-
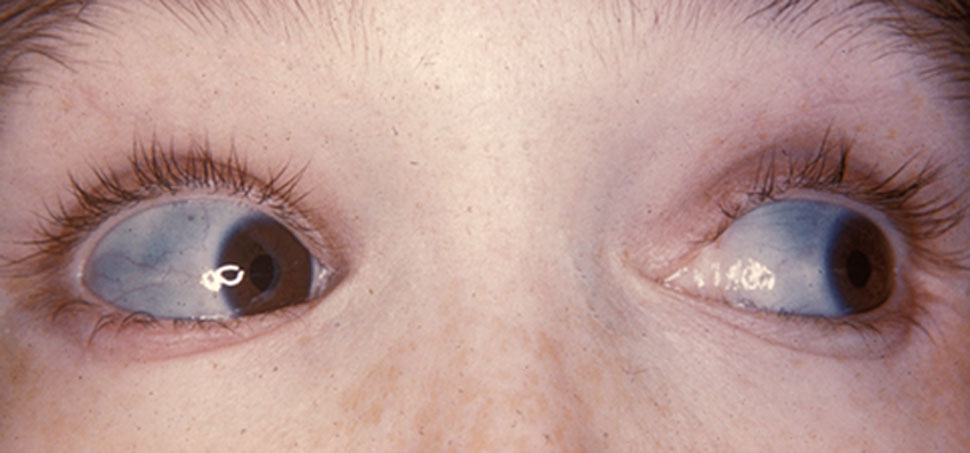
Buphthalmos
Wikipedia
It is sometimes referred to as buphthalmia (plural buphthalmias). [2] It usually appears in the newborn period or the first 3 months of life. [3] and in most cases indicates the presence of congenital (infantile) glaucoma , which is a disorder in which elevated pressures within the eye lead to structural eye damage and vision loss. ... Retrieved 26 September 2017 . ^ http://www.merriam-webster.com/medical/Buphthalmos Merriam-Webster online medical dictionary ^ Merriam-Webster: Over 80% of cases are evident by 3 years of age. ^ The Schlemm's canal is usually collapsed (Merriam-Webster) ^ Yanoff & Duker: Ophthalmology, 3rd ed. 2008 ^ http://www.swingmusic.net/Ray_Charles_Biography.html Swing Music website, page for Ray Charles External links [ edit ] Classification D ICD - 10 : Q15.0 ICD - 9-CM : 743.2 OMIM : 231300 MeSH : D006871 SNOMED CT : 413728006 v t e Congenital malformations and deformations of eyes Adnexa Eyelid Ptosis Ectropion Entropion Distichia Blepharophimosis Ablepharon Marcus Gunn phenomenon Lacrimal apparatus Congenital lacrimal duct obstruction Globe Entire eye Anophthalmia ( Cystic eyeball , Cryptophthalmos ) Microphthalmia Lens Ectopia lentis Aphakia Iris Aniridia Anterior segment Axenfeld–Rieger syndrome Cornea Keratoglobus Megalocornea Other Buphthalmos Coloboma ( Coloboma of optic nerve ) Hydrophthalmos Norrie disease

-
Pemphigus Foliaceus
Wikipedia
You can help by adding to it . ( August 2017 ) Epidemiology [ edit ] Pemphigus is endemic in the rural areas of Brazil, especially along inland riverbeds. [1] History [ edit ] Pierre Louis Alphée Cazenave first described the disease in 1844. [4] See also [ edit ] List of cutaneous conditions Pemphigus References [ edit ] ^ a b Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine . (6th ed.). Page 558–562. McGraw-Hill. ISBN 0-07-138076-0 . ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).C3, DSG1, DSG3, HLA-DRB1, RBM45, TNF, CTLA4, ICOS, LAIR2, KIR3DL1, IL6, VEGFA, GEM, TNFSF13B, CD86, LILRB1, NKG7, FOXP3, CD226, KLRG1, EIF2AK3, KRT20, SPAG16, TRAF2, PRRT2, LENG8, MAPK1, PRKCB, MIR338, PRKCA, KIR3DL2, LTA, LAIR1, CD19, MS4A1, CD40, CD40LG, CD59, CR1, EGFR, ENG, EPHB2, FCAR, HLA-A, HLA-C, HLA-DQA1, HSPA1L, HSPA4, CD1D, KIR3DS1, MIR584
-
Developmental Delay With Variable Intellectual Impairment And Behavioral Abnormalities
OMIM
Description Developmental delay with variable intellectual impairment and behavioral abnormalities (DDVIBA) is an autosomal dominant neurodevelopmental disorder. Most patients have impaired intellectual development with speech difficulties, and many have behavioral abnormalities, most commonly autism spectrum disorder (ASD), defects in attention, and/or hyperactivity. ... Inheritance Although the vast majority of TCF20 mutations occur de novo, Vetrini et al. (2019) observed 4 unrelated families with DDVIBA who showed autosomal dominant inheritance. Molecular Genetics In a 25-year-old woman (family 6) with DDVIBA, Babbs et al. (2014) identified a de novo heterozygous frameshift mutation in the TCF20 gene (603107.0001). ... The mutations were found by trio-based whole-exome sequencing and confirmed by Sanger sequencing. The patients were ascertained from a cohort of 313 individuals with intellectual disability who underwent trio-based whole-exome sequencing. In 28 patients from 27 unrelated families, including a set of monozygotic twins (patients 27 and 28), with DDVIBA, Vetrini et al. (2019) identified 25 heterozygous mutations in the TCF20 gene (see, e.g., 603107.0004-603107.0006). ... Several patients carried possible pathogenic variants