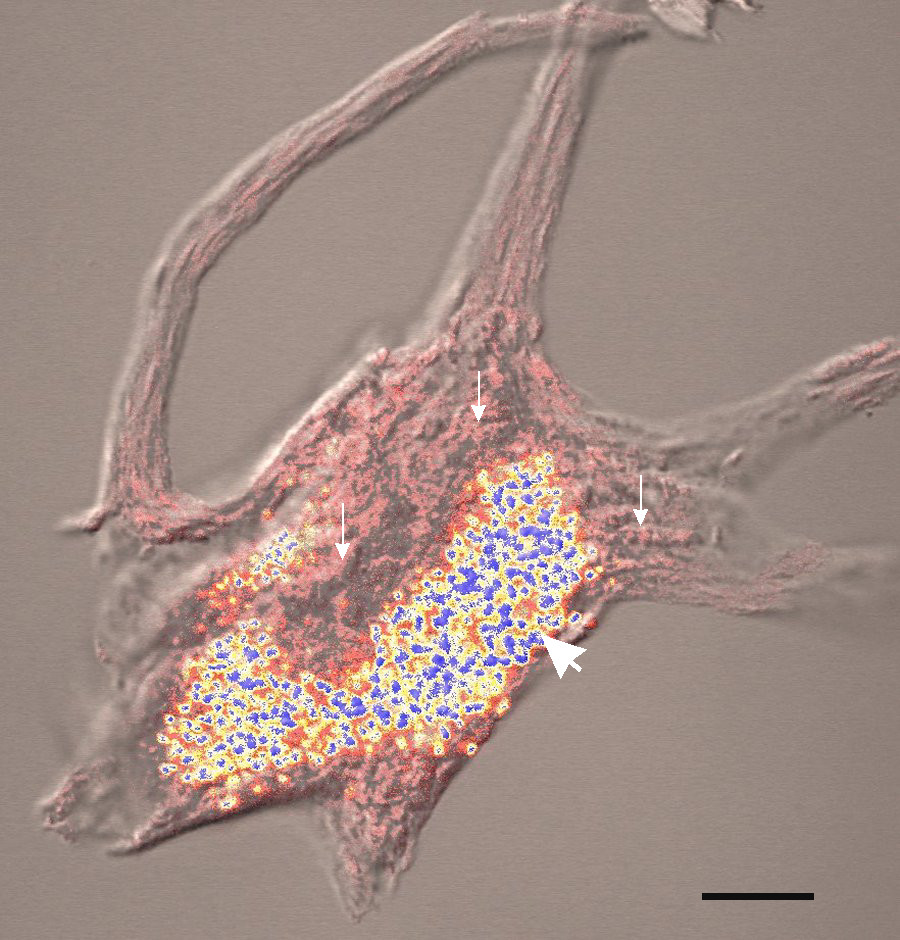
-
Glycogen Storage Disease Ii
OMIM
Francesconi and Auff (1982) described Wolff-Parkinson-White syndrome (194200) and second-degree atrioventricular block in a patient with the adult form of glycogenosis II. ... INHERITANCE - Autosomal recessive HEAD & NECK Ears - Hearing loss Mouth - Macroglossia CARDIOVASCULAR Heart - Cardiomegaly - Shortened P-R interval on EKG - Huge QRS complexes - Wolf-Parkinson-White syndrome Vascular - Cerebral artery aneurysm RESPIRATORY - Respiratory failure due to muscle weakness - Dyspnea - Respiratory infections CHEST Ribs Sternum Clavicles & Scapulae - Diaphragmatic paralysis ABDOMEN Liver - Hepatomegaly Spleen - Splenomegaly MUSCLE, SOFT TISSUES - Weakness - Proximal muscle weakness - Myopathic pattern on EMG - Firm muscles NEUROLOGIC Central Nervous System - Hypotonia - Abnormal brain myelination Peripheral Nervous System - Absent deep tendon reflexes METABOLIC FEATURES - Fever of central origin LABORATORY ABNORMALITIES - Elevated serum creatine kinase - Elevated AST and LDH, especially infantile-onset - Presence of vacuoles on muscle biopsy - Deficiency of alpha-1,4-glucosidase (acid maltase) MISCELLANEOUS - Two presentations - rapid, fatal disorder of infancy and slowly progressive muscular disorder of childhood - Patients with later onset have better prognosis - Incidence of 1 in 40,000 infants worldwide MOLECULAR BASIS - Caused by mutation in the alpha-1,4-glucosidase gene (GAA, 606800.0002 ) ▲ CloseGAA, TNNT2, SI, VEGFA, HTRA1, CFH, MGAM, ARMS2, AMD1, AMD1P2, KDR, PLXNA2, APOE, CCL2, SERPINF1, PGF, CFB, RPE, CRP, CCR2, FLT1, PRKAG2, ELN, ELF3, IGF2R, G6PC, CX3CR1, C3, SKIV2L, IL10, HMCN1, SOD1, SOD2, IL6, TFEB, PON1, MAN2B1, SERPING1, GLA, NFE2L2, IGF1, AAVS1, ACE, IDUA, ELOVL4, TNF, TLR3, RIPK1, FZD4, NELFE, CXCL12, SLC6A8, VLDLR, CXCR4, ZFP36, VWF, TFR2, VIM, UGT1A, TTR, STC1, TRAF6, TNXB, STBD1, TIMP3, NAT2, CFHR4, ADIPOQ, PLEKHA1, UGT1A5, UGT1A9, UGT1A4, UGT1A1, UGT1A3, CCDC40, APOM, PNPLA2, SLURP1, FKBPL, ABCG1, CIAO3, ARMC9, ACAD10, COL18A1, NLRP3, C1QTNF5, ASPM, TEC, C20orf181, UGT1A6, UGT1A7, UGT1A8, UGT1A10, SEMA3E, ATP6AP2, AKR1A1, ARIH2, CXCL13, SLCO1B1, CXCL6, PPARGC1A, KERA, IKZF2, DKK1, SLC16A8, SLC17A5, HPLH1, HPGDS, TBK1, PIK3R4, ANGPTL4, DDIT4, CXCL5, NPPB, CCL11, RYR1, CNTF, COL8A1, CP, CPT2, DES, DHFR, NQO1, EPO, ERCC6, F2, F10, EFEMP1, FCGR3A, FCGR3B, FECH, FHL1, FN1, CCR3, CCR1, CETP, ANG, ACTB, ACTN3, ADM, PARP1, AGL, AHR, ALB, ANPEP, CD59, XIAP, APOA1, APRT, ARSB, BDNF, CALCR, CALR, FPR1, FUT1, GALC, ND5, LRP5, MAP2, MFAP1, MFGE8, CXCL9, MME, MMP9, MYP2, LIPC, NGF, SERPINA1, PPARA, PPARG, MAPK3, RAD51B, PRPH2, LPA, LAMP2, GAS6, CFHR2, GBE1, MSTN, SFN, CXCL1, GSTP1, GSTZ1, GYG1, HIF1A, ITGAM, IDS, CFI, IGFALS, IL1B, CXCL8, IL17A, CXCL10, LINC01672
-
Monoclonal B-Cell Lymphocytosis
Wikipedia
