-
Bethlem Myopathy
Wikipedia
. ^ a b Jobsis GJ, Boers JM, Barth PG, de Visser M (1999). "Bethlem myopathy: a slowly-progressive congenital muscular dystrophy with contractures" . ... PMC 1736127 . PMID 16141002 . ^ a b Okada M et al (2007) Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. ... ISSN 0004-282X . ^ Norwood, Fiona L. M.; Harling, Chris; Chinnery, Patrick F.; Eagle, Michelle; Bushby, Kate; Straub, Volker (2009). ... PMID 19767415 . ^ Lampe, Anne Katrin; Flanigan, Kevin M.; Bushby, Katharine Mary; Hicks, Debbie (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E.COL6A1, COL6A3, COL6A2, COL12A1, ANO5, DMD, LMNA, CAPN3, VWF, FKRP, ADIPOQ, LAMB1, SGCG, LEP, HSPG2, DAG1, COL6A5

-
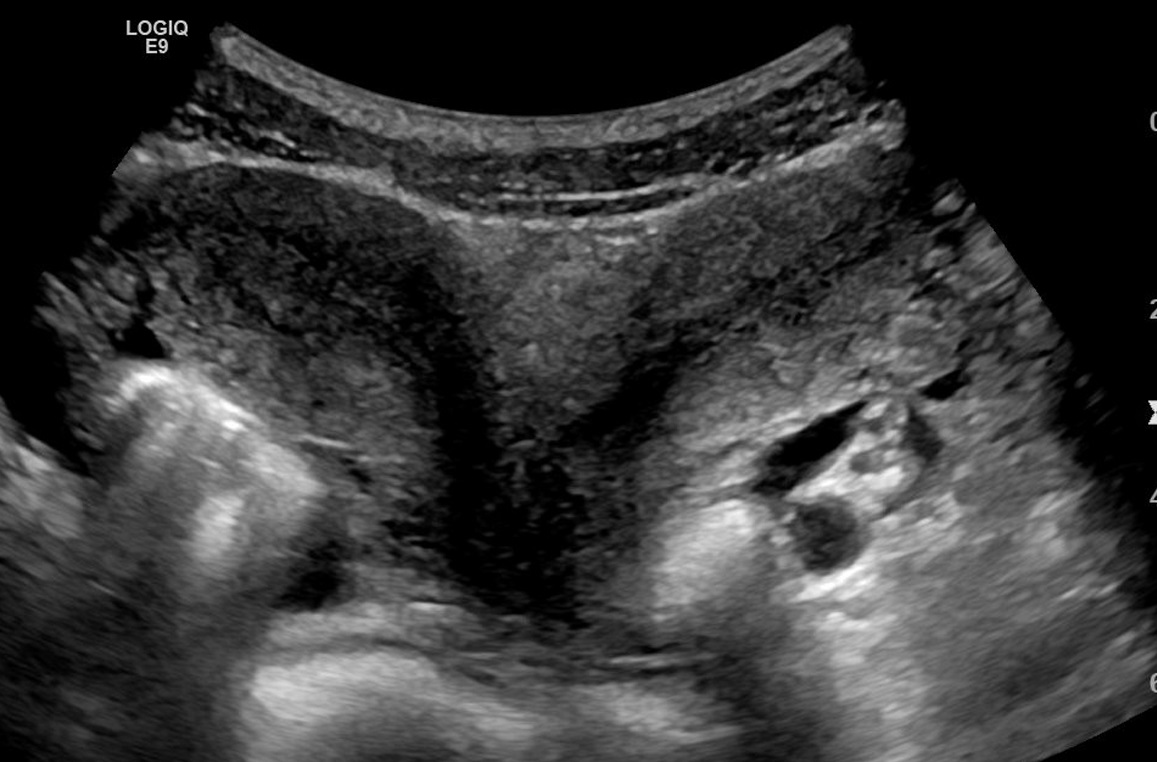
Uterus Didelphys
Wikipedia
PMID 6479426 . ^ Grimbizis, G. F.; Camus, M; Tarlatzis, BC; Bontis, JN; Devroey, P (2001). ... PMID 11284660 . ^ Madureira, A. J.; Mariz, C. M.; Bernardes, J. C.; Ramos, I. M. (2006). ... PMID 11755560 . S2CID 37650526 . ^ a b Pui, M (2004). "Imaging diagnosis of congenital uterine malformation".