in other genes, which may have contributed to the phenotype. INHERITANCE - Autosomal dominant GROWTH Height - Tall stature (in some patients) Weight - Obesity (in some patients) Other - Somatic overgrowth (in some patients) HEAD & NECK Head - Macrocephaly (in some patients) - Brachycephaly - Plagiocephaly Face - Dysmorphic facial features, variable - Midface hypoplasia - Long face - Full face - Frontal bossing - Tall forehead Ears - Low-set ears - Posteriorly rotated ears Eyes - Deep-set eyes - Epicanthal folds - Strabismus - Myopia Nose - Depressed nasal bridge - Short nose - Bulbous nose Mouth - Thin upper lip - Tented upper lip - Full low lip - Open mouth - Downturned corners of the mouth CHEST Breasts - Inverted nipples ABDOMEN Gastrointestinal - Feeding difficulties - Constipation SKELETAL Spine - Scoliosis Hands - Tapering fingers - Fifth finger clinodactyly Feet - Foot deformities MUSCLE, SOFT TISSUES - Hypotonia NEUROLOGIC Central Nervous System - Global developmental delay - Impaired intellectual development, mild to moderate - Motor delay - Delayed walking - Speech delay - Speech difficulties - Seizures (in some patients) - Sleep disturbances - Abnormal movements (in some patients) - Ataxia - Spasticity - Jerky movements - Dyspraxia - Poor coordination - Structural brain abnormalities, mild, non-specific Behavioral Psychiatric Manifestations - Autism spectrum disorder - Attention deficit-hyperactivity (ADHD) - Hyperactivity - Obsessive-compulsive disorder - Anxiety - Aggressive behavior - Food-seeking behavior MISCELLANEOUS - Highly variable phenotype - De novo mutation (in most patients) MOLECULAR BASIS - Caused by mutation in the transcription factor 20 gene (TCF20, 603107.0001 ) ▲ Close
-
Alexander Disease
Wikipedia
Rare genetic disorder of the white matter of the brain Alexander disease Brain of a 4-year-old boy with Alexander disease showing macroencephaly and periventricular leukomalacia (note brownish discoloration around the cerebral ventricles ) Specialty Endocrinology , neurology Alexander disease is a very rare autosomal dominant leukodystrophy , which are neurological conditions caused by anomalies in the myelin which protects nerve fibers in the brain. The most common type is the infantile form that usually begins during the first 2 years of life. Symptoms include mental and physical developmental delays, followed by the loss of developmental milestones, an abnormal increase in head size and seizures. ... Symptoms of the adult form may also resemble multiple sclerosis . [2] No more than 500 cases have been reported. [2] See also [ edit ] The Myelin Project The Stennis Foundation References [ edit ] ^ "Alexander Disease Information Page" . National Institute of Neurological Disorders and Stroke. 2018. ... Retrieved 2016-11-08 . ^ "Alexander Disease Information Page: National Institute of Neurological Disorders and Stroke (NINDS)" . www.ninds.nih.gov .GFAP, DES, CRYAB, HSPB2, HSPB3, HSPB1, NFE2L2, GABPA, TARDBP, SYNM, WDHD1, BTG3, TRH, SNCA, SLC1A2, PLEC, AVP, KRT8, ITPR2, CASP3, MTOR, MAPK14, COL4A1, CHI3L1, CD38, CASP6, HSPB8

-
Focal Dystonia
Wikipedia
Liona Boyd , Canadian classical guitarist, publicized as the "First Lady of the Guitar", retired from the concert stage for six years in 2003, due to focal dystonia that affected her right hand. ... In the 2000s, he regained use of his right hand and recommenced performing and recording with two hands. Dominic Frasca , guitarist Reinhard Goebel , Baroque violinist, switched to playing left-handed. ... "Man of constant sorrow: Charlie Parr's quiet battle to stay alive - City Pages" . City Pages . Retrieved 23 April 2018 .
