-
Nosebleed
Wikipedia
Pronunciation Epistaxis / ˌ ɛ p ɪ ˈ s t æ k s ɪ s / EP -ih- STAK -sis Specialty Otorhinolaryngology Symptoms Bleeding from the nose [1] Usual onset Less than 10 and over 50 years old [2] Risk factors Trauma, Excessive Nose-picking , Certain infections , blood thinners , high blood pressure , alcoholism , seasonal allergies , dry weather [3] Diagnostic method Direct observation [1] Differential diagnosis Bleeding from the lungs , esophageal varices [1] Prevention Petroleum jelly in the nose [4] Treatment Pressure over the lower half of the nose, nasal packing , endoscopy [5] Medication Tranexamic acid [6] Frequency 60% at some point in time [7] Deaths Rare [3] A nosebleed , also known as epistaxis , is bleeding from the nose . [1] Blood can also flow down into the stomach and cause nausea and vomiting . [8] In more severe cases blood may come out of both nostrils . [9] Rarely bleeding may be so significant low blood pressure occurs. [1] Rarely the blood can come up the nasolacrimal duct and out from the eye. [10] Risk factors include trauma including putting the finger in the nose, blood thinners , high blood pressure , alcoholism , seasonal allergies , dry weather, and inhaled corticosteroids . [3] There are two types: anterior, which is more common; and posterior, which is less common but more serious. [3] Anterior nosebleeds generally occur from Kiesselbach's plexus while posterior bleeds generally occur from the sphenopalatine artery . [3] The diagnosis is by direct observation. [1] Prevention may include the use of petroleum jelly in the nose. [4] Initially treatment is generally by applying pressure for at least five minutes over the lower half of the nose. [5] If this is not sufficient nasal packing may be used. [5] Tranexamic acid may also be helpful. [6] If bleeding episodes continue endoscopy is recommended. [5] About 60% of people have a nosebleed at some point in their life. [7] About 10% of nosebleeds are serious. [7] Nosebleeds are rarely fatal, accounting for only 4 of the 2.4 million deaths in the U.S. in 1999. [11] Nosebleeds most commonly affect those younger than 10 and older than 50. [2] Contents 1 Cause 2 Pathophysiology 3 Prevention 4 Treatment 4.1 Nasal packing 4.2 Tranexamic acid 4.3 Cauterization 4.4 Surgery 4.5 Other 5 Society and culture 5.1 Etymology 6 References 7 External links Cause [ edit ] Two children boxing , the one on the right having a nosebleed due to a punch to the face. ... PMID 24439881 . ^ a b c d Tunkel, David E.; Anne, Samantha; Payne, Spencer C.; Ishman, Stacey L.; Rosenfeld, Richard M.; Abramson, Peter J.; Alikhaani, Jacqueline D.; Benoit, Margo McKenna; Bercovitz, Rachel S.; Brown, Michael D.; Chernobilsky, Boris; Feldstein, David A.; Hackell, Jesse M.; Holbrook, Eric H.; Holdsworth, Sarah M.; Lin, Kenneth W.; Lind, Meredith Merz; Poetker, David M.; Riley, Charles A.; Schneider, John S.; Seidman, Michael D.; Vadlamudi, Venu; Valdez, Tulio A.; Nnacheta, Lorraine C.; Monjur, Taskin M. (7 January 2020). ... Retrieved January 31, 2010 . ^ J. F. Lubianca Neto; F. D. Fuchs; S. R. Facco; M. Gus; L. Fasolo; R. Mafessoni; A. ... E.; Williams, R. J.; Kuhn, I.; Carrie, S. (December 2017). "Intranasal packs and haemostatic agents for the management of adult epistaxis: systematic review". ... Classification D ICD - 10 : R04.0 ICD - 9-CM : 784.7 MeSH : D004844 DiseasesDB : 18327 External resources MedlinePlus : 003106 eMedicine : emerg/806 ent/701 Patient UK : Nosebleed v t e Symptoms and signs relating to the respiratory system Auscultation Stethoscope Respiratory sounds Stridor Wheeze Crackles Rhonchi Stertor Squawk Pleural friction rub Fremitus Bronchophony Terminal secretions Elicited findings Percussion Pectoriloquy Whispered pectoriloquy Egophony Breathing Rate Apnea Prematurity Dyspnea Hyperventilation Hypoventilation Hyperpnea Tachypnea Hypopnea Bradypnea Pattern Agonal respiration Biot's respiration Cheyne–Stokes respiration Kussmaul breathing Ataxic respiration Other Respiratory distress Respiratory arrest Orthopnea / Platypnea Trepopnea Aerophagia Asphyxia Breath holding Mouth breathing Snoring Other Chest pain In children Precordial catch syndrome Pleurisy Nail clubbing Cyanosis Cough Sputum Hemoptysis Epistaxis Silhouette sign Post-nasal drip Hiccup COPD Hoover's sign asthma Curschmann's spirals Charcot–Leyden crystals chronic bronchitis Reid index sarcoidosis Kveim test pulmonary embolism Hampton hump Westermark sign pulmonary edema Kerley lines Hamman's sign Golden S sign Authority control GND : 4171181-6 NDL : 00560692ACVRL1, DNAH11, ZBTB16, WIPF1, WAS, VWF, TERT, TERC, TBXAS1, TBXA2R, STIM1, STAT5B, STAT3, RARA, PRTN3, PRKAR1A, PRKACG, PRF1, PML, GFI1B, FCGR2C, CACNA1D, RASGRP2, IRF2BP2, SLFN14, MCFD2, HPS4, DTNBP1, FIP1L1, HPS6, TBL1XR1, NABP1, P2RY12, BCOR, TET2, GP6, SBDS, PTPN22, NBEAL2, HPS5, PLAU, NUMA1, NPM1, MYH9, FGB, FGA, F13B, F13A1, F11, F10, F7, F5, F2, ETV6, EPOR, ENG, CYP11B2, CYP11B1, CTLA4, LYST, RUNX1, FGG, FYB1, GATA1, IFNG, MYD88, MPL, SMAD4, KCNJ5, JAK2, ITGB3, ITGA2B, HPS1, GBA, HLA-DPB1, HLA-DPA1, GP9, GP1BB, GP1BA, GGCX, GDF2, BLOC1S3

