-
Congenital Mirror Movement Disorder
Wikipedia
Retrieved 2017-12-06 . ^ a b c d e f g h i j k l m n Méneret, Aurélie; Trouillard, Oriane; Depienne, Christel; Roze, Emmanuel (1993). ... Seattle (WA): University of Washington, Seattle. PMID 25763452 . ^ a b c d e f g h i Galléa, Cécile; Popa, Traian; Billot, Ségolène; Méneret, Aurélie; Depienne, Christel; Roze, Emmanuel (November 2011). ... PMID 21633904 . ^ a b c d e Schott, G D; Wyke, M A (July 1981). "Congenital mirror movements" . ... Retrieved 2017-12-06 . ^ a b c d e f g h i Méneret, Aurélie; Depienne, Christel; Riant, Florence; Trouillard, Oriane; Bouteiller, Delphine; Cincotta, Massimo; Bitoun, Pierre; Wickert, Julia; Lagroua, Isabelle (2014-06-03). ... ISSN 1388-2457 . PMID 12948795 . ^ Leinsinger, G L; Heiss, D T; Jassoy, A G; Pfluger, T; Hahn, K; Danek, A (1997-05-01).CDH2, DCC, RAD51, ANOS1, DNAL4, NTN1, FGFR1, FGF8, FEZF1, CCDC141, PROKR2, KISS1R, SPRY4, PROK2, KNL1, WDR11, CHD7, IL17RD, NSMF, FLRT3, CEP152, LINC01917, SEMA3A, CREBBP, HS6ST1, FGF17, HESX1, TACR3, SOX10, DUSP6, EP300, RAD51B, GDF6, MEF2C, SMN2, SCN8A, POMK, SMN1, NTNG1

-
Kounis Syndrome
Wikipedia
Three variants of Kounis syndrome were found and a study concluded that type 1 variant was most commonly seen followed by type 2 and 3 respectively. [7] References [ edit ] ^ a b c d e f g Kounis NG (1 October 2016). "Kounis syndrome: an update on epidemiology, pathogenesis, diagnosis and therapeutic management". ... PMID 23232800 . S2CID 32619275 . ^ a b c d e f g h i j k l m Giovannini M, Koniari I, Mori F, Ricci S, Simone LD, Favilli S, Trapani S, Indolfi G, Kounis N, Novembre E (2020-05-28). ... PMID 32547613 . ^ a b c d Kounis NG, Mazarakis A, Tsigkas G, Giannopoulos S, Goudevenos J (November 2011). ... Journal für Kardiologie - Austrian Journal of Cardiology . 19 : 118–122. ^ Kounis NG, Koniari I, Velissaris D, Tzanis G, Hahalis G (2019-07-11). "Kounis Syndrome—not a Single-organ Arterial Disorder but a Multisystem and Multidisciplinary Disease" .
-
Innate Resistance To Hiv
Wikipedia
FEMS Microbiology Letters . 241 (1): 1–12. doi : 10.1016/j.femsle.2004.09.040 . PMID 15556703 . ^ Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, Schneider T, Hofmann J, Kücherer C, Blau O, Blau IW, Hofmann WK, Thiel E (February 2009). ... PMID 19213682 . ^ Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, Schneider T (March 2011). ... PMID 24597871 . ^ Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL, June CH (March 2014).
-
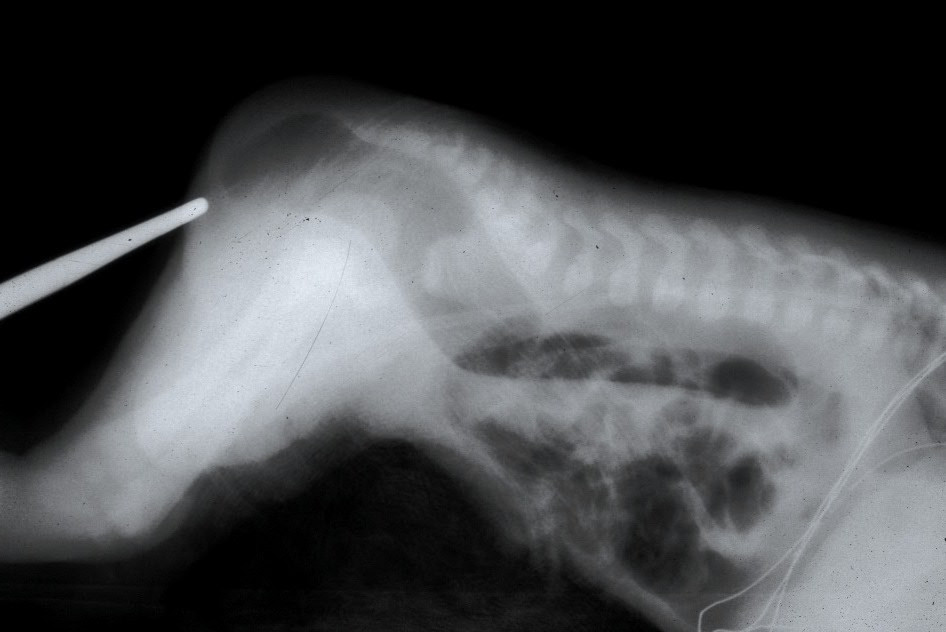
Currarino Syndrome
Wikipedia
PMID 16639328 . ^ Samuel, M.; Hosie, G.; Holmes, K. (December 2000). "Currarino triad—Diagnostic dilemma and a combined surgical approach". ... PMID 4418917 . ^ Belloni, E; Martucciello, G; Verderio, D; Ponti, E; Seri, M; Jasonni, V; Torre, M; Ferrari, M; Tsui, LC; Scherer, SW (January 2000). ... PMID 16254195 . ^ Emans PJ, van Aalst J, van Heurn EL, Marcelis C, Kootstra G, Beets-Tan RG, Vles JS, Beuls EA (2006). ... CS1 maint: multiple names: authors list ( link ) ^ Samuel M, Hosie G, Holmes K (Dec 2000). "Currarino triad--diagnostic dilemma and a combined surgical approach". ... CS1 maint: multiple names: authors list ( link ) ^ Chakhalian D, Gunasekaran A, Gandhi G, Bradley L, Mizell J, Kazemi N (2017).