-
Thiourea Tasting
OMIM
They concluded that variability in thresholds is controlled by a major locus with incomplete dominance, as well as by a multifactorial component. ... They performed a genome screen by using 1,324 markers with an average spacing of 4 cM. Analyses were first carried out with a recessive genetic model that had traditionally been assumed for the trait, and a threshold score of 8.0 delineating tasters from nontasters. ... Drayna et al. (2003) found evidence for other possible quantitative loci on chromosomes 1 (lod = 2.31 at 344 cM) and 16 (lod = 2.01 at 14 cM). A subsequent 2-locus whole genome scan conditional on the chromosome 7 quantitative trait locus identified the chromosome 16 locus (2-locus lod = 3.33 at 14 cM). ... History Nebert (1997) suggested that the first example of pharmacogenetics was the phenylthiourea nontaster trait first described by Snyder (1932). ... Endocrine - Relationship between PTC nontasting to cretinism Misc - Variation in ability to taste PTC Inheritance - Autosomal dominant - possibly two loci involved ▲ Close
-
Hypohidrotic Ectodermal Dysplasia
GeneReviews
HED is inherited in an autosomal dominant, autosomal recessive, or X-linked manner. ... Hypodontia (congenital absence of teeth): An average of nine permanent teeth – typically the canines and first molars –develop in individuals with classic HED [Lexner et al 2007]. ... Sequence analysis of EDA is performed first, followed by gene-targeted deletion/duplication analysis of EDA if no pathogenic variant is found. ... Sequence analysis of genomic DNA cannot detect a (multi)exon or whole-gene deletion on an X chromosome of carrier females. 6. ... Mild HED Females with XLHED and males and females with autosomal dominant HED (ADHED) typically have mild HED.EDA, EDARADD, EDAR, EDA2R, TRAF6, IKBKG, TNF, HAND2, GJB6, WNT10A, NFKBIA, FN1, LEF1, CD38, XIST, MADCAM1, CXCR4, NR4A3, GOLPH3, KDF1, BMS1, ORAI1, TNFRSF1A, TNFRSF13B, TAB2, ACACA, STAT1, GH1, CDC42, CTNNB1, EFNB1, EGFR, ELANE, G6PD, GFI1, IFNG, FAS, IFNGR1, KRT19, LTB, PGK1, PGK1P1, PKP1, PTGS2, RAC1

-
Simpson-Golabi-Behmel Syndrome Type 1
GeneReviews
Note: Lack of amplification by PCR prior to sequence analysis can suggest a putative (multi)exon or whole-gene deletion on the X chromosome in affected males; confirmation requires additional testing by gene-targeted deletion/duplication analysis. Sequence analysis of GPC3 is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Lack of amplification by PCR prior to sequence analysis can suggest a putative (multi)exon or whole-gene deletion on the X chromosome in affected males; confirmation requires additional testing by gene-targeted deletion/duplication analysis. 5. ... Gene-targeted methods will detect single-exon up to whole-gene deletions; however, breakpoints of large deletions and/or deletion of adjacent genes may not be determined.
