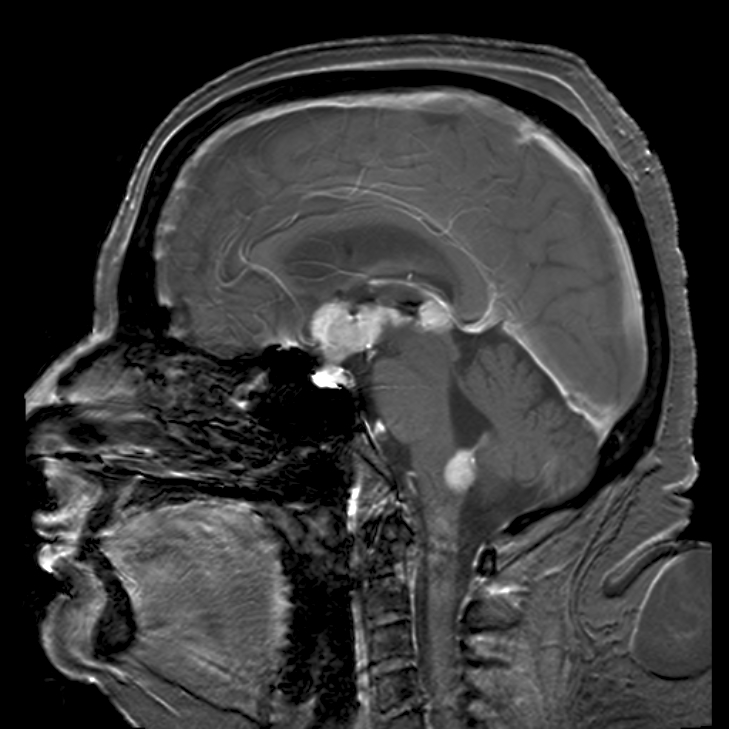
It is recommended that TSH levels are maintained below 2.5 mU/l in the first trimester of pregnancy and below 3 mU/l in later pregnancy. [22] The recommended maintenance dose of thyroxine in pregnancy is about 2.0-2.4 µg/kg daily. ... Red cell zinc may also be useful in differentiating the two. [25] Hyperthyroidism due to Graves’ disease may worsen in the first trimester of pregnancy, remit in later pregnancy, and subsequently relapse in the postpartum. [ citation needed ] Risks of hyperthyroidism on fetal and maternal well-being [ edit ] Uncontrolled hyperthyroidism in pregnancy is associated with an increased risk of severe pre-eclampsia and up to a four-fold increased risk of low birth weight deliveries. Some of these unfavourable outcomes are more marked in women who are diagnosed for the first time in pregnancy. A recent study has also shown that already high normal maternal FT4 levels are associated with a decrease in child IQ and gray matter and cortex volumes, similar to the effects of hypothyroidism. [5] Uncontrolled and inadequately treated maternal hyperthyroidism may also result in fetal and neonatal hyperthyroidism [26] due to the transplacental transfer of stimulatory TSH receptor antibodies (TRAbs). [27] Clinical neonatal hyperthyroidism occurs in about 1% of infants born to mothers with Graves’ disease. ... If a woman is already receiving carbimazole , a change to propylthiouracil (PTU) is recommended but this should be changed back to carbimazole after the first trimester. This is because carbimazole can rarely be associated with skin and also mid line defects in the fetus but PTU long term also can cause liver side effects in the adult. Carbimazole and PTU are both secreted in breast milk but evidence suggests that antithyroid drugs are safe during lactation. [31] There are no adverse effects on IQ or psychomotor development in children whose mothers have received antithyroid drugs in pregnancy.Current guidelines suggest that a pregnant patient should be on PTU during the first trimester of pregnancy due to lower tetragenic effect and then be switched to methimazole during the second and third trimester due to lower liver dysfunction side effects. [ citation needed ] Postpartum thyroiditis [ edit ] Main article: Postpartum thyroiditis Postpartum thyroid dysfunction (PPTD) is a syndrome of thyroid dysfunction occurring within the first 12 months of delivery as a consequence of the postpartum immunological rebound that follows the immune tolerant state of pregnancy.