Individuals with high-count MBL (studies based primarily on the CLL/SLL phenotype) are at an increased risk for developing: 1) cancers of the breast, lung, and gastrointestinal tract in up to 13% of all cases; 2) autoimmune hemolytic anemia and immune thrombocytopenic purpura ; 3) unexplained kidney disease as manifested by chronic kidney disease and/or the nephrotic syndrome ; and 4) serious infections. [2] While earlier studies suggested that only very high-count MBL (i.e. >10x10 9 B-cells/L) was associated with a decrease in survival, [2] more recent studies indicate that high-count MBL (i.e. ... PMID 16042682 . v t e Leukaemias , lymphomas and related disease B cell ( lymphoma , leukemia ) (most CD19 CD20 ) By development/ marker TdT+ ALL ( Precursor B acute lymphoblastic leukemia/lymphoma ) CD5 + naive B cell ( CLL/SLL ) mantle zone ( Mantle cell ) CD22 + Prolymphocytic CD11c+ ( Hairy cell leukemia ) CD79a + germinal center / follicular B cell ( Follicular Burkitt's GCB DLBCL Primary cutaneous follicle center lymphoma ) marginal zone / marginal zone B-cell ( Splenic marginal zone MALT Nodal marginal zone Primary cutaneous marginal zone lymphoma ) RS ( CD15 +, CD30 +) Classic Hodgkin lymphoma ( Nodular sclerosis ) CD20+ ( Nodular lymphocyte predominant Hodgkin lymphoma ) PCDs / PP ( CD38 +/ CD138 +) see immunoproliferative immunoglobulin disorders By infection KSHV ( Primary effusion ) EBV Lymphomatoid granulomatosis Post-transplant lymphoproliferative disorder Classic Hodgkin lymphoma Burkitt's lymphoma HCV Splenic marginal zone lymphoma HIV ( AIDS-related lymphoma ) Helicobacter pylori ( MALT lymphoma ) Cutaneous Diffuse large B-cell lymphoma Intravascular large B-cell lymphoma Primary cutaneous marginal zone lymphoma Primary cutaneous immunocytoma Plasmacytoma Plasmacytosis Primary cutaneous follicle center lymphoma T/NK T cell ( lymphoma , leukemia ) (most CD3 CD4 CD8 ) By development/ marker TdT+ : ALL ( Precursor T acute lymphoblastic leukemia/lymphoma ) prolymphocyte ( Prolymphocytic ) CD30+ ( Anaplastic large-cell lymphoma Lymphomatoid papulosis type A ) Cutaneous MF+variants indolent: Mycosis fungoides Pagetoid reticulosis Granulomatous slack skin aggressive: Sézary disease Adult T-cell leukemia/lymphoma Non-MF CD30 -: Non-mycosis fungoides CD30− cutaneous large T-cell lymphoma Pleomorphic T-cell lymphoma Lymphomatoid papulosis type B CD30 +: CD30+ cutaneous T-cell lymphoma Secondary cutaneous CD30+ large-cell lymphoma Lymphomatoid papulosis type A Other peripheral Hepatosplenic Angioimmunoblastic Enteropathy-associated T-cell lymphoma Peripheral T-cell lymphoma not otherwise specified ( Lennert lymphoma ) Subcutaneous T-cell lymphoma By infection HTLV-1 ( Adult T-cell leukemia/lymphoma ) NK cell / (most CD56 ) Aggressive NK-cell leukemia Blastic NK cell lymphoma T or NK EBV ( Extranodal NK-T-cell lymphoma / Angiocentric lymphoma ) Large granular lymphocytic leukemia Lymphoid+ myeloid Acute biphenotypic leukaemia Lymphocytosis Lymphoproliferative disorders ( X-linked lymphoproliferative disease Autoimmune lymphoproliferative syndrome ) Leukemoid reaction Diffuse infiltrative lymphocytosis syndrome Cutaneous lymphoid hyperplasia Cutaneous lymphoid hyperplasia