-
Leber Congenital Amaurosis
Wikipedia
. ^ a b Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, et al. (August 2009). ... P.; Ardinger, H. H.; Pagon, R. A.; Wallace, S. E.; Bean LJH; Stephens, K.; Amemiya, A. (1993). ... PMID 20301475 . ^ Perrault I, Rozet JM, Calvas P, Gerber S, Camuzat A, Dollfus H, et al. (December 1996). ... Retrieved 2007-09-21 . ^ Dharmaraj S, Li Y, Robitaille JM, Silva E, Zhu D, Mitchell TN, et al. ... S2CID 27501557 . ^ Perrault I, Hanein S, Zanlonghi X, Serre V, Nicouleau M, Defoort-Delhemmes S, et al.RPGRIP1, CRX, CRB1, NMNAT1, RPE65, AIPL1, CEP290, IQCB1, LCA5, SPATA7, TULP1, KCNJ13, LRAT, IMPDH1, RD3, GUCY2D, RDH12, USP45, GDF6, MERTK, IFT140, PCYT1A, MPP5, ABCA4, CLUAP1, USH2A, SLC38A8, CDHR1, GRM6, RP2, AHI1, PDE6A, CNGB3, GIGYF2, ZFYVE26, NR2E3, RGS9, CRB2, LCA10, OTX2, CLTA, PTPRC, MYO7A, CNGA3, CCT2, GUCA1A, PRPH2, IMPG1, CABP4, GUCY2F, SLC7A14, GUCA1B, LPCAT1, HSD17B6, ENO2, CNTLN, PSMD13, PRPH, GUCY2EP, BCL2, MIR204, EGF, SP4, RPGR, NUB1, ADH7, NRL, TUB, ND4, FST, SLC19A2, PRPF8, PNPLA6, CILK1, NINL, SERPINF1, RPGRIP1L, MEF2C, TAP2, MEF2A, ELOVL4, IMPG2, PDE6B
-
Tropical Disease
Wikipedia
TDR history The current TDR disease portfolio includes the following entries: [5] Historical TDR disease portfolio Disease When added Pathogen Primary vector Primary endemic areas Frequency Annual deaths Symptoms Complications Malaria 1975 Plasmodium falciparum and four other Plasmodium species of protazoa Anopheles mosquitoes throughout the tropics 228 million (2018) 405,000 (2018) fever , tiredness , vomiting , headache yellow skin , seizures , coma , death Schistosomiasis / ˌ ʃ ɪ s t ə s ə ˈ m aɪ ə s ɪ s / [6] [7] (snail fever, bilharzia, "schisto") 1975 Schistosoma flatworms (blood flukes) freshwater snails throughout the tropics 252 million (2015) 4,400–200,000 abdominal pain , diarrhea , bloody stool , blood in the urine . ... Liver damage , kidney failure , infertility , bladder cancer Lymphatic filariasis 1975 Wuchereria bancrofti , Brugia malayi , and Brugia timori filarial worms mosquitoes throughout the tropics 38.5 million (2015) few lymphoedema , elephantiasis , hydrocele Onchocerciasis / ˌ ɒ ŋ k oʊ s ɜːr ˈ k aɪ ə s ɪ s , - ˈ s aɪ -/ [8] [9] (river blindness) 1975 Onchocerca volvulus filarial worms [10] Simuliidae black flies sub-Saharan Africa 15.5 million (2015) 0 itching , papules edema , lymphadenopathy , visual impairment , blindness Chagas disease (American trypanosomiasis) 1975 Trypanosoma cruzi protozoa Triatominae kissing bugs South America 6.2 million (2017) 7,900 (2017) fever , swollen lymph nodes , headache heart failure , enlarged esophagus , enlarged colon African trypanosomiasis (sleeping sickness) 1975 Trypanosoma brucei gambiense and T. b. rhodesiense protozoa Glossina tsetse flies sub-Saharan Africa 11,000 (2015) 3,500 (2015) first stage: fever , headache , itchiness , joint pain second stage: insomnia , confusion , ataxia , hemiparesis , paralysis anemia , endocrine disfunction , cardiac disfunction , kidney dysfunction , coma , death Leishmaniasis 1975 Leishmania protozoa Phlebotominae sandflies throughout the tropics 4–12 million 24,200 (2015) skin ulcers fever , anemia , enlarged liver , enlarged spleen , death Leprosy † (Hansen's disease) 1975 Mycobacterium leprae and M. lepromatosis mycobacteria extensive contact (probably airborne disease ) throughout the tropics 209,000 (2018) few skin lesions , [11] numbness permanent damage to the skin, nerves, limbs, and eyes Dengue fever 1999 dengue virus Aedes aegypti and other Aedes mosquitoes tropical Asia 390 million (2020) 40,000 fever , headache , muscle and joint pain , rash , vomiting , diarrhea low levels of blood platelets , hypotension , hemorrhage , shock Tuberculosis † (TB, consumption) 1999 Mycobacterium tuberculosis mycobacteria airborne disease worldwide 10 million (active, 2018), 2 billion (latent, 2018) 1.5 million (2018) chronic cough , fever , cough with bloody mucus , weight loss death TB-HIV coinfection ‡ 1999 HIV + Mycobacterium tuberculosis sexual contact + airborne disease Africa 1.2 million (2015) 251,000 (2018) Sexually transmitted infections (notably syphilis , gonorrhoea , chlamydia , trichomoniasis , hepatitis B , HSV , HIV , and HPV ) 2000 bacteria, parasite, viruses sexual contact worldwide various various † Although leprosy and tuberculosis are not exclusively tropical diseases, their high incidence in the tropics justifies their inclusion. ‡ People living with HIV are 19 (15-22) times more likely to develop active TB disease than people without HIV.