Morisaki et al. (1992) reported an 18-year-old German woman who first noted calf pain at 4 years of age, usually related to exercise. ... Morisaki et al. (2000) and Abe et al. (2000) reported a 46-year-old Japanese woman who had first noted muscle weakness of the legs at 40 years of age. ... Castro-Gago et al. (2011) reported a 6-month-old Spanish girl who presented with weakness and hypotonia from the first month of life. She was conceived by in vitro fertilization with sperm donation.
Adenosine monophosphate deaminase 1 (AMPD1) deficiency is an inherited condition that can affect the muscles used for movement (skeletal muscles). Many people with AMPD1 deficiency do not have symptoms. People who do have symptoms typically have muscle pain (myalgia), cramping, and weakness after exercise, and often get tired faster than others. Some affected people appear to have more severe symptoms. AMPD1 deficiency is caused by changes (mutations) in the AMPD1 gene and is inherited in an autosomal recessive manner. Other types of AMPD deficiency include the acquired type (due to a muscle or joint condition), and the coincidental inherited type (due to both mutations in the AMPD1 gene and a separate muscle or joint disorder).
A number sign (#) is used with this entry because complete erythrocyte AMP deaminase deficiency is caused by homozygous mutation in the AMPD3 gene (102772) on chromosome 11p15. Description Complete deficiency of erythrocyte AMP deaminase is a clinically benign disorder (Ogasawara et al., 1987; Zydowo et al., 1989). Clinical Features Ogasawara et al. (1987) observed 6 related individuals with complete deficiency of erythrocyte AMP deaminase (isozyme E). All were healthy and had no hematologic disorders. The ATP level was approximately 150% higher in AMP-deficient red cells compared to the level in the control cells. Degradation of adenine nucleotide was slower in the deficient erythrocytes than in the control erythrocytes.
Onset occurs at or soon after birth, and features can include growth retardation, microcephaly, hypertonicity, axial hypotonia, encephalopathy, cardiomyopathy, and liver dysfunction. Death usually occurs in the first weeks or years of life (summary by Smits et al., 2011). ... Smits et al. (2011) reported a girl, born of consanguineous parents, with COXPD1 characterized by onset in the first days of life of axial hypotonia, spasticity, refractory seizures, feeding problems, and increased serum and CSF lactate. ... INHERITANCE - Autosomal recessive GROWTH Other - Intrauterine growth retardation HEAD & NECK Head - Microcephaly, mild Eyes - Poor eye contact - Nystagmus ABDOMEN Liver - Fulminant hepatic failure (in 2 sibs) - Liver necrosis - Cholestasis - Hepatomegaly Gastrointestinal - Feeding problems NEUROLOGIC Central Nervous System - 'Stiffness' - Decreased spontaneous movement - Delayed motor development - Axial hypotonia - Spasticity - Hyperreflexia - Seizures, refractory - Hypoplasia of the corpus callosum - Delayed myelination - Generalized brain atrophy - Cystic lesions in the basal ganglia METABOLIC FEATURES - Metabolic acidosis, severe LABORATORY ABNORMALITIES - Increased serum lactate - Increased cerebrospinal fluid lactate - Increased serum direct bilirubin - Fibroblasts show decreased activity of mitochondrial respiratory complex I, complex III, complex IV, and complex V MISCELLANEOUS - Onset at birth - Death within first months or years of life - Four patients have been reported (as of July 2011) MOLECULAR BASIS - Caused by mutation in the mitochondrial elongation factor G1 gene (GFM1, 606639.0001 ) ▲ Close
Combined oxidative phosphorylation deficiency 1 is a severe condition that primarily impairs neurological and liver function. Most people with combined oxidative phosphorylation deficiency 1 have severe brain dysfunction (encephalopathy) that worsens over time; they also have difficulty growing and gaining weight at the expected rate (failure to thrive). In some cases, affected individuals have abnormal muscle tone (increased or decreased), developmental delay, seizures, loss of sensation in the limbs (peripheral neuropathy), and an unusually small head (microcephaly ). Liver disease is common in people with combined oxidative phosphorylation deficiency 1, with individuals quickly developing liver failure. Individuals with this condition also usually have a potentially life-threatening buildup of a chemical called lactic acid in the body (lactic acidosis).