-
Wolcott–rallison Syndrome
Wikipedia
. ^ a b c d e f g h i Spehar Uroić A, Mulliqi Kotori V, Rojnić Putarek N, Kušec V, Dumić M (April 2014). "Primary hypothyroidism and nipple hypoplasia in a girl with Wolcott-Rallison syndrome". ... PMID 15772126 . ^ Senée V, Vattem KM, Delépine M, Rainbow LA, Haton C, Lecoq A, Shaw NJ, Robert JJ, Rooman R, Diatloff-Zito C, Michaud JL, Bin-Abbas B, Taha D, Zabel B, Franceschini P, Topaloglu AK, Lathrop GM, Barrett TG, Nicolino M, Wek RC, Julier C (July 2004). ... S2CID 12244157 . ^ a b Julier C, Nicolino M (November 2010). "Wolcott-Rallison syndrome" .

-
Auto-Brewery Syndrome
Wikipedia
. ^ Kaji H, Asanuma Y, Yahara O, Shibue H, Hisamura M, Saito N, et al. (1984). "Intragastrointestinal alcohol fermentation syndrome: report of two cases and review of the literature". ... ISSN 1595-689X . ^ a b Hafez EM, Hamad MA, Fouad M, Abdel-Lateff A (May 2017). "Auto-brewery syndrome: Ethanol pseudo-toxicity in diabetic and hepatic patients". ... PMID 31037230 . ^ a b Saverimuttu J, Malik F, Arulthasan M, Wickremesinghe P (October 2019). ... PMC 1295472 . PMID 1744875 . ^ Simic M, Ajdukovic N, Veselinovic I, Mitrovic M, Djurendic-Brenesel M (March 2012). ... PMID 31423320 . ^ Spinucci G, Guidetti M, Lanzoni E, Pironi L (July 2006).
-
Alopecia Universalis
Wikipedia
.; Messenger, Andrew G.; Christiano, Angela M.; Sundberg, John P. (2017-03-16). ... PMID 12833016 . ^ Clynes, Raphael; Christiano, Angela M.; Vaughan, Roger; Furniss, Megan; Ulerio, Grace; Clark, Charlotte; Cerise, Jane E.; Nguyen, Nhan; Jabbari, Ali (2016-09-22). ... PMID 27699253 . ^ Morris, Gabriela M.; Nahmias, Zachary P.; Kim, Brian S. (2018-07-01). ... PMID 30023415 . ^ Navarini, Alexander A.; French, Lars E.; Trüeb, Ralph M.; Kamarachev, Jivko; Maul, Julia-Tatjana; Anzengruber, Florian (2016). ... PMID 27194979 . ^ Clynes, Raphael; Christiano, Angela M.; Mackay-Wiggan, Julian; Petukhova, Lynn; Singh, Pallavi; Rothman, Lisa; DeStefano, Gina M.; Harel, Sivan; Jong, Annemieke de (September 2014).HR, VDR, AHSG, LMNA, DSP, WDR11, PTPN22, FOXN1, XIST, TNF, LGALS8, AIRE, IL1RN, IL1B, HMGB1, HLA-DRB1, HLA-A, ACE, RBM45

-
Experimental Autoimmune Encephalomyelitis
Wikipedia