-
Gastric Antral Vascular Ectasia
Wikipedia
. ^ a b c d e f g h i j k l m n o p q r s Tuveri, Massimiliano; Borsezio, Valentina; Gabbas, Antonio; Mura, Guendalina (2007). ... PMID 21160630 . ^ a b El-Gendy, Hala; Shohdy, Kyrillus S.; Maghraby, Gehad G.; Abadeer, Kerolos; Mahmoud, Moustafa (2017-02-01). ... PMID 10205216 . ^ Krstić, M; Alempijević, T; Andrejević, S; Zlatanović, M; Damjanov, N; Ivanović, B; Jovanović, I; Tarabar, D; Milosavljević, T (2010). ... PMC 7045742 . PMID 17054131 . ^ a b c Saab S, Nieto JM, Lewis SK, Runyon BA (2006). ... PMID 14585429 . ^ Wells, C; Harrison, M; Gurudu, S; Crowell, M; Byrne, T; Depetris, G; Sharma, V (2008).

-
Disseminated Intravascular Coagulation
Wikipedia
. ^ a b Robbins, Stanley L.; Cotran, Ramzi S.; Kumar, Vinay; Collins, Tucker (1999). ... PMID 17108099 . ^ Grewal, PK; Uchiyama, S; Ditto, D; Varki, N; Le, DT; Nizet, V; Marth, JD (June 2008). ... Thrombosis and Haemostasis . 86 (5): 1327–30. doi : 10.1055/s-0037-1616068 . PMID 11816725 . S2CID 39696424 . ^ Gando, S (2012). ... Lancet . 350 (9091): 1590–1593. doi : 10.1016/s0140-6736(97)06356-3 . PMID 9393338 . ^ Gando, S (1999). "Disseminated intravascular coagulation and sustained systemic inflammatory response syndrome predict organ dysfunctions after trauma: application of clinical decision analysis" . ... PMC 1191617 . PMID 9923809 . ^ Sallah, S (2001). "Disseminated intravascular coagulation in solid tumors: clinical and pathologic study".F3, F2, THBD, PROC, SERPINC1, OXT, F7, SERPINE1, IL6, TFPI, PLAT, FGA, PROS1, SERPIND1, NUMA1, NPM1, CFI, BCOR, NLRC4, CFH, NABP1, CD46, STAT5B, PML, TBL1XR1, ZBTB16, STAT3, FIP1L1, PRKAR1A, HELLPAR, RARA, IRF2BP2, HMGB1, PC, TNF, F10, MBL2, MPO, COX8A, BTBD8, OTOR, SLC25A10, ALB, TLR4, ZFP36, NCAM1, PLG, ADAMTS13, F5, ANGPT2, CD34, COLEC11, EGF, IL10, SH3BP4, SLC17A5, FCRL6, RN7SL263P, TEK, CABIN1, PGR-AS1, C20orf181, MIR122, ASPG, VEGFA, VTN, TAT, VWF, IL4I1, PROCR, RNF34, CLDN10, ARTN, DNAI2, RHOF, MBL3P, AGRP, TAL1, STIL, HGF, GPT, F13A1, F2R, EPHB2, EGFR, MAPK14, CRP, CPB2, COL4A2, CEACAM8, CD14, VPS51, BRAF, APOH, ANXA5, ALK, HLA-DOA, HMOX1, IFNG, PRH2, PMEL, SELL, SDC1, PTAFR, MASP1, MAPK8, MAPK1, PRH1, IL1B, PLD1, NRAS, MUC1, MMP10, ATXN3, MB, IL7, LINC02605

-
Antiphospholipid Syndrome
Wikipedia
Lupus anticoagulant (LAC) antibodies bind to prothrombin , thus increasing its cleavage to thrombin , its active form. [ citation needed ] In APS there are also antibodies binding to protein S , which is a co-factor of protein C. Thus, anti-protein S antibodies decrease protein C efficiency. [8] Annexin A5 forms a shield around negatively charged phospholipid molecules, thus reducing their availability for coagulation. ... Res . 67 (4): 355–65. doi : 10.1016/0049-3848(92)90266-d . PMID 1329261 . ^ a b Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, DE Groot PG, Koike T, Meroni PL, Reber G, Shoenfeld Y, Tincani A, Vlachoyiannopoulos PG, Krilis SA (February 2006). ... Retrieved 2020-08-31 . ^ de Jong PG, Goddijn M, Middeldorp S (2013). "Antithrombotic therapy for pregnancy loss" . ... PMID 6414579 . ^ Erkan D, Derksen R, Levy R, Machin S, Ortel T, Pierangeli S, Roubey R, Lockshin M (2011).APOH, PPARG, FRMD4A, TSHR, F5, F3, SYCP2L, F2, PTPRO, GPI, ANXA5, TLR4, SH2B2, KLK3, ANXA2, TNF, AGER, CPB2, MTOR, MTHFR, F10, PLG, HT, TFPI, PLAT, SELPLG, ACR, SERPINE1, MOK, LRP8, VWF, HMGB1, VIM, SELP, RAB4A, CCL2, CXCL12, ATXN2, RO60, S100A10, TRIM21, ABCA1, SSB, THBD, TNFRSF1B, NR1I2, ADIPOQ, SH2B3, PROCR, ADAMTS13, TREX1, PTPN22, FOXP3, SLC52A1, IL21, ANXA8, ANXA8L1, PROS1, NOS3, PON1, PLSCR1, HLA-DPB1, GP1BA, GCY, FGA, FCGR2A, F2RL1, EMD, EDN1, DECR1, CRP, CD36, CD1D, CALR, B2M, SERPINC1, AQP4, AMH, HLA-DRB1, HRES1, IDS, MBL2, PF4, PC, SERPINB2, TNFRSF11B, MYD88, MSN, MPL, LPA, IFNG, LGALS9, LCT, CXCL10, CXCL8, IL1B, IGFBP1, IGF1, C20orf181

-
Latent Autoimmune Diabetes In Adults
Wikipedia
S2CID 2179462 . ^ a b c d e f g h Carlsson S. (2019). Environmental (Lifestyle) Risk Factors for LADA. ... P., Tuomi, T., Åsvold, B. O., & Carlsson, S. (2019). Interaction Between Overweight and Genotypes of HLA, TCF7L2, and FTO in Relation to the Risk of Latent Autoimmune Diabetes in Adults and Type 2 Diabetes. ... -O.; Dorkhan, M.; Groop, L.; Martinell, M.; Tuomi, T.; Wolk, A.; Carlsson, S. (October 2014). "Fatty fish consumption and risk of latent autoimmune diabetes in adults" . ... PMID 8425674 . ^ Hagopian, W A; Karlsen, A E; Gottsäter, A; Landin-olsson, M; Grubin, C E; Sundkvist, G; Petersen, J S; Boel, E; Dyrberg, T; Lernmark, A (January 1993). ... Sources [ edit ] Eisenbarth, George S., ed. (2010). Immunoendocrinology: Scientific and Clinical Aspects .