Hepatoencephalopathy due to combined oxidative phosphorylation deficiency type 1 is a rare, inherited mitochondrial disorder due to a defect in mitochondrial protein synthesis characterized by intrauterine growth retardation, metabolic decompensation with recurrent vomiting, persistent severe lactic acidosis, encephalopathy, seizures, failure to thrive, severe global developmental delay, poor eye contact, severe muscular hypotonia or axial hypotonia with limb hypertonia, hepatomegaly and/or liver dysfunction and/or liver failure, leading to fatal outcome in severe cases. Neuroimaging abnormalities may include corpus callosum thinning, leukodystrophy, delayed myelination and basal ganglia involvement.
Balci et al. (1991) reported the disorder in Turkish brother and sister with affected first-cousin parents. Two earlier-born male infants had died 1 hour after birth with the same clinical appearance. ... Yang et al. (1991) reported the case of a male fetus aborted at 30 weeks' gestation because of abnormalities visualized on sonography. This was the first instance of polydactyly associated with this syndrome. ... Referring to this as type IV short-rib syndrome, Cideciyan et al. (1993) reported the case of an infant who died of respiratory insufficiency during the first day of life. Findings evident on physical examination included bilateral anophthalmia, midline cleft lip and palate, bilateral single transverse palmar creases, and single umbilical artery.
A rare ciliopathy with major skeletal involvement characterized by short ribs and hypoplastic thorax, small iliac bones, short tubular bones with smooth metaphyseal margins, and bowed radii and ulnae. The tibiae are relatively well tubulated and longer than the fibulae. There is a high frequency of brain defects, while post-axial polydactyly is rare. Additional features may include cleft lip, absence of internal genitalia, and renal, biliary, and pancreatic cysts, among others.
Nazzaro et al. (1988) reported 4 sibs with Papillon-Lefevre syndrome, ranging in age from 2 to 11 years. The parents were double first cousins. Hattab et al. (1995) reported 4 cases of PLS affecting 2 Jordanian families with a total of 8 children. The patients were between 4.5 and 12 years of age and their parents, who were first cousins, were not affected. In all patients, there was a relationship between increased severity of skin lesions and seasonal variations and intensified periodontal destruction. ... Palmoplantar keratosis, varying from mild psoriasiform scaly skin to overt hyperkeratosis, typically develops within the first 3 years of life. Keratosis also affects other sites such as the elbows and knees.
Hyperkeratosis of palms and soles of feet appear in first few years of life. Destructions of periodontium follows almost immediately after the eruption of last molar tooth.
Clinical description Diffuse palmoplantar keratoderma (see this term) with erythematous plaques develops between the first and fourth years of life, with the soles being usually more severely affected than the palms.
Some patients present with esophageal candidiasis as a first presentation of systemic candidiasis . ... A severe case of candidiasis H&E stain of esophagus showing Candida hyphae within the lamina propria Treatment [ edit ] The current first-line treatment is fluconazole , 200 mg. on the first day, followed by daily dosing of 100 mg. for at least 21 days total.
Provided there is no hernia present, it goes away without treatment in the first year. Primary hydroceles may develop in adulthood, particularly in the elderly and in hot countries, by slow accumulation of serous fluid. ... (exception - funicular hydrocele, encysted hydrocele) Treatment [ edit ] Most hydroceles appearing in the first year of life seldom require treatment as they resolve without treatment. [3] Hydroceles that persist after the first year or occur later in life require treatment only in selected cases, such as patients who are symptomatic with pain or a pressure sensation, or when the scrotal skin integrity is compromised from chronic irritation; the treatment of choice is surgery and the operation is conducted via an open access technique aiming to excise the hydrocele sac. [4] [5] Anesthesia is required for the operation and general anesthesia is of choice in children, while spinal anesthesia is usually sufficient in adults.