with bandlike and perivascular patterns with nodular pattern Jessner lymphocytic infiltrate of the skin General Hematological malignancy leukemia Lymphoproliferative disorders Lymphoid leukemiasNOTCH2, KRT20, MS4A1, LOC102724971, IGH, MBL2, LOC102723407, IGHV3OR16-7, IGHV3-69-1, CD19, LEF1, ITGA4, NOTCH1, IGF2BP1, IL22, MBL3P, CD274, IL21, AICDA, XPO1, HM13, CLEC12A, IMMP1L, MIR155, MIR15A, MIR21, LINC01672, COLEC10, BCL2, VIM, TP53, CXCR5, CD22, CD38, CD79B, CDK6, CCR6, IL2, IMPA1, JAK2, LAIR1, MME, NTF3, PIK3C2B, SELL, SIM1, BCL6, TNF, SPN
-
Fish Allergy
Wikipedia
. ^ a b Ridolo E, Martignago I, Senna G, Ricci G (October 2016). "Scombroid syndrome: it seems to be fish allergy but... it isn't". ... External links [ edit ] Classification D ICD - 10 : T78.0 , T78.1 , L23.6 , L27.2 , Z91.0 v t e Allergic conditions Respiratory system Allergic rhinitis (hay fever) Asthma Hypersensitivity pneumonitis Eosinophilic pneumonia Eosinophilic granulomatosis with polyangiitis Allergic bronchopulmonary aspergillosis Farmer's lung Laboratory animal allergy Skin Angioedema Urticaria Atopic dermatitis Allergic contact dermatitis Hypersensitivity vasculitis Blood and immune system Serum sickness Circulatory system Anaphylaxis Digestive system Coeliac disease Eosinophilic gastroenteritis Eosinophilic esophagitis Food allergy Egg allergy Milk intolerance Nervous system Eosinophilic meningitis Genitourinary system Acute interstitial nephritis Other conditions Drug allergy Allergic conjunctivitis Latex allergy v t e Hypersensitivity and autoimmune diseases Type I / allergy / atopy ( IgE ) Foreign Atopic eczema Allergic urticaria Allergic rhinitis (Hay fever) Allergic asthma Anaphylaxis Food allergy common allergies include: Milk Egg Peanut Tree nut Seafood Soy Wheat Penicillin allergy Autoimmune Eosinophilic esophagitis Type II / ADCC IgM IgG Foreign Hemolytic disease of the newborn Autoimmune Cytotoxic Autoimmune hemolytic anemia Immune thrombocytopenic purpura Bullous pemphigoid Pemphigus vulgaris Rheumatic fever Goodpasture syndrome Guillain–Barré syndrome " Type V "/ receptor Graves' disease Myasthenia gravis Pernicious anemia Type III ( Immune complex ) Foreign Henoch–Schönlein purpura Hypersensitivity vasculitis Reactive arthritis Farmer's lung Post-streptococcal glomerulonephritis Serum sickness Arthus reaction Autoimmune Systemic lupus erythematosus Subacute bacterial endocarditis Rheumatoid arthritis Type IV / cell-mediated ( T cells ) Foreign Allergic contact dermatitis Mantoux test Autoimmune Diabetes mellitus type 1 Hashimoto's thyroiditis Multiple sclerosis Coeliac disease Giant-cell arteritis Postorgasmic illness syndrome Reactive arthritis GVHD Transfusion-associated graft versus host disease Unknown/ multiple Foreign Hypersensitivity pneumonitis Allergic bronchopulmonary aspergillosis Transplant rejection Latex allergy (I+IV) Autoimmune Sjögren syndrome Autoimmune hepatitis Autoimmune polyendocrine syndrome APS1 APS2 Autoimmune adrenalitis Systemic autoimmune disease