-
Anaplastic Large-Cell Lymphoma
Wikipedia
. ^ Kempf W, Pfaltz K, Vermeer MH, Cozzio A, Ortiz-Romero PL, Bagot M, Olsen E, Kim YH, Dummer R, Pimpinelli N, Whittaker S, Hodak E, Cerroni L, Berti E, Horwitz S, Prince HM, Guitart J, Estrach T, Sanches JA, Duvic M, Ranki A, Dreno B, Ostheeren-Michaelis S, Knobler R, Wood G, Willemze R. ... Retrieved 26 November 2018 . ^ Miranda RN, Aladily TN, Prince HM, Kanagal-Shamanna R, de Jong D, Fayad LE, Amin MB, Haideri N, Bhagat G, Brooks GS, Shifrin DA, O'Malley DP, Cheah CY, Bacchi CE, Gualco G, Li S, Keech JA Jr, Hochberg EP, Carty MJ, Hanson SE, Mustafa E, Sanchez S, Manning JT Jr, Xu-Monette ZY, Miranda AR, Fox P, Bassett RL, Castillo JJ, Beltran BE, de Boer JP, Chakhachiro Z, Ye D, Clark D, Young KH, Medeiros LJ. ... S2CID 19099328 . ^ a b Miranda RN, Aladily TN, Prince HM, Kanagal-Shamanna R, de Jong D, Fayad LE, Amin MB, Haideri N, Bhagat G, Brooks GS, Shifrin DA, O'Malley DP, Cheah CY, Bacchi CE, Gualco G, Li S, Keech JA Jr, Hochberg EP, Carty MJ, Hanson SE, Mustafa E, Sanchez S, Manning JT Jr, Xu-Monette ZY, Miranda AR, Fox P, Bassett RL, Castillo JJ, Beltran BE, de Boer JP, Chakhachiro Z, Ye D, Clark D, Young KH, Medeiros LJ. ... Invest . 88 (1): 48–57. doi : 10.1038/labinvest.3700696 . PMID 17965727 . ^ Park SJ, Kim S, Lee DH, et al. (August 2008). "Primary systemic anaplastic large cell lymphoma in Korean adults: 11 years' experience at Asan Medical Center" .ALK, STAT3, HSP90AA1, TWIST1, ACVRL1, NPM1, TP53, SLPI, TNFRSF8, IRF4, MYC, JUNB, TNF, JUN, CD274, CDKN1B, BCL2, DUSP22, COIL, IL1R1, WDR48, CDKN2A, NOS1, JUND, RTEL1, FOS, FOSB, NOS2, PDLIM7, NCAM1, BCL3, CEBPB, IGH, ATIC, KIT, TPM3, MCL1, PDGFRB, LGALS1, BATF3, NFKB1, MSN, RPE65, FAS, CLU, MAPK3, EZH2, IFNG, NTRK1, ANPEP, JAK3, CKAP4, AKT1, UVRAG, CIB1, GZMB, IL10, TP63, IL6, NOTCH1, IL5, PWWP3A, MIR155, MUC1, INSR, ISG20, IL17A, EML4, PDGFRA, KRT20, SMUG1, TFG, HERPUD1, BCL10, NR0B2, WAS, MAGEH1, VEGFA, IL22, FOXP3, TIMP1, TIA1, TRBV20OR9-2, STAT1, PIK3CA, IL2RA, SOAT1, SNAP25, CLIP1, PTPRC, PTPN11, PTPN6, MAPK8, PART1, PRF1, PNLIP, PIK3CD, PIK3CB, SPN, PIK3CG, IL2, CDKN2B, CASP3, MS4A1, CCR3, CLTC, TNFSF8, BSG, COL15A1, ETFA, DPP4, CDK6, CDKN1A, CD96, CHP1, NAPSA, CXCL14, CAPG, BCL2L11, TRAP1, EBI3, BTK, CALM2, BATF, MRPL28, CARS1, GNLY, SUB1, PSIP1, GRAP2, CFLAR, DOK2, ZAP70, TRAF1, TRAF2, CD44, TYK2, CD40, CD28, WIPF1, YY1, CLTCL1, SOCS3, CCNT1, CASP8, SKAP1, RIPK1, IL18R1, NRP1, ZHX2, ARHGEF7, ZNF652, TRIM32, CDC42, MIR146A, PDCD1LG2, BIRC2, TP53INP1, BMS1P20, IL17F, TCHHL1, RPTN, RHOV, RGL4, HRNR, MIR150, PDLIM2, AHR, MIR29A, MIR17HG, MIR135B, MIR494, LINC01013, CERNA3, H3P9, H3P42, H3P10, CARS2, TSPYL2, SMG1, BAK1, TBC1D9, BRAF, CADM1, PRDM1, BCL6, BCL2L1, NXT1, CCND1, BLNK, BAX, ASCC1, RBAK, GDE1, BACH1, ARNT, CROT, RHOA, FASLG, PRDM8, SLC12A9, BIRC3, RNF213, CD68, TMOD1, TNFRSF1B, FOXO1, FRA7H, MYH9, FN1, NFATC1, FLT4, NFKB2, NGF, FLT3, FOXO3, FKBP4, MTAP, FGFR4, PAEP, PAX5, PCYT1A, PDCD1, PTK2B, ETS1, ABCB1, SERPINA1, MTOR, FUT1, ERBB4, HDAC1, ID2, IL9, ICAM1, HSPA1B, HSPA1A, HOXC6, HOXC5, ITK, HDAC2, GTF2H4, CD99, GRB2, CXCR3, LDLR, GLI1, TACSTD2, MCC, FUT4, CD46, MDK, SERPINB9, EPHB2, TNFRSF1A, CCL22, RPS6KB1, MAPK14, S100A1, S100A10, S100B, SCT, CCL5, CCL11, CCL17, SHH, RPL18A, ST6GAL1, MAP3K8, CCR8, SQLE, CD52, STAT5B, TGFB1, CDK9, IGF1, RPS6, CSF1R, ADARB1, PTBP1, PKM, EGFR, E2F2, MAPK1, DUSP5, MAP2K7, PRSS1, PRSS2, PRSS3, PTEN, RORC, PTN, CYP51A1, CTNNB1, CSF2, RAF1, RAG1, RAG2, RB1, RENBP, H3P40