If your child is affected, the provider might ask: When did you first notice this swelling? Has it increased over time? ... If you're affected, your provider might ask: When did you first notice the swelling? Have you had any discharge from your penis or blood in your semen?
Hydrocele testis The right testis , exposed by laying open the tunica vaginalis . (Tunica vaginalis is labeled at upper right.) Specialty Urology A hydrocele testis is an accumulation of clear fluid within the cavum vaginale , the potential space between the layers of the tunica vaginalis of the testicle . A primary hydrocele causes a painless enlargement in the scrotum on the affected side and is thought to be due to the defective absorption of fluid secreted between the two layers of the tunica vaginalis (investing membrane). A secondary hydrocele is secondary to either inflammation or a neoplasm in the testis. A hydrocele usually occurs on one side, but can also affect both sides.
The most common problems arise in the skin and eyes , and later in blood vessels in the form of premature atherosclerosis . [2] [3] [4] PXE is caused by autosomal recessive mutations in the ABCC6 gene on the short arm of chromosome 16 (16p13.1). [3] [5] [6] Contents 1 Signs and symptoms 2 Genetics 3 Pathophysiology 4 Diagnosis 4.1 Differential diagnosis 5 Treatment 6 Epidemiology 7 History 8 Images 9 See also 10 References 11 External links Signs and symptoms [ edit ] Usually, pseudoxanthoma elasticum affects the skin first, often in childhood or early adolescence. [7] Small, yellowish papular lesions form and cutaneous laxity mainly affect the neck, axillae (armpits), groin, and flexural creases (the inside parts of the elbows and knees). [3] [8] Skin may become lax and redundant. Many individuals have "oblique mental creases" (horizontal grooves of the chin) [9] PXE first affects the retina through a dimpling of the Bruch membrane (a thin membrane separating the blood vessel-rich layer from the pigmented layer of the retina ), that is only visible during ophthalmologic examinations. [10] This is called peau d'orange (a French term meaning "skin of the orange"). ... Although there have been reports of autosomal dominant inheritance, the inheritance is typically autosomal recessive (both parents need to be carriers, and there is a 25% chance that a child will inherit both abnormal copies of the gene and therefore develop the condition). [3] Strong genetic linkage was found with mutations in the ABCC6 gene, which codes for the ABCC6 protein, which is a membrane transporter from the large ATP-binding cassette transporter family. ... The disease occurs in all ethnicities, but Afrikaners are more likely to have PXE as a result of a founder effect (i.e., higher prevalence in the small group of people from whom Afrikaners descend). [36] History [ edit ] The first description of PXE that distinguished it from other xanthoma conditions was by Dr Ferdinand-Jean Darrier in 1896. [37] The eponym "Grönblad-Strandberg syndrome" is used in older literature, after two physicians who made further discoveries in the disease manifestations. [38] PXE has the distinction of being the only disease for which a layperson is the discover of the mutated gene.
Another way to diagnosis 48,XXXY is by chromosomal microarray showing the presence of extra X chromosomes. [3] Chromosomal microarray (CMA) is used to detect extra or missing chromosomal segments or whole chromosomes. CMA uses microchip-based testing to analyze many pieces of DNA.
Etiology The most likely etiology is the non disjunction of homologous chromosomes (during the first meiotic division) or sister chromatids (during the second meiotic division) in the parental germ cells.
48,XXXY syndrome is a type of chromosome abnormality characterized by the presence of 2 extra X chromosomes in males. It is sometimes referred to as a variant of Klinefelter syndrome , but differs from Klinefelter syndrome in many ways and is more severe. Signs and symptoms of 48,XXXY syndrome can vary but may include learning difficulties; intellectual disability; low muscle tone (hypotonia); hypogonadism; delayed growth; distinctive facial features; and a variety of birth defects that may affect the genital and musculoskeletal systems. Many also have poorly developed social skills and delayed language development. This condition is not inherited and likely results from a random error in cell division.