EAE was motivated by observations during the convalescence from viral diseases by Thomas M. Rivers, D. H. Sprunt and G. P. Berry in 1933. ... PMID 16315280 . S2CID 12141651 . ^ MANNIE, M., R. H. SWANBORG and J. A. STEPANIAK, 2009,"Experimental autoimmune encephalomyelitis in the rat." ... Neuro Oncol . 18 : iv25. doi : 10.1093/neuonc/now188.085 . ^ a b Höftberger R, Leisser M, Bauer J, Lassmann H (Dec 2015). "Autoimmune encephalitis in humans: how closely does it reflect multiple sclerosis?" ... Neurology , April 2014. ^ Desai RA, Davies AL, Tachrount M, Kasti M, Laulund F, Golay X, Smith KJ (2016). ... CS1 maint: multiple names: authors list ( link ) ^ Cristofanilli M, Rosenthal H, Cymring B, Gratch D, Pagano B, Xie B, Sadiq SA (2014).MOG, MBP, PLP1, EPO, FOXO3, NFE2L2, SIRT1, PPARA, CBLB, CCL3, CCL2, CCL1, PTN, PTGS2, PTGS1, PDGFRB, CCL11, PDGFRA, PDGFB, NTRK1, NGFR, NGF, NEFL, MMP9, MMP7, CCL5, CX3CL1, CIITA, VCAM1, HAVCR2, IL21, GHRL, IL22, RANGRF, AKAP12, QKI, IL18R1, VEGFA, TNF, SHH, TLR4, TLR2, TIMP3, TGFB2, TGFB1, ADAM17, STAT4, SNCB, SLPI, MMP2, A2M, MDK, CASP9, CCR5, CCR1, CTSC, ENTPD1, CD86, CD80, CD28, CD4, CAV3, CASP8, AIF1, CASP3, CACNA1B, C6, C3, BDNF, RHOA, AQP4, APOE, ANXA1, CNP, CPB2, CSPG4, CX3CR1, MAPT, LTA, JAK3, ITGA4, IL18, IL17A, IL16, IL12B, IL10RA, IL10, IL6, IL4, IL2RA, HMGCR, HLA-DQB1, HLA-DQA1, CXCR3, FGF2, DAB2, CCR2
-
Normal Pressure Hydrocephalus
Wikipedia
PMID 14303656 . ^ Krauss JK, Faist M, Schubert M, Borremans JJ, Lucking CH, Berger W (2001). "Evaluation of Gait in Normal Pressure Hydrocephalus Before and After Shunting". In Ruzicka E, Hallett M, Jankovic J (eds.). Gait Disorders . ... PMC 5619317 . PMID 29213984 . ^ a b Kiefer M, Unterberg A (January 2012). "The differential diagnosis and treatment of normal-pressure hydrocephalus" . ... PMID 20477715 . ^ Marmarou A, Bergsneider M, Klinge P, Relkin N, Black PM (September 2005). ... Neurology . 42 (1): 54–59. doi : 10.1212/wnl.42.1.54 . PMID 1734324 . ^ Poca MA, Mataró M, Del Mar Matarín M, Arikan F, Junqué C, Sahuquillo J (May 2004).CFAP43, NPHP1, CSF2, TUBB3, LAMC2, PMPCA, ERCC6, ERCC8, NXPH1, ACE, NPHP4, NEK8, MUC1, AQP4, ANKS3, APOE, UMOD, INVS, ALDH3A2, MLYCD, DNAJC13, ALB, NPHP3, CPVL, GLIS2, ANKS6, MAPKBP1, TYRP1, TBPL1, PER2, PROM1, VIM, TRPC1, TNF, TAZ, PSMD10, PSEN1, PRNP, PRKD1, PAX2, EPO, C3, BCL2, MIR4274

-
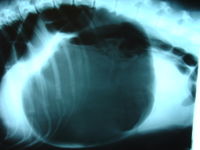
Gastric Dilatation Volvulus
Wikipedia