-
Interstitial Granulomatous Drug Reaction
Wikipedia
Interstitial granulomatous drug reaction Specialty Dermatology Interstitial granulomatous drug reaction is an uncommon, yet under-recognized, pattern of adverse reactions to medication . [1] : 707 See also [ edit ] Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Cutaneous Pasteurella Hemolytica Infection
Wikipedia
Cutaneous Pasteurella hemolytica infection Specialty Infectious disease Cutaneous Pasteurella hemolytica infections may occur in patients with skin injury and exposure Pasteurella hemolytica . [1] : 281 See also [ edit ] Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Ked Itch
Wikipedia
Ked itch Specialty Dermatology Ked itch is a cutaneous condition caused by sheep ked ( Melophagus ovinus ) which feed by thrusting their sharp mouth parts into the skin and sucking blood. [1] : 448 See also [ edit ] Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Xanthomatous Biliary Cirrhosis
Wikipedia
Xanthomatous biliary cirrhosis Other names Obstructive liver disease , Xanthomatous biliary cirrhosis , is a condition in which there is hyperlipoproteinemia due to liver disease resulting in plane xanthomas . [1] : 534 See also [ edit ] Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: Clinical Dermatology .
-
Mucosal Lentigines
Wikipedia
Mucosal lentigines Other names Labial, penile, and vulvar melanosis , and Melanotic macules Specialty Dermatology Mucosal lentigines is a cutaneous condition characterized by light brown macules on mucosal surfaces . [1] : 686 See also [ edit ] Lentigo Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Sowda
Wikipedia
Sowda Specialty Dermatology Sowda is a cutaneous condition, a localized type of onchocerciasis . [1] : 440 This is mostly seen in patients with the disease from Yemen, Saudi Arabia, East and West Africa See also [ edit ] Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Candidal Intertrigo
Wikipedia
Candidal intertrigo Intertrigo interdigital Specialty Infectious disease Candidal intertrigo is an infection of the skin by Candida albicans , more specifically located between intertriginous folds of adjacent skin. [1] : 309 See also [ edit ] Candidiasis Intertrigo Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .

-
Postencephalitic Trophic Ulcer
Wikipedia
Postencephalitic trophic ulcer is an ulceration of the nose similar to trigeminal trophic lesions , and has been reported following epidemic encephalitis and herpes zoster of the trigeminal nerve . [1] : 65 See also [ edit ] List of cutaneous conditions References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Metastatic Calcinosis Cutis
Wikipedia
Metastatic calcinosis cutis Specialty Dermatology Metastatic calcinosis cutis is a cutaneous condition characterized by calcification of the skin resulting from the deposition of calcium and phosphorus , and associated with an internal malignancy . [1] : 528 See also [ edit ] Calcinosis cutis Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Disseminated Coccidioidomycosis
Wikipedia
Disseminated coccidioidomycosis Other names Coccidioidal granuloma Specialty Infectious disease Disseminated coccidioidomycosis is a systemic infection with Coccidioides immitis , in which 15-20% of people develop skin lesions . [1] : 315 See also [ edit ] Coccidioidomycosis List of cutaneous conditions References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: Clinical Dermatology .
-
Medication-Induced Hyperlipoproteinemia
Wikipedia
Medication-induced hyperlipoproteinemia Specialty Dermatology Medication-induced hyperlipoproteinemia is a condition that results from the decreasing of lipoprotein lipase activity resulting in eruptive xanthomas . [1] : 535 See also [ edit ] Normolipoproteinemic xanthomatosis Cerebrotendinous xanthomatosis Skin lesion References [ edit ] ^ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology .
-
Pentosuria
Orphanet
Pentosuria is an inborn error of metabolism which is characterized by the excretion of 1 to 4 g of the pentose L-xylulose in the urine per day.

-
Hypertriglyceridemia, Familial
OMIM
The serum triglyceride level was significantly different among the genotypic groups (G/G 92.5 +/- 37.8 mg/dl, G/T 106.6 +/- 34.8 mg/dl, T/T 183.0 mg/dl, p = 0.014) in control subjects. ... Jointly, these variants explained 49% of the genetic variance in triglyceridemia; however, only the SLC25A40 variant was significantly associated with triglyceride levels (p = 0.0001). The c.374A-G transition in exon 7 of the SLC25A40 gene results in a highly disruptive tyr125-to-cys (Y125C) substitution at a highly conserved residue just outside the second helical transmembrane region of the inner mitochondrial membrane transport protein.
-
Oculomotor Apraxia
Wikipedia
. ^ a b Criscuolo, C, Chessa, L, Di Giandomenico, S, Mancini, P, Saccà F,, Grieco, G, Piane, M, Barbieri, F, De Michele, G, Banfi, S, Pierelli, F, Rizzuto, N, Santorelli, F, Gallosti, L, Filla, A, Casali, C. ... Retrieved 28 December 2019 ^ a b Klivényi, P, Nemeth, D, Sefcsik, T, Janacsek, K, Hoffmann, I, Haden, G, Londe, Z, Vecsei, L. Cognitive functions in ataxia with oculomotor apraxia type 2. Frontiers in Neuro-ophthalmology 3 (2012):125. ^ Le Ber, I, Bouslam, N, Rivaud-Péchoux, S, Guimarães, J, Benomar, A, Chamayou, C, Goizet, C, Moreira, MC, Klur, S, Yahyaoui, M, Agid, Y, Koenig, M, Stevanin, G, Brice A, Dürr A. Brain 127 (2004):759-67. ^ Saunders-Pullman, R, Raymond, D, Stoessl,, A, Hobson, D, Nakamura, T, Pullman, S, Lefton, D, Okun, M, Uitti, R, Sachdev, R, Stanley, K, San Luciano, M, Hagenah, J, Gatti, R, Ozelius, L, Bressman, S.SETX, APTX, PNKP, AHI1, NPHP1, MRE11, XRCC1, INPP5E, PIK3R5, MKS1, ZC4H2, NGLY1, PIGV, KIAA0556, CWF19L1, RPGRIP1L, EXOSC3, TMEM216, B9D1, PGAP2, TREM2, CC2D2A, TMEM138, UFC1, SUFU, AFG3L2, DHX30, CPLANE1, PIGW, HYLS1, ARL13B, CEP120, TOE1, CEP41, PGAP3, TMEM67, PIGY, PIGO, TMEM107, KIAA1109, ARMC9, CEP290, TCTN2, CSPP1, TCTN1, TMEM231, FA2H, TMEM237, LAMA1, PSEN2, GRID2, PIBF1, PSEN1, MAPK1, PDE6D, MYO9A, MAPT, ITPR1, GBA, ATM, FOXG1, DLAT, ARL3, CRKL, TPP1, ERCC8, BRAF, PEX2, RORA, ATXN2, SORL1, SLC30A9, TOMM40, TUBB3, ABCA7, STUB1, DNM1L, KIAA0586, CEP104, PIGL, CACNA1G, SQSTM1, GPAA1, TYROBP, APP, SPR, BCR, AFP, CSTB, TPO, ALDH3A2, ADCY5
-
Congenital Stenosis Of Vena Cava
Wikipedia
. ^ Harrison, D; Sullivan, P; Christman, G; Takao, C (April 2017). "Intravascular Stent Implantation for Refractory Chylothorax Secondary to Congenital Superior Vena Cava Stenosis in an Infant" (PDF) . ... PMC 5795860 . PMID 29391034 . ^ a b c d e f g h i Halparin J, Mongale P, Newall F (April 2015). ... Department of Health and Human Services. August 2016. ^ a b c d e f g h i j Harrison DJ, Sullivan PM, Christman G, Takao C (April 2017).

-
Multifactorial Diseases
Wikipedia
Cite journal requires |journal= ( help ) ^ Carpenter, Geoffrey (December 1982). "Copeland, John G. et al. Telemundo: A Basic Reader. New York: Random House, Inc., 1980; Freeman, G. Ronald. Intercambios: An Activities Manual. New York: Random House, Inc., 1980Copeland, John G. et al. Telemundo: A Basic Reader. New York: Random House, Inc., 1980. Pp. 264.Freeman, G. Ronald. Intercambios: An Activities Manual.