Overview A muscle cramp is a sudden, unexpected tightening of one or more muscles. Sometimes called a charley horse, a muscle cramp can be very painful. Exercising or working hard, especially in heat, can lead to muscle cramps. Some medicines and illnesses also might cause muscle cramps. Muscle cramps aren't usually harmful. Self-care measures can treat most muscle cramps. Symptoms Muscle cramps occur mostly in leg muscles, most often in the calf.
This is also true for some cases of chronic fatigue syndrome , where objective post-exertion muscle weakness with delayed recovery time has been measured and is a feature of some of the published definitions. [4] [5] [6] [7] [8] [9] Asthenia vs. myasthenia [ edit ] Asthenia ( Greek : ἀσθένεια , lit lack of strength but also disease ) is a medical term referring to a condition in which the body lacks or has lost strength either as a whole or in any of its parts. It denotes symptoms of physical weakness and loss of strength .
Green leafy vegetables may help prevent development of subsequent SCC and multiple studies found that raw vegetables and fruits are significantly protective against SCC risk. [17] [18] On the other hand, consumption of whole milk, yogurt, and cheese may increase SCC risk in susceptible people. [19] In addition, meat and fat dietary pattern can increase the risk of SCC in people without a history of SCC, but the association is again more prominent in people with a history of skin cancer. [20] Tobacco smoking and a dietary pattern characterized by high beer and liquor intake also increase the risk of SCC significantly. [21] [17] References [ edit ] ^ a b "NCI Dictionary of Cancer Terms" .
Extended survival has been seen, however, in a subgroup of AIDS patients with CD4 counts of more than 200 and no concurrent opportunistic infections, who can tolerate aggressive therapy consisting of either methotrexate monotherapy or vincristine , procarbazine, or whole brain radiotherapy. These patients have a median survival of 10–18 months.
Primary central nervous system lymphoma (primary CNS lymphoma) is a rare form of non-Hodgkin lymphoma in which cancerous cells develop in the lymph tissue of the brain and/or spinal cord. Because the eye is so close to the brain, primary CNS lymphoma can also start in the eye (called ocular lymphoma). The signs and symptoms vary based on which parts of the central nervous system are affected, but may include nausea and vomiting; seizures; headaches; arm or leg weakness; confusion; double vision and/or hearing loss. The exact underlying cause of primary CNS lymphoma is poorly understood; however, people with a weakened immune system (such as those with acquired immunodeficiency syndrome ) or who have had an organ transplant appear to have an increased risk of developing the condition. Treatment varies based on the severity of the condition and location of the cancerous cells.
Primary central nervous system lymphoma (PCNSL) is a rare nervous system tumor, predominantly due to diffuse large B-cell lymphoma, that involves brain, leptomeninges, eyes, or rarely spinal cord, in the absence of systemic diffusion at the time of diagnosis. It is characterized by a solitary tumor that, depending on its location, can lead to a variety of symptoms such as headache, nausea, vomiting (and other signs of raised intracranial pressure), focal neurologic deficits, neuropsychiatric and ocular symptoms, seizures and personality changes.
Clinical Features Stuve and Wiedemann (1971) reported 2 sisters and a first-cousin male with congenital bowing of the long bones. ... One patient died after 10 days of life from respiratory insufficiency with apnea; her sister developed hyperthermia with temperatures as high as 41 degrees centigrade and died on day 5 of life. The male first cousin to these 2 girls had identical congenital flexion contractures of the fingers and toes and died of respiratory insufficiency as a neonate. ... Raas-Rothschild et al. (2003) reported 2 sibs with Stuve-Wiedemann syndrome who died of respiratory failure due to pulmonary hypertension. The parents were first-cousin Muslim Arabs originating from the Jerusalem area. ... Al-Gazali et al. (2003) reported 3 children from 2 inbred Arab families with Stuve-Wiedemann syndrome who had survived the first year of life; their ages were 6, 2.8, and 2 years. ... This report confirmed that survival in this syndrome is possible and that the prognosis improves after the first year of life. It also supported the existence of a characteristic phenotype in Stuve-Wiedemann syndrome survivors that includes neurologic symptoms reminiscent of dysautonomia in addition to the skeletal abnormalities and distinctive radiologic features.