-
Fecal–oral Route
Wikipedia
. ^ Kal, K and Chambers, R (2008) Handbook on Community-led Total Sanitation Archived 2015-04-10 at the Wayback Machine , Plan UK Accessed 2015-02-26 ^ Hale TL, Keusch GT (1996). Baron S, et al. (eds.). Shigella in: Baron's Medical Microbiology (4th ed.). ... (via NCBI Bookshelf) . ^ Giannella RA (1996). Baron S; et al. (eds.). Salmonella :Epidemiology in: Baron's Medical Microbiology (4th ed.). ... (via NCBI Bookshelf) . ^ Finkelstein RA (1996). Baron S; et al. (eds.). Cholera, Vibrio cholerae O1 and O139, and Other Pathogenic Vibrio s in: Baron's Medical Microbiology (4th ed.). ... Retrieved 2016-04-18 . ^ Zuckerman AJ (1996). Baron S; et al. (eds.). Hepatitis Viruses in: Baron's Medical Microbiology (4th ed.). ... Retrieved 2020-02-04 . ^ Meyer EA (1996). Baron S; et al. (eds.). Other Intestinal Protozoa and Trichomonas Vaginalis in: Baron's Medical Microbiology (4th ed.).
-
Innate Resistance To Hiv
Wikipedia
This is attributed to individuals being heterozygous for the mutation, which prevents the delta mutation from effectively prohibiting HIV from entering immune cells. [23] See also [ edit ] Viruses portal Discovery and development of CCR5 receptor antagonists Entry inhibitor HIV tropism Timothy Ray Brown Stephen Crohn References [ edit ] ^ Scutti S (2014-11-20). "Why Some People Are Naturally Immune To HIV" . ... HIVPlusMag . Retrieved 2015-01-20 . ^ Nolen S (2007-05-27). "Staying alive: the women who are immune to Aids" . the Guardian . ... PMID 15556703 . ^ Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, Schneider T, Hofmann J, Kücherer C, Blau O, Blau IW, Hofmann WK, Thiel E (February 2009). ... Blood . 117 (10): 2791–99. doi : 10.1182/blood-2010-09-309591 . PMID 21148083 . ^ Zhen A, Kitchen S (December 2013). "Stem-cell-based gene therapy for HIV infection" . ... PMC 4084652 . PMID 24597865 . ^ Rodríguez-Mora S, De Wit F, García-Perez J, Bermejo M, López-Huertas MR, Mateos E, Martí P, Rocha S, Vigón L, Christ F, Debyser Z, Jesús Vílchez J, Coiras M, Alcamí J (August 2019).
-
Pityriasis Alba
Wikipedia
.; King, Richard A.; Oetting, William S.; Ortonne, Jean-Paul (2008). The Pigmentary System: Physiology and Pathophysiology . ... PMID 2978289 . ^ Rigopoulos D, Gregoriou S, Charissi C, Kontochristopoulos G, Kalogeromitros D, Georgala S (2006). ... Journal of the European Academy of Dermatology and Venereology : JEADV . 16 (5): 463–468. doi : 10.1046/j.1468-3083.2002.00494.x . PMID 12428838 . ^ Dogra S, Kumar B (2003). "Epidemiology of skin diseases in school children: a study from northern India". ... PMID 14651562 . ^ Faye O, N'Diaye HT, Keita S, Traoré AK, Hay RJ, Mahé A (2005). ... PMID 10354028 . ^ Inanir I, Sahin MT, Gündüz K, Dinç G, Türel A, Oztürkcan S (2002). "Prevalence of skin conditions in primary school children in Turkey: differences based on socioeconomic factors".