-
Corpus Callosum, Agenesis Of
OMIM
Formation of the corpus callosum begins as early as 6 weeks' gestation, with the first fibers crossing the midline at 11 to 12 weeks' gestation, and completion of the basic shape by age 18 to 20 weeks (Schell-Apacik et al., 2008). ... Lynn et al. (1980) reported affected father and son, suggesting autosomal dominant inheritance. Naritomi et al. (1997) reported 3 sibs with agenesis of corpus callosum and severe psychomotor retardation, 2 of whom were dizygotic twins. ... They suggested that agenesis of the corpus callosum, when transmitted as an autosomal dominant trait, is clinically relatively mild as compared with autosomal or X-linked recessive forms and may be more common than generally thought. ... Schell-Apacik et al. (2008) noted that ACC and dysgenesis of the corpus callosum has been associated with at least 7 autosomal dominant, 23 autosomal recessive, and 12 X-linked complex genetic syndromes. ... The variants, which were found by whole-exome sequencing and confirmed by Sanger sequencing, segregated with the disorder in the family.DCC, PAK3, SIN3A, L1CAM, ARX, SLC12A6, SOX2, BUB1B, CREBBP, TUBA1A, FOXG1, ZBTB18, DISC1, ARID1B, PYCR1, EPG5, CYP11A1, VAX1, RPGRIP1L, FGFR1, DHCR24, NFIA, TUBB2B, EHMT1, GPX4, CDK5RAP2, MKS1, GLI2, GLI3, SLC25A1, KAT6B, KRAS, RAB3GAP2, NDE1, KIF4A, LETM1, PIGN, PPP2R3C, ADNP, SUFU, SPECC1L, GLDC, GPC3, WDR60, NCAPD3, ANKLE2, CEP152, FANCI, RTTN, MBTPS2, AUTS2, PHGDH, DACT1, HNRNPU, TUBA8, TRAPPC12, HCCS, MRPS16, HSPA9, BCL11A, CDON, POMT2, GMPPB, RAB3GAP1, PYCR2, IGBP1, GTPBP2, IGF2, SETD2, DLL1, AHI1, B9D1, HIBCH, TDGF1, MYB, MID1, ZIC1, FOXH1, HESX1, DCHS1, RAD51, KCNAB2, OFD1, EOMES, ALX1, FZD3, SALL1, ZNF148, ZIC2, WNT3, LARGE1, SHH, STIL, SIX3, SKI, NELFA, NSD2, SMO, TP53, SOX3, NKX2-1, TGIF1, ABAT, PTCH1, PROP1, POLR3A, OTX2, CIT, B4GAT1, GCSH, POMT1, TUBB3, RXYLT1, NFIB, NFIX, NODAL, DNAL4, NPHP1, SIX6, MED12, COPB2, KIF14, PDHA1, ARNT2, FIG4, KIAA0753, PTDSS1, BMS1, CEP135, PRRX1, POU1F1, NTN1, RECQL4, GDF1, NDUFB11, TMLHE, CSF1R, WDR34, CDK5, CDK6, LHX4, CENPF, MAP11, COL4A1, COX7B, DISP1, CPT2, POMGNT2, MFSD2A, FKRP, POMK, CTNNB1, DDX59, CEP63, SPG11, PGAP1, DAG1, NARS2, DCX, DYNC2H1, MCPH1, TICRR, CEP41, ADAT3, C12orf57, ACACA, TMLHE-AS1, ACTB, RNU4ATAC, CRPPA, ALX3, AMT, KIF7, RERE, ATRX, RSPO2, WDR62, ASPM, HYLS1, ASXL1, BMP4, SASS6, CEP120, B3GALNT2, B3GLCT, DIS3L2, PROKR2, BORCS5, FAT4, SHANK3, TAF13, ALX4, CENPJ, EMX2, SLC25