Contents 1 Types 1.1 Type 1: distal 1.2 Type 2: proximal 1.3 Type 3: combined proximal and distal 1.4 Type 4: absolute hypoaldosteronism or aldosterone insensitivity 2 History 3 See also 4 References 5 External links Types [ edit ] An overview of types 1, 2, and 4 is presented below (type 3 is usually excluded from modern classifications): Type Type 1 Type 2 Type 4 Location Collecting Tubules, distal tubules Proximal tubules Adrenal Acidemia Yes (very severe) Yes Mild when present Potassium Hypokalemia Hypokalemia Hyperkalemia Pathophysiology Failure of α intercalated cells to secrete H + and reclaim K + Failure of proximal tubular cells to reabsorb H C O − 3 Deficiency of aldosterone , or a resistance to its effects, ( hypoaldosteronism or pseudohypoaldosteronism ) Type 1: distal [ edit ] Main article: Distal renal tubular acidosis Distal RTA (dRTA) is the classical form of RTA, being the first described. Distal RTA is characterized by a failure of H+ secretion into lumen of nephron by the alpha intercalated cells of the medullary collecting duct of the distal nephron . ... Patient's with mutations in ATP6V1B1 and ATP6V0A4 will present with symptoms within the first year of life, while those with mutation of the SLC4A1 have delayed onset around 10 years of age. [4] Electrolyte imbalances remain the same, while in severe cases symptoms can advance to amino aciduria and hyperammonemia . [5] In a large Asian series of Distal renal Tubular Acidosis in Sjogren's Syndrome, late diagnosis is a rule in spite of overt hypokalemic periodic paralysis in a vast majority of them [6] dRTA is the most common form of RTA diagnosed in Western countries, and can be classified as either hereditary (primary) or acquired (secondary). ... Hereditary dRTA generally presents as failure to thrive during the first several months of life. Other common clinical manifestations in children include a variety of gastrointestinal and urinary symptoms, including polyuria, polydipsia, constipation, diarrhea, bouts of dehydration, and decreased appetite. [8] Type 2: proximal [ edit ] Radiograph of a child with rickets , a complication of both proximal and, less commonly, distal RTA. ... Its cardinal feature is hyperkalemia , and measured urinary acidification is normal, hence it is often called hyperkalemic RTA or tubular hyperkalemia. [17] Causes include: Aldosterone deficiency ( hypoaldosteronism ): Primary vs. hyporeninemic (including diabetic nephropathy) Aldosterone resistance Drugs: NSAIDs , ACE inhibitors and ARBs , Eplerenone , Spironolactone , Trimethoprim , Pentamidine Pseudohypoaldosteronism History [ edit ] Renal tubular acidosis was first described in 1935 by Lightwood and 1936 by Butler et al. in children. [18] [19] Baines et al. first described it in adults in 1945. [20] Donald L.