-
Nail Clubbing
Wikipedia
Cite journal requires |journal= ( help ) ^ a b c d e Burcovschii, S; Aboeed, A (January 2019). "Nail Clubbing". ... Archived from the original (PDF) on 2003-11-01. ^ Epstein O, Dick R, Sherlock S (1981). "Prospective study of periostitis and finger clubbing in primary biliary cirrhosis and other forms of chronic liver disease" . ... PMID 11466101 . ^ Shah K, Ferrara TM, Jan A, Umair M, Irfanullah, Khan S, Ahmad W, Spritz RA (August 2017). ... Dermatol . 177 (2): 546–548. doi : 10.1111/bjd.15094 . PMID 27681482 . ^ a b Uppal S, Diggle CP, Carr IM, et al. (June 2008). ... PMID 2891996 . ^ Schamroth L (February 1976). "Personal experience". S. Afr. Med. J . 50 (9): 297–300. PMID 1265563 .

-
Cryptogenic Organizing Pneumonia
Wikipedia
References [ edit ] ^ " bronchiolitis obliterans with organizing pneumonia " at Dorland's Medical Dictionary ^ White, Eric J. Stern, Charles S. (1999). Chest radiology companion . ... Retrieved 23 November 2012 . ^ Levy, Barry S.; Wegman, David H.; Baron, Sherry L.; Sokas, Rosemary K., eds. (2011). ... Retrieved June 23, 2015 . ^ Mukhopadhyay, Sanjay; Mehrad, Mitra; Dammert, Pedro; Arrossi, Andrea V; Sarda, Rakesh; Brenner, David S; Maldonado, Fabien; Choi, Humberto; Ghobrial, Michael (2019). ... ISSN 0002-9173 . PMID 31621873 . ^ Nogi, S; Nakayama, H; Tajima, Y; Okubo, M; Mikami, R; Sugahara, S; Akata, S; Tokuuye, K (2014).UNC119, CD68, CRP, NR3C1, HDAC2, CXCL8, TNF, TNFRSF1A, TNFRSF1B, CD163, ING4, COP1, HT, TOMM5, WG, SFTPA1

-
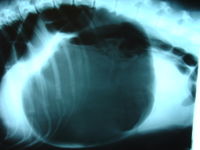
Gastric Dilatation Volvulus
Wikipedia
. ^ a b Beck J, Staatz A, Pelsue D, Kudnig S, MacPhail C, Seim H, Monnet E (2006). ... Retrieved 2007-04-17 . ^ a b Parton A, Volk S, Weisse C (2006). "Gastric ulceration subsequent to partial invagination of the stomach in a dog with gastric dilatation-volvulus". ... Assoc . 216 (1): 40–5. doi : 10.2460/javma.2000.216.40 . PMID 10638316 . ^ Braun L, Lester S, Kuzma A, Hosie S (1996). "Gastric dilatation-volvulus in the dog with histological evidence of preexisting inflammatory bowel disease: a retrospective study of 23 cases". ... Retrieved 2007-04-17 . ^ a b Rawlings C, Mahaffey M, Bement S, Canalis C (2002). "Prospective evaluation of laparoscopic-assisted gastropexy in dogs susceptible to gastric dilatation". ... Retrieved 15 October 2017 . ^ a b Mackenzie G, Barnhart M, Kennedy S, DeHoff W, Schertel E (March–April 2010).
-
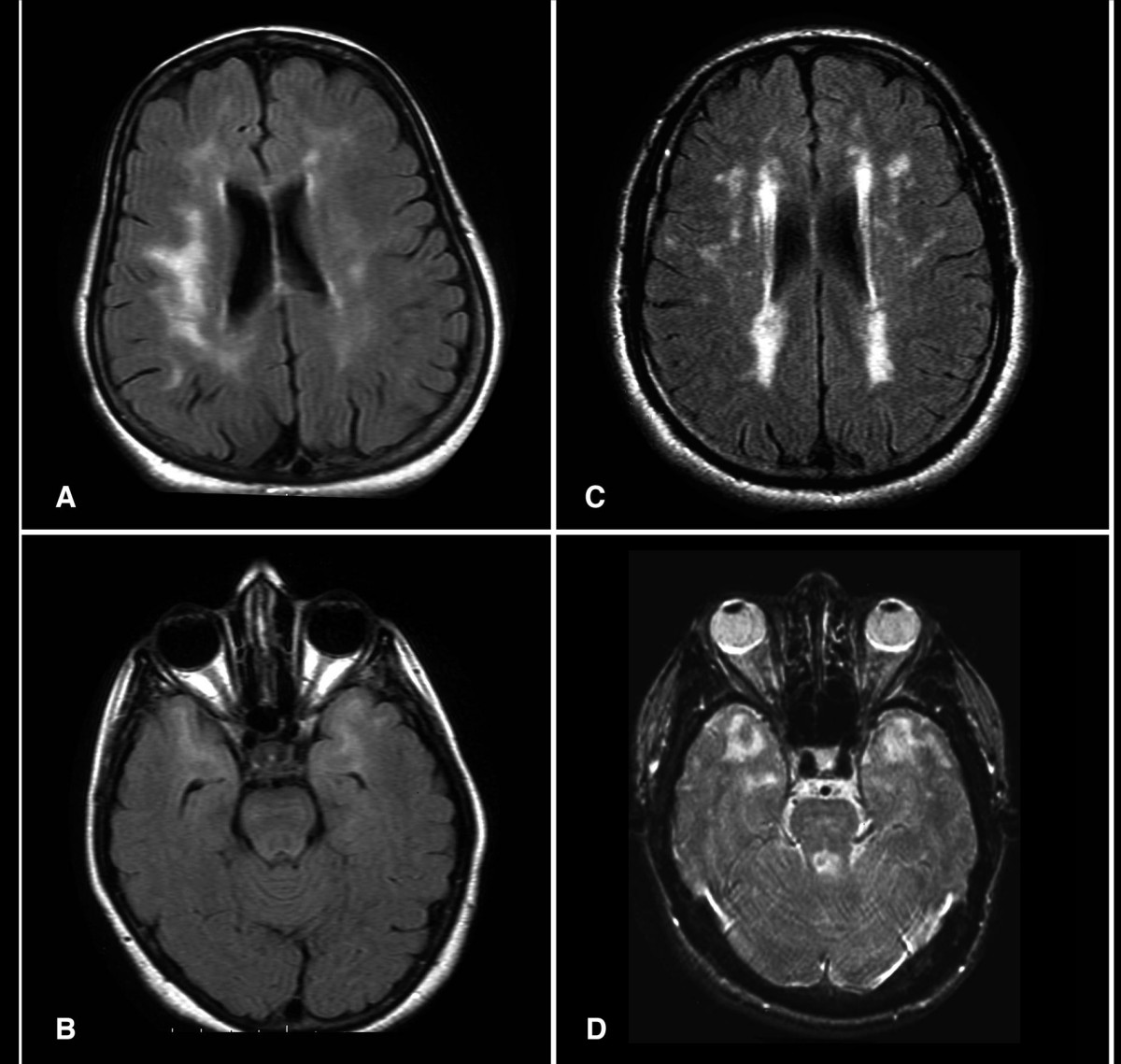
Cadasil
Wikipedia
The disease progresses to subcortical dementia associated with pseudobulbar palsy and urinary incontinence . [ citation needed ] Ischemic strokes are the most frequent presentation of CADASIL, with approximately 85% of symptomatic individuals developing transient ischemic attacks or stroke(s). The mean age of onset of ischemic episodes is approximately 46 years (range 30–70). ... As with other individuals, people with CADASIL should be encouraged to quit smoking. [20] In one small study, around 1/3 of patients with CADASIL were found to have cerebral microhemorrhages (tiny areas of old blood) on MRI . [15] L-arginine, a naturally occurring amino acid, has been proposed as a potential therapy for CADASIL, [21] but as of 2017 there are no clinical studies supporting its use. [18] Donepezil , normally used for Alzheimer's Disease, was not shown not to improve executive functioning in CADASIL patients. [22] In popular culture [ edit ] John Ruskin has been suggested to have suffered from CADASIL. [23] Ruskin reported in his diaries having visual disturbances consistent with the disease, and