. ^ a b Glickman L, Glickman N, Schellenberg D, Raghavan M, Lee T (2000). "Incidence of and breed-related risk factors for gastric dilatation-volvulus in dogs". ... PMID 8784718 . ^ Glickman L, Glickman N, Schellenberg D, Raghavan M, Lee T (2000). "Non-dietary risk factors for gastric dilatation-volvulus in large and giant breed dogs". ... Retrieved 2007-04-17 . ^ Bright, Ronald M. (2004). "Gastric dilatation-volvulus: risk factors and some new minimally invasive gastropexy techniques" . ... Retrieved 15 October 2017 . ^ a b Mackenzie G, Barnhart M, Kennedy S, DeHoff W, Schertel E (March–April 2010). ... Assoc . 204 (9): 1465–71. PMID 8050972 . ^ Ward M, Patronek G, Glickman L (2003). "Benefits of prophylactic gastropexy for dogs at risk of gastric dilatation-volvulus".
-
Abetalipoproteinemia
Wikipedia
PMID 20953537 . ^ a b Moutzouri E, Elisaf M, Liberopoulos EN (March 2011). "Hypocholesterolemia". ... PMID 21827896 . ^ Hussain MM, Rava P, Walsh M, Rana M, Iqbal J (February 2012). ... PMC 3337244 . PMID 22353470 . ^ Najah M, Youssef SM, Yahia HM, Afef S, Awatef J, Saber H, et al. ... PMID 23556456 . ^ Magnolo L, Najah M, Fancello T, Di Leo E, Pinotti E, Brini I, et al. ... PMID 23043934 . ^ a b Pons V, Rolland C, Nauze M, Danjoux M, Gaibelet G, Durandy A, et al.MTTP, APOB, PANK2, VPS13A, SAR1B, GATA1, ABL1, KCNN4, XK, CAD, CYP4F22, HADHB, MT1B, BCR, HADHA, EVPL, RN7SL263P, GPT, TNFSF11, MPO, LGALS1, TNFRSF11A, MRPL28, IL18R1, WASF1, EBI3, SORBS2, FAM107B, SMR3B, SUB1, CRLF2, PTP4A3, PART1, NXT1, TEK, NOX4, HDL3, JPH3, PCSK9, MYB, CCL2, HIF1A, ACTB, ALK, APOE, CBL, CD34, CDKN2B, EPHA4, ETV6, FABP1, GFI1, IL10, PTPN1, IL11, JAK2, LDLR, MAP3K1, KMT2A, ABL2, NPM1, TNFRSF11B, PIK3C2A, PRL, H3P9

-
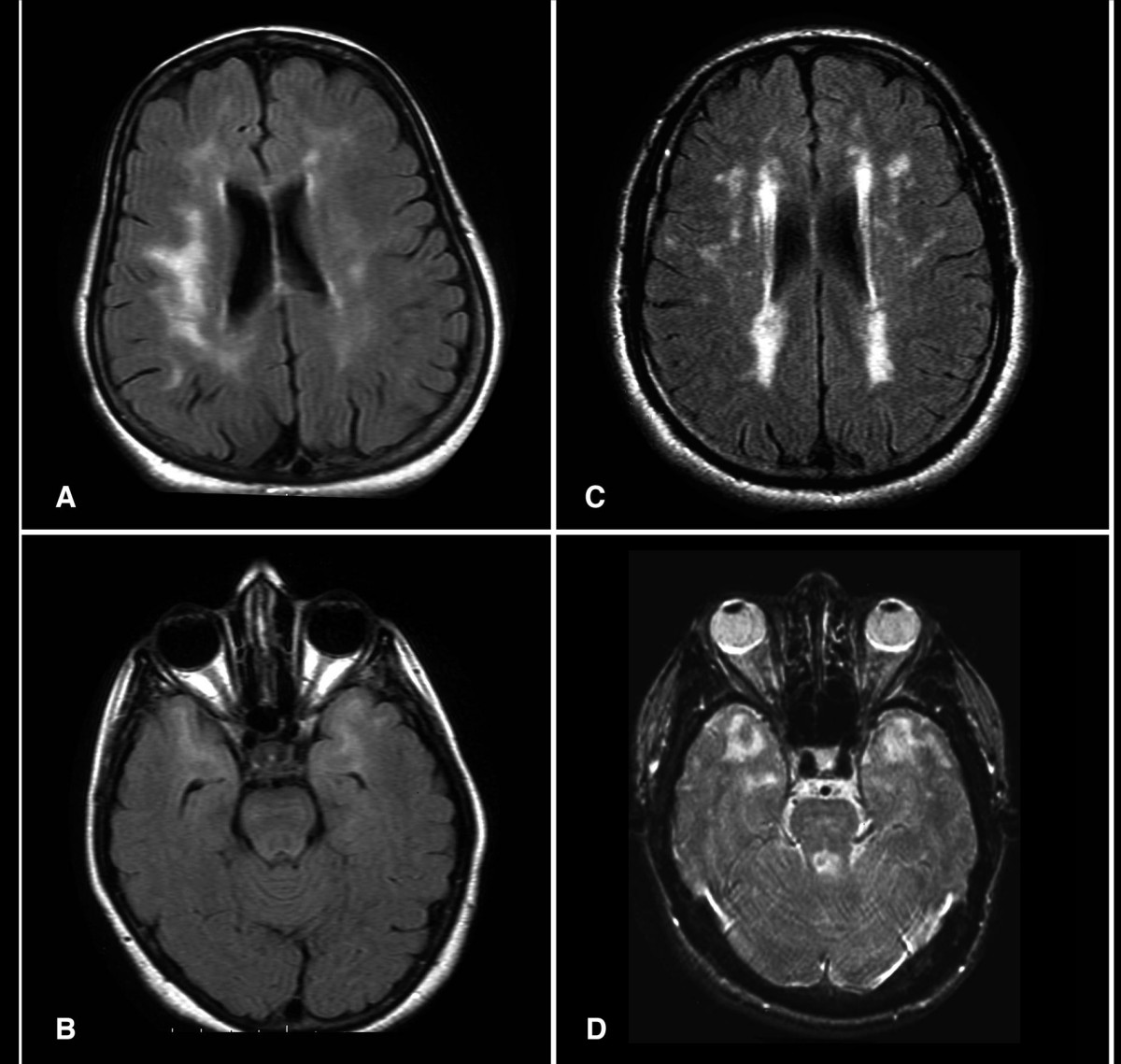
Cadasil
Wikipedia
.; Joutel, A.; Vahedi, K.; Iba-Zizen, M. T.; Tournier-Lasserve, E.; Bousser, M. ... PMID 7676806 . ^ Vlachakis D, Champeris Tsaniras S, Ioannidou K, Papageorgiou L, Baumann M, Kossida S (October 2014). "A series of Notch3 mutations in CADASIL; insights from 3D molecular modelling and evolutionary analyses". ... Lancet . 358 (9298): 2049–51. doi : 10.1016/S0140-6736(01)07142-2 . PMID 11755616 . ^ Ueda M, Nakaguma R, Ando Y (March 2009). ... PMID 20464302 . ^ a b Lesnik Oberstein, S. A.; van den Boom, R.; van Buchem, M. A.; van Houwelingen, H. C.; Bakker, E.; Vollebregt, E.; Ferrari, M. D.; Breuning, M. H.; Haan, J. (2001-09-25). "Cerebral microbleeds in CADASIL".NOTCH3, HTRA1, EGF, COL4A1, COL18A1, NOTCH1, JAG1, APOE, APP, LPA, TREX1, GFAP, PLXNA2, CTSA, PROC, ACTB, SOD1, TGFB1, NOTCH2, LAP, MRS2, RNF213, NOX5, THBD, KDR, NEFL, ACTG2, RBPJ, FN1, FBN1, ELN, DVL1, DCN, CSF3, CSF1R, CLU, CACNA1A, BGN, APCS, LINC01191
-
Enteritis
Wikipedia