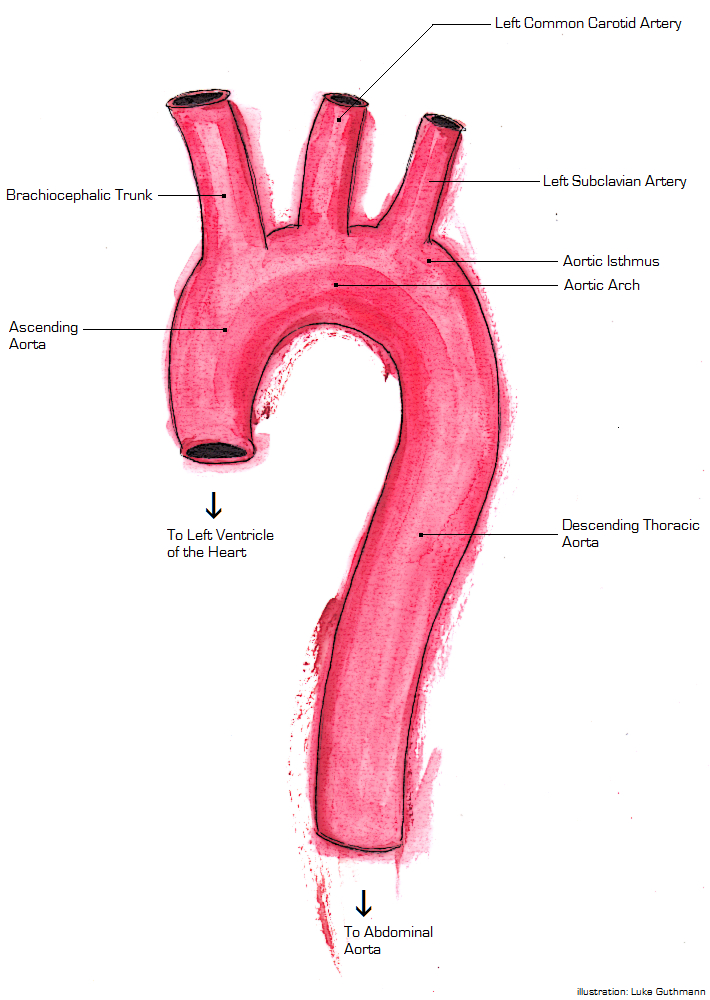
A more recently proposed mechanism is that the aorta can be compressed between bony structures (such as the manubrium , clavicle , and first rib ) and the spine . In the ascending aorta (the portion of the aorta which is almost vertical), one mechanism of injury is torsion (a two-way twisting). [5] There are clinical predictors of an aortic injury. [6] The predictors include if a patient is older than 50, was an unrestrained patient, has hypotension, has a thoracic injury requiring thoracotomy, has a spinal injury, or has a head injury. [6] If four of these criteria are met their likelihood for an aortic injury is 30% [6] The aortic wall is made up of three different components the inner layer (intima), the muscle layer (media), and the outer layer (adventitia). ... If esophageal injury is expected, the patient has a facial injury, or if the patient has difficulty maintaining their away then the trans esophageal echo is contraindicated. [8] Treatment [ edit ] The first line treatment for patients with thoracic aortic injury is maintaining the patient's airway with intubation and treating secondary injuries such as a hemothorax. [4] After ensuring the patient has a patent airway and other life-threatening injuries are treated then treatment for the aortic injury can be started. ... A patient can either undergo endovascular repair or surgical repair. [9] Endovascular repair is the current gold standard due to increased success rates and lower complications. [9] [1] Patients that are able to undergo endovascular repair without contraindications should proceed with it. [1] Repair should be delayed if there is life-threatening intra-abdominal or intracranial bleeding or if the patient is at risk for infection. [9] Endovascular Repair [ edit ] Main article: Endovascular aneurysm repair Endovascular repair is done by first gaining vascular access usually through the femoral artery. [8] A catheter is inserted to the point of injury and a luminal stent is deployed. [1] Blood is then able to be pumped through the stent and prevent the aortic wall from rupturing. [1] Open Surgical Repair [ edit ] Main article: Open aortic surgery Surgical repair is done by way of a thoracotomy or opening of the chest wall. [8] From this point multiple methods can be used, but the most successful methods enable distal perfusion to prevent ischemia. [8] When the surgery is performed a constant check of blood flow to the parts of the body away from the injury should be monitored to know if oxygenation is occurring. [8] Medical Management [ edit ] While waiting for surgery careful regulation of blood pressure and heart rate is necessary. [3] Systolic blood pressure should be maintained between 100 and 120 mmHg allowing for perfusion distal to the injury but decreasing the risk of rupture while the heart rate should be kept under 100 beats per minute. Esmolol is first choice to maintain blood pressure and heart rate due to its short time of action, but if the blood pressure is not within range adding nitroprusside sodium can be added as a second agent. [9] The treatment is similar to what is done for aortic dissections. [7] If the patient has minimal aortic injury then the patient can be managed non surgically. [8] Rather the patient can be followed with serial images. If the patient does develop a more severe injury including a full thickness injury through the media layer then the patient should be treated with surgery. [8] Outcomes [ edit ] Thoracic aortic injury is the 2nd leading cause of death involving both blunt trauma. 80% of patients that have a thoracic aortic injury will die immediately. [4] Of the patients that do make it to be evaluated only 50% will survive 24 hours. [1] Of the patients that do survive the first 24 hours 14% develop paraplegia. [6] Epidemiology [ edit ] Thoracic aortic injury is most commonly caused by a penetrating trauma in up to 90% of cases. [10] Of these cases around 28% are confined to the thoracic portion of the aorta including the ascending aorta, aorta arch, and the descending aorta. [10] Of the thoracic aortic injuries the ligament arteriosum is the most common location followed by the portion of the aorta after the origin of the left subclavian artery. [10] The most common mechanism leading to thoracic aortic injury is a motor vehicle collision.