it has also been suggested that it might have been a factor in causing him to describe James Whistler 's Nocturne in Black and Gold – The Falling Rocket as "ask[ing] two hundred guineas for throwing a pot of paint in the public's face". ... PMID 25824603 . ^ Joutel A, Andreux F, Gaulis S, et al. (March 2000). "The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients" . ... PMID 7676806 . ^ Vlachakis D, Champeris Tsaniras S, Ioannidou K, Papageorgiou L, Baumann M, Kossida S (October 2014). ... PMID 20464302 . ^ a b Lesnik Oberstein, S. A.; van den Boom, R.; van Buchem, M.NOTCH3, HTRA1, EGF, COL4A1, COL18A1, NOTCH1, JAG1, APOE, APP, LPA, TREX1, GFAP, PLXNA2, CTSA, PROC, ACTB, SOD1, TGFB1, NOTCH2, LAP, MRS2, RNF213, NOX5, THBD, KDR, NEFL, ACTG2, RBPJ, FN1, FBN1, ELN, DVL1, DCN, CSF3, CSF1R, CLU, CACNA1A, BGN, APCS, LINC01191
-
Cavitary Myiasis
Orphanet
Clinical presentation is variable depending on the affected site(s) and degree of infestation and include foreign-body sensation (with or without movement sensation), hemorrhage, pain, edema, sensory loss, malodor, and pruritus, among others.
-
Leukoplakia
GARD
Removing the source of irritation may cause the condition to go away, but surgery to remove the sore(s) may be necessary in some cases.CTTN, TP53, MDM2, GSTT1, GSTM1, XRCC1, EGFR, BCL2, GSTM3, ERBB2, CYP1A1, DEFB4A, CDKN1A, CCND1, TGFB1, POLRMT, CIB2, PAX5, ABCB6, DLEC1, TP63, PABPN1, MFAP5, PRKDC, TFAM, PTGS2, RPE65, UVRAG, TP73, S100A4, PDPN, TLR4, TGM3, NAT1, RRAS2, CKAP4, POLG2, SIK1B, DEFB4B, MIR31, MIR29A, MIR21, GSTK1, LTO1, SIK1, SLCO6A1, DERL3, ARHGAP24, MTUS1, SLC12A9, EIF5A2, PIWIL2, TLR9, CD274, HPGDS, NOTCH1, NRAS, MMP13, NOS2, NELL2, FHIT, FGFR2, FGFR3, FGF2, ERCC2, ECT2, DNMT3B, DES, DEFB1, CYP2E1, COL4A1, CDKN2A, CDKN1C, CASP10, CASP7, CASP1, BSG, BRCA1, BAG1, GFAP, GSTP1, HOXC9, MCC, COX2, MSH2, MRE11, ATM, MMP9, MMP2, MLH1, MCL1, MAL, HOXC13, LCN2, LAMC2, KRT19, KRT13, KRT4, KRAS, IL10, IL6, MTCO2P12

-
Griscelli Syndrome
Wikipedia
. ^ Rath, Sanjeev; Jain, Vivek; Marwaha, R. K.; Trehan, Amita; Rajesh, L. S.; Kumar, Vijay (February 2004). "Griscelli syndrome". ... External links [ edit ] Classification D ICD - 10 : E70.3 OMIM : 214450 607624 609227 DiseasesDB : 32776 External resources eMedicine : derm/926 Orphanet : 381 Griscelli syndrome type 1 at NIH 's Office of Rare Diseases OMIM: 214450 Griscelli syndrome type 2 at NIH 's Office of Rare Diseases OMIM: 607624 Griscelli syndrome type 3 at NIH 's Office of Rare Diseases OMIM: 609227 v t e Pigmentation disorders / Dyschromia Hypo- / leucism Loss of melanocytes Vitiligo Quadrichrome vitiligo Vitiligo ponctué Syndromic Alezzandrini syndrome Vogt–Koyanagi–Harada syndrome Melanocyte development Piebaldism Waardenburg syndrome Tietz syndrome Loss of melanin / amelanism Albinism Oculocutaneous albinism Ocular albinism Melanosome transfer Hermansky–Pudlak syndrome Chédiak–Higashi syndrome Griscelli syndrome Elejalde syndrome Griscelli syndrome type 2 Griscelli syndrome type 3 Other Cross syndrome ABCD syndrome Albinism–deafness syndrome Idiopathic guttate hypomelanosis Phylloid hypomelanosis Progressive macular hypomelanosis Leukoderma w/o hypomelanosis Vasospastic macule Woronoff's ring Nevus anemicus Ungrouped Nevus depigmentosus Postinflammatory hypopigmentation Pityriasis alba Vagabond's leukomelanoderma Yemenite deaf-blind hypopigmentation syndrome Wende–Bauckus syndrome Hyper- Melanin / Melanosis / Melanism Reticulated Dermatopathia pigmentosa reticularis Pigmentatio reticularis faciei et colli Reticulate acropigmentation of Kitamura Reticular