PMID 23904840 . ^ Epple, H.-J.; Zeitz, M. (2011-09-01). "[Infectious enteritis]". ... PMC 2732465 . PMID 11927019 . ^ Colles, F. M.; McCarthy, N. D.; Howe, J. C.; Devereux, C. L.; Gosler, A. G.; Maiden, M. C. J. (2009-01-01). "Dynamics of Campylobacter colonization of a natural host, Sturnus vulgaris (European Starling)" . ... PMID 15367586 . ^ a b c Petrillo, T. M.; Beck-Sagué, C. M.; Songer, J. G.; Abramowsky, C.; Fortenberry, J. D.; Meacham, L.; Dean, A. G.; Lee, H.; Bueschel, D. M. (2000-04-27). "Enteritis necroticans (pigbel) in a diabetic child".TNFSF15, ICAM1, MPO, CXCL1, CXCL8, IL10, IL6, FOXP3, CPB2, CST2, TLR2, TLR4, AIMP2, RFXANK, ST8SIA2, AGER, ZYX, XBP1, GLP2R, TSC2, TIMP1, TGFB2, TACR1, TAC1, STAT5B, BECN1, SLC9A6, GRAP2, EGLN1, MIR148B, MIR32, MIR30A, SLC38A5, SYVN1, NOD2, HAMP, IL17D, SPI1, IL22, ICOS, POLDIP2, RNF19A, KLRK1, CAMKK2, AHSA1, STAT5A, NFATC1, SPARC, FDPS, HSPA5, CFH, GCG, LRRC32, FOSB, FOS, FES, DUSP1, MAPK1, DEFB1, MAPK14, CRK, COL3A1, CD40LG, CASP8, ALOX15, TNC, IFNA1, IFNA13, IFNG, PLA2G4A, SERPINE1, ALB, MMP9, MMP1, SMAD3, LYZ, LTF, JUND, JUNB, JUN, IL15, IL11, IL4, IL1B, KLRC4-KLRK1
-
Dysmetria
Wikipedia
Cerebellum . 6 (3): 254–67. doi : 10.1080/14734220701490995 . PMID 17786822 . ^ Manto M (2009). "Mechanisms of human cerebellar dysmetria: experimental evidence and current conceptual bases" . ... PMID 10377369 . ^ a b c d e f g h i j k l m n o p q Vahdat S, Maghsoudi A, Haji Hasani M, Towhidkhah F, Gharibzadeh S, Jahed M (October 2006). ... Lett . 325 (3): 211–5. doi : 10.1016/S0304-3940(02)00268-9 . PMID 12044658 . ^ Manto, M.; Godaux, E.; Jacquy, J. (January 1994). ... ISSN 0364-5134 . PMID 8285591 . ^ Leggio, M.; Molinari, M. (February 2015). "Cerebellar sequencing: a trick for predicting the future".RFC1, SNX14, SCN8A, RPL27A, STUB1, ATXN10, TTC19, GTPBP2, TMEM106B, PTRH2, AP5Z1, TIMMDC1, PNPLA8, TBL2, SACS, ATP6AP2, HIBCH, ABHD12, PLD3, WARS2, PIK3R5, ADPRS, CAMTA1, PMPCA, NFASC, SPART, SETX, POLR3A, AFG3L2, KIF1C, YME1L1, NOP56, SYNE1, ABCB7, PUM1, LNPK, TGM6, JMJD8, EBF3, BRAT1, SAMD9L, VWA3B, PRICKLE1, WDR81, SFXN4, MARS2, NDUFAF2, SLC25A46, REPS1, TCTN2, ANO10, FA2H, COA7, IRF2BPL, PIEZO2, MYORG, GJC2, COQ8A, ERMARD, NGLY1, POLR3B, CWF19L1, DARS2, MSTO1, TECPR2, PMPCB, GTF2IRD1, PEX2, PRKCG, PPP2R2B, PLP1, PDYN, OPHN1, OPA1, NEU1, MRE11, MAG, LIMK1, KCNC1, ITPR1, IFRD1, GTF2I, GRM1, GRID2, GJB1, B4GALNT1, FXN, FMR1, ELN, ATN1, CRAT, TPP1, ERCC8, CAV1, KIF1A, PRNP, RARS1, ATP1A3, RFC2, ATG5, PEX16, ADGRG1, CACNA2D2, CTDP1, BAZ1B, CACNA1G, SQSTM1, GPAA1, DEGS1, PLA2G6, ACOX2, LAGE3, XRCC4, XRCC1, CLIP2, VLDLR, UCHL1, TTPA, TOP3A, TBP, SPTBN2, SLC9A1, SCN2A, ATXN7, ATXN2, ATXN1, CCDC88C