If left untreated, these patients can develop ischemic injury of vital organs , leading to multi-system organ failure . [4] The first factor to be considered is whether the hypovolemic shock has resulted from hemorrhage or fluid losses , as this will dictate treatment. ... The body compensates with increased sympathetic tone resulting in increased heart rate , increased cardiac contractility , and peripheral vasoconstriction . The first changes in vital signs seen in hypovolemic shock include an increase in diastolic blood pressure with narrowed pulse pressure . [4] As volume status continues to decrease, systolic blood pressure drops. ... In the undifferentiated patient with shock, septic shock and toxic causes are also on the differential. [3] Management [ edit ] The first step in managing hemorrhagic shock is recognition. ... However, patients that received the more balanced ratio of 1:1:1 were less likely to die as a result of exsanguination in 24 hours and were more likely to achieve hemostasis Additionally, reduction in time to first plasma transfusion has shown a significant reduction in mortality in damage control resuscitation. [3] In addition to blood products, products that prevent the breakdown of fibrin in clots, or antifibrinolytics, have been studied for their utility in the treatment of hemorrhagic shock in the trauma patient. ... The CRASH-2 study was a randomized control trial of tranexamic acid versus placebo in trauma has been shown to decrease overall mortality when given in the first eight hours of injury. [3] Follow-up analysis shows additional benefit to tranexamic acid when given in the first three hours after surgery. [3] Damage control resuscitation is to occur in conjunction with prompt intervention to control the source of bleeding. [3] Strategies may differ depending on proximity to definitive treatment. [3] For patients in hemorrhagic shock, early use of blood products over crystalloid resuscitation results in better outcomes.
In individuals with hearing loss following aminoglycoside exposure, molecular testing for the pathogenic variants m.1555A>G and m.1494C>T in MT-RNR1 and m.7445A>C/T/G in MT-TS1 can be done first. A multigene panel that includes the mitochondrial genes listed in Table 1a and other genes of interest (see Table 1b and Differential Diagnosis) may also be considered. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
Clinical Features Holmes et al. (1995) described 3 sibs, 1 female and 2 male, with absence or hypoplasia of the tibia in association with other malformations. The parents were first cousins once removed. The girl had unilateral cleft lip, absence of the diaphragm, and postaxial polydactyly of the feet.
Tibia absent - polydactyly - arachnoid cyst syndrome is a very rare constellation of multiple anomalies, including absence or hypoplasia of the tibia. Epidemiology It has been described in 3 sibs (two males and one female). Clinical description The syndrome is characterized by absence or hypoplasia of the tibia, pre and postaxial polydactyly of the hands and/or feet, syndactyly of the toes, shortening and bowing of other long bones, and retrocerebellar arachnoid cyst. Genetic counseling Parental consanguinity reported in the family suggests an autosomal recessive pattern of inheritance.
Some of the affected individuals had development of first and second molars. In some cases, it appeared that the remaining teeth were not permanent teeth; instead, they were primary and sometimes remained until the forties.