pigmented anomaly of the flexures Naegeli–Franceschetti–Jadassohn syndrome Dyskeratosis congenita X-linked reticulate pigmentary disorder Galli–Galli disease Revesz syndrome Diffuse/ circumscribed Lentigo / Lentiginosis : Lentigo simplex Liver spot Centrofacial lentiginosis Generalized lentiginosis Inherited patterned lentiginosis in black persons Ink spot lentigo Lentigo maligna Mucosal lentigines Partial unilateral lentiginosis PUVA lentigines Melasma Erythema dyschromicum perstans Lichen planus pigmentosus Café au lait spot Poikiloderma ( Poikiloderma of Civatte Poikiloderma vasculare atrophicans ) Riehl melanosis Linear Incontinentia pigmenti Scratch dermatitis Shiitake mushroom dermatitis Other/ ungrouped Acanthosis nigricans Freckle Familial progressive hyperpigmentation Pallister–Killian syndrome Periorbital hyperpigmentation Photoleukomelanodermatitis of Kobori Postinflammatory hyperpigmentation Transient neonatal pustular melanosis Other pigments Iron Hemochromatosis Iron metallic discoloration Pigmented purpuric dermatosis Schamberg disease Majocchi's disease Gougerot–Blum syndrome Doucas and Kapetanakis pigmented purpura / Eczematid-like purpura of Doucas and Kapetanakis Lichen aureus Angioma serpiginosum Hemosiderin hyperpigmentation Other metals Argyria Chrysiasis Arsenic poisoning Lead poisoning Titanium metallic discoloration Other Carotenosis Tar melanosis Dyschromia Dyschromatosis symmetrica hereditaria Dyschromatosis universalis hereditaria See also Skin color Skin whitening Tanning Sunless Tattoo removal Depigmentation v t e Inherited disorders of trafficking / vesicular transport proteins Vesicle formation Lysosome / Melanosome : HPS1 – HPS7 Hermansky–Pudlak syndrome LYST Chédiak–Higashi syndrome COPII : SEC23A Cranio-lenticulo-sutural dysplasia COG7 CDOG IIE APC: AP1S2 X-linked intellectual disability AP3B1 Hermansky–Pudlak syndrome 2 AP4M1 CPSQ3 Rab ARL6 BBS3 RAB27A Griscelli syndrome 2 CHM Choroideremia MLPH Griscelli syndrome 3 Cytoskeleton Myosin : MYO5A Griscelli syndrome 1 Microtubule : SPG4 Hereditary spastic paraplegia 4 Kinesin : KIF5A Hereditary spastic paraplegia 10 Spectrin : SPTBN2 Spinocerebellar ataxia 5 Vesicle fusion Synaptic vesicle : SNAP29 CEDNIK syndrome STX11 Hemophagocytic lymphohistiocytosis 4 Caveolae : CAV1 Congenital generalized lipodystrophy 3 CAV3 Limb-girdle muscular dystrophy 2B , Long QT syndrome 9 Vacuolar protein sorting : VPS33B ARC syndrome VPS13B Cohen syndrome DYSF Distal muscular dystrophy See also vesicular transport proteins

-
Canine Degenerative Myelopathy
Wikipedia
Retrieved 2008-07-25 . ^ Awano, T.; Johnson, G. S.; Wade, C. M.; Katz, M. L.; Johnson, G. ... F.; Perloski, M.; Biagi, T.; Baranowska, I.; Long, S.; March, P. A.; Olby, N. J.; Shelton, G. D.; Khan, S.; O'Brien, D. P.; Lindblad-Toh, K.; Coates, J. ... Saunders Company, 2004, pp 147-9. ^ Kathmann I, I; Cizinauskas S; Doherr MG; Steffen F; Jaggy A. (July–August 2006).

-
Long Face Syndrome
Wikipedia
Therefore, individual variations in response should be expected from the alteration of a long face syndrome patient's breathing mode." [6] References [ edit ] ^ Carano, Aldo; Siciliani, Giuseppe; Bowman, S. Jay (September 2005). "Treatment of skeletal open bite with a device for rapid molar intrusion: a preliminary report" . ... CS1 maint: DOI inactive as of January 2021 ( link ) ^ Schendel, S. A.; Eisenfeld, J.; Bell, W. H.; Epker, B. ... PMID 1067758 . ^ Taub, Daniel I.; Jacobs, Jordan M. S.; Jacobs, Jonathan S. (2013). "Chapter 16: Anthropometry, cephalometry, and orthognathic surgery" .