-
Anaphylaxis
Wikipedia
PMC 3122150 . PMID 21682750 . ^ Worm, M (2010). "Epidemiology of anaphylaxis". ... Retrieved 2014-01-16 . ^ a b Simons FE, Ebisawa M, Sanchez-Borges M, Thong BY, Worm M, Tanno LK, Lockey RF, El-Gamal YM, Brown SG, Park HS, Sheikh A (2015). "2015 update of the evidence base: World Allergy Organization anaphylaxis guidelines" . ... PMID 20684869 . ^ Simons, FE; Ardusso, LR; Dimov, V; Ebisawa, M; El-Gamal, YM; Lockey, RF; Sanchez-Borges, M; Senna, GE; Sheikh, A; Thong, BY; Worm, M; World Allergy, Organization. (2013). ... PMID 20543673 . S2CID 205435146 . ^ Halbrich, M; Mack, DP; Carr, S; Watson, W; Kim, H (2015). ... ISBN 978-1-4200-1756-4 . ^ a b Tejedor-Alonso M, A; Moro-Moro, M; Múgica-García, MV (2015).KNG1, ASPG, LTB4R, NLRP3, BTK, PTGS1, PTGS2, ADRB1, VCAM1, TPSD1, ACE, F9, VWF, CD63, FCGR2B, HP, MRGPRX2, FCGR3A, FCGR2A, STK11, MILR1, CCL2, IL10, IL33, TNF, FCGR1A, OGA, FCER1A, IL6, CD33, SIRT1, IL9, FCGR3B, IL4, MCAT, IL5, MCPH1, STAT3, IGHE, SLC16A1, ADRB2, COX8A, PTPN1, VEGFA, RHD, TRPV1, ABHD16A, NR0B2, TRPC1, SCN5A, SAA1, PVALB, RPE65, TM7SF2, STXBP2, RGS13, STAT6, BHLHE40, SPG7, CCL7, SAA2, CCL3, PTPN6, ABL1, SOCS1, NT5C3A, TNFRSF12A, ARHGEF40, WDR11, CYSLTR2, AICDA, AHRR, CABS1, TSLP, SESTD1, C20orf181, LINC01672, LOC102723407, LOC102723971, LOC102724971, LINC02210-CRHR1, BFAR, CPA4, TNFSF10, IGHV3-69-1, HDAC3, SOCS3, HACD1, PCLAF, NR1H4, IL18BP, CAP1, PROC, SORBS1, MASP2, BRD4, PART1, CHIC2, SGSM3, IGHV3OR16-7, PTAFR, MTTP, PLSCR1, RCAN1, CPN1, CRH, CRHR1, CSF3, CTAA1, CTSD, DNASE1, ELAVL2, CHRM3, EPO, ESR1, ESR2, F8, F10, MS4A2, FCGR1B, CPA3, CDH15, PITX2, APOA1, ACADM, ADAM10, JAG1, AGT, AGXT, AHR, ANGPT1, FASLG, CD36, ARSA, AVP, BAAT, BRAF, TSPO, CD80, CD86, FLG, GAPDH, GATA1, NFKB1, LNPEP, LTC4S, LYZ, MITF, MPO, MRC1, ABO, NHS, GATA2, NOS3, NOTCH1, PAEP, PAFAH1B2, PECAM1, PFAS, SERPINB6, KIT, ISG20, IL18, IL17A, GCG, GPT, HDC, HLA-DQB1, NR4A1, HSPA4, HTR3A, ICAM1, IFNG, IGH, IL1A, IL1B, IL2RA, IL4R, IL13, PERCC1
-
Thrombotic Thrombocytopenic Purpura
Wikipedia
Med . 339 (22): 1629–31. doi : 10.1056/NEJM199811263392210 . PMID 9828253 . ^ Furlan M, Robles R, Galbusera M, et al. (1998). ... J Pediatr Hematol Oncol ^ Schulman I, Pierce M, Lukens A, Currimbhoy Z (July 1960). ... ISBN 978-0781765077 . ^ Kokame, K.; Matsumoto, M; Soejima, K; Yagi, H; Ishizashi, H; Funato, M; Tamai, H; Konno, M; Kamide, K; Kawano, Y; Miyata, T; Fujimura, Y (14 August 2002). ... PMID 21051740 . ^ a b Caprioli J, Noris M, Brioschi S, et al. (August 2006). ... PMC 3159000 . PMID 20058209 . ^ Bitzan M, Schaefer F, Reymond D (September 2010).ADAMTS13, TFPI, F3, THBD, CFH, VWF, HLA-DRB1, PLG, SH3BP4, FLT4, ZFP36, HLA-DQB1, RBM45, CASP1, ABO, CRISP2, THBS1, DGKE, TNFSF10, CFLAR, PKD2L1, MAPKAPK2, CFHR3, TGFB1, WAS, ABCA1, PLA2G15, CCL2, MIR133B, PRSS55, RMDN2, KCNH8, TNFRSF13C, IL33, ACCS, SPZ1, CD248, ACSS2, ADAMTSL4, KRT20, TLR9, CLEC1B, PTPN22, STAT3, SERPINF2, S100B, MS4A1, EPHB1, ELAVL1, SLC25A10, CSF3, CRP, CD27, CD19, GHRH, CASP8, CASP3, CACNA1S, C3, APOH, APOA1, F2, BRF1, PTX3, CD46, PTGS2, PHEX, SERPINF1, SERPINE1, NRAS, COX2, JAK2, CFHR1, IL1B, IFNG, IFNB1, HP, HLA-DRB4, HLA-DRB3, MTCO2P12
-
Acute Inhalation Injury
Wikipedia
Proc Am Thorac Soc. 7: 253 ^ Kennedy SM, Enarson DA, Janssen RG, Chan-Yeung M. (1991) Lung health consequences of reported accidental chlorine gas exposures among pulpmill workers. ... Postgrad Med. 112:133-40. ^ Lalić H, Djindjić-Pavicić M, Kukuljan M. (2009) Ammonia intoxication on workplace--case report and a review of literature. ... Clin Chest Med. 15:103-16. ^ Witschi H. (1977) Environmental agents altering lung biochemistry. Fed Proc. 36:1631-4. ^ Adelipour M, Imani Fooladi AA, Yazdani S, Vahedi E, Ghanei M, Nourani MR. (2011) Smad molecules expression pattern in human bronchial airway induced by sulfur mustard. Iran J Allergy Asthma Immunol. 10:147-54. ^ Ghabili K, Agutter PS, Ghanei M, Ansarin K, Panahi Y, Shoja MM. (2011) Sulfur mustard toxicity: history, chemistry, pharmacokinetics, and pharmacodynamics. ... Crit Care Clin. 2:455-70. ^ Uchida T, Makita K. (2008) Acute lung injury and alveolar epithelial function. Masui. 57:51-9. ^ Tang PS, Mura M, Seth R, Liu M. (2008) Acute lung injury and cell death: how many ways can cells die?
-
Chytridiomycosis
Wikipedia
JSTOR 3761366 . ^ Martel, A.; Spitzen-van der Sluijs, A.; Blooi, M.; Bert, W.; Ducatelle, R.; Fisher, M. ... T.; Rachowicz L. J.; Knapp R. A.; Stice M. J.; Tunstall T.; Bingham R. E.; Parker J. ... Retrieved 14 October 2013 . ^ McMahon, T. A.; Brannelly, L. A.; Chatfield, M. W.; Johnson, P. T.; Joseph, M. B.; McKenzie, V. ... Ecological Applications . 11 (2): 464–479. doi : 10.1890/1051-0761(2001)011[0464:DOTCRL]2.0.CO;2 . ^ Hayes TB, Case P, Chui S, Chung D, Haeffele C, Haston K, Lee M, Mai VP, Marjuoa Y, Parker J, Tsui M (April 2006). ... L.; Schrenzel M. D.; Pessier A. P. (2012). "Treatment of chytridiomycosis with reduced-dose itraconazole" .