A19, EP300, INPP5E, KIF15, FH, FGFR2, TMEM237, KNL1, ERCC6, FGF8, GPC4, FKTN, ZSWIM6, WDR35, FLI1, IFT80, PORCN, VAC14, FRMD4A, FLVCR2, PIEZO2, PRDM16, BCL11B, POMGNT1, GAS1, PHC1, EML1, CWF19L1, GABRD, EFNB1, MYBL1, EGFR, KIT, MIR483, NOTCH1, PRKAB1, ZEB2, PRKAA1, PRKAA2, CDKN2A, IGF1, ERBB2, NR5A1, IGF1R, HTC2, CXCR4, ACACB, BCL2A1, VEGFA, SOX10, ZNRF3, LOC110806263, CCND1, ELAVL2, RUNX3, ESR1, PGR, TWIST1, VIM, APC, RASSF1, MTOR, SLC12A7, PIK3CA, MDM2, TERT, SST, MC2R, POMC, HIF1A, AKT3, BRCA1, ERCC1, SCARNA22, POTEM, ACOT7, TYMS, TOP2A, MIR497, ARMC5, VAV2, MIR503, MALAT1, MIR335, POTEKP, ENOPH1, FHL5, CD274, LONP1, SLC9A3R2, PTTG1, STK11, SCAF11, PROM1, ZNF462, BECN1, DKK3, BBC3, GEMIN2, ACTBL2, MIR139, MIR184, MIR195, MIR210, SMUG1, SERPINA3, AQP1, ESRRA, IL6, RARRES2, XIAP, MSH2, INHA, ATM, PTEN, BCL2, MAPK8, CYP19A1, PRKAR1A, PPARG, STMN1, PIK3CG, PIK3CD, PIM1, PIK3CB, ABCB1, PECAM1, SMAD4, MEN1, NEFL, MET, CDK2, MRC1, HSP90AA1, CDH2, FABP7, GNAS, FOXO1, FSCN1, SMARCA1, SLC2A1, SKP2, FOXM1, ACTG1, JAG1, SMARCA2, FN1, EZH2, ACTG2, ACKR3, CTLA4, FGFR3, KREMEN1, SESN2, CYP2B6, MED25, ETV6, CTBS, DIABLO, MINDY4, CYP2C19, MAML2, PBK, VCAN, CRH, FOXO3, CP, CGA, CDKN1B, FATE1, FOLH1, ZNF160, FOS, PDCD1LG2, FCGR1B, NDRG2, SARDH, TRPV4, EN1, CTTN, GAS5, EIF4EBP1, EIF4E, EGF, E2F1, NDRG4, PINK1, DNAH8, RTN4R, BIRC7, CYP11B1, DOCK6, CORO7, TIMM8A, DECR1, DDT, LIN28A, DAPK1, FCGR1A, DAB1, ARHGAP31, EPHB2, CYP17A1, STAT3, EARS2, TMEM67, CDH1, AKT1, ALCAM, MIR511, MIR431, ABCD1, ALOX15B, MIR17HG, MIR34A, MIR222, MIR214, AMPD2, MIR21, MIR205, BIRC2, BIRC3, SNORA21, AGT, UCA1, MIR1202, H3P23, ACADVL, MTCO2P12, MYB-AS1, ASIC2, TMED7-TICAM2, MIR1275, ADK, PRINS, FCGR1CP, ZGLP1, MIR675, SNORA51, SCARNA4, MIR183, MIR155, MIR150, CD44, CCNB1, PRICKLE1, AMER1, CD14, CD36, GAA, TWIST2, CASP3, TXNRD3, CYP2U1, AZIN2, CDA, CDK1, MRAP2, KCTD11, CAD, MIRLET7D, EOGT, SBSN, AQP3, TICAM2, ARSA, BARD1, GADL1, HMSD, BRCA2, BCL9, PCSK9, SKA1, HCN2, BDNF, BNIP3, FOSB, GUSB, GARS1, NECTIN1, AIFM1, PTGS2, BCL10, SPHK1, PTK7, PTPRC, NCOA1, MAPK1, ALDH18A1, CDK10, DGKZ, PPM1D, RASGRF1, RFX3, PSMD9, PRKD1, AXIN2, PCLAF, NOS2, NUMA1, PA2G4, PCNA, MFN2, MELK, PLAG1, PRKACA, MAD2L1BP, APOBEC3B, PCYT1B, PMAIP1, MED27, DDX23, RPS6KB1, AXIN1, GLYAT, TFAP2C, SOS1, SOX4, TLR4, SPN, SPOCK1, TGFA, SPP1, SORD, SRC, TCF21, ZEB1, TBX1, STAR, SYP, TSC1, SOAT1, MLRL, S100B, RRM1, NR4A3, RRM2, GHS, RAB7A, S100A1, SCN7A, UBC, SDHB, SFPQ, SF1, SGTA, VEGFC, KDM6A