-
Von Willebrand Disease
Wikipedia
Haemophilia . 14 (2): 171–232. doi : 10.1111/j.1365-2516.2007.01643.x . PMID 18315614 . ^ Lavin M, Aguila S, Schneppenheim S, Dalton N, Jones KL, O'Sullivan JM, O'Connell NM, Ryan K, White B, Byrne M, Rafferty M, Doyle MM, Nolan M, Preston RJ, Budde U, James P, Di Paola J, O'Donnell JS (November 2017). ... PMID 28916584 . ^ "Von Willebrand Disease" . hemophilia.org . 4 March 2014 . Retrieved 4 April 2018 . ^ Lavin M, Aguila S, Dalton N, Nolan M, Byrne M, Ryan K, White B, O'Connell NM, O'Sullivan JM, Di Paola J, James PD, O'Donnell JS (July 2018). ... Kouides: Inherited Bleeding Disorders in Women P., ISBN 1-4051-6915-X ^ Favaloro EJ, Bonar R, Kershaw G, Sioufi J, Baker R, Hertzberg M, Street A, Marsden K (July 2006). ... Retrieved 27 March 2020 . ^ "Canine von Willebrand Disease - Breed Summaries" . ahdc.vet.cornell.edu . 2019-02-08. ^ "Canine von Willebrand Disease" . vetgen.com . ^ Lehner S, Ekhlasi-Hundrieser M, Detering C, Allerkamp H, Pfarrer C, von Depka Prondzinski M (February 2018). ... The New England Journal of Medicine . 351 (7): 683–94. doi : 10.1056/NEJMra040403 . PMID 15306670 . Laffan M, Brown SA, Collins PW, Cumming AM, Hill FG, Keeling D, Peake IR, Pasi KJ (May 2004).VWF, P2RY12, F8, COX8A, ADAMTS13, GP1BA, F9, AK3, ABO, IL11, F11, ACTB, VWA8, ITIH1, TLR5, ITIH5, ANGPT2, POU2F3, CHRD, ALKBH1, CLEC4M, C20orf181, MUC5B, ATP6V0A2, SMPX, RABGEF1, CRISPLD2, SEC1P, PRB2, GP6, BMPER, TERF2IP, FAM20C, STXBP5, ANTXR1, ANO2, RAP1A, THPO, PDIA3, AGT, AVP, CANX, CD40, CLCA1, CSHL1, COCH, EPHA3, F2, F3, IFNA2, THBS1, RBPJ, ITGA2, ITGA2B, LRPAP1, MTHFR, CCN3, SERPINE1, PLG, POMC, SET, BOP
-
Intravascular Lymphomas
Wikipedia
PMID 31966830 . ^ a b c d e f g h i j k l m n o p q r s t Ponzoni M, Campo E, Nakamura S (October 2018). ... PMID 19717091 . ^ a b c d e f g h i j k Zanelli M, Mengoli MC, Del Sordo R, Cagini A, De Marco L, Simonetti E, Martino G, Zizzo M, Ascani S (November 2018). ... PMID 28461685 . ^ a b Komeno Y, Akiyama M, Okochi Y, Tokuda H, Abe K, Iihara K, Ryu T (2019). ... PMID 29737107 . ^ Saito T, Matsuya T, Takahashi C, Kaneta K, Ohishi Y, Uehara J, Hashimoto M, Honma M, Ishida-Yamamoto A (March 2017). ... PMID 27422850 . ^ Ferreri AJ, Campo E, Seymour JF, Willemze R, Ilariucci F, Ambrosetti A, Zucca E, Rossi G, López-Guillermo A, Pavlovsky MA, Geerts ML, Candoni A, Lestani M, Asioli S, Milani M, Piris MA, Pileri S, Facchetti F, Cavalli F, Ponzoni M (October 2004).BCL6, BCL2, TNFRSF8, CD274, SMUG1, ACP3, LAMC2, KRT20, PRTN3, MME, KMT2A, IGH, IRF4, ALK, CSF3, CSF2, CRP, MS4A1, PWWP3A

-
Wernicke Encephalopathy
Wikipedia
PMID 11304070 . ^ a b c d e f g h i j k l m Galvin R, Bråthen G, Ivashynka A, Hillbom M, Tanasescu R, Leone MA (December 2010). ... PMID 20050893 . ^ Kondo, K.; Fujiwara, M.; Murase, M.; Kodera, Y.; Akiyama, S.; Ito, K.; Takagi, H. (1996). ... ISBN 978-1-58890-191-0 . ^ Ishiko T, Taguchi T, Takeguchi M, Saito H, Nanri K (September 2009). ... ISBN 978-9701057070 . ^ a b Vasconcelos, M. M.; Silva, K. P.; Vidal, G.; Silva, A. ... PMID 22093426 . ^ Passemard, S.; Kaindl, A. M.; Verloes, A. (2013). "Microcephaly".
-
Devil Facial Tumour Disease
Wikipedia
PMID 19021786 . ^ Hawkins CE, McCallum H, Mooney N, Jones M, Holdsworth M. (2009). Sarcophilus harrisii . ... Version 2009.1. [ full citation needed ] ^ Janeway CA, Travers P, Walport M, Shlomchik M. (2001). Immunobiology. ... PMID 20219455 . ^ Epstein B, Jones M, Hamede R, Hendricks S, McCallum H, Murchison EP, et al. ... Retrieved 2016-08-30 . ^ Epstein B, Jones M, Hamede R, Hendricks S, McCallum H, Murchison EP, et al. ... Retrieved 2018-01-27 . ^ Cheng Y, Makara M, Peel E, Fox S, Papenfuss AT, Belov K (2019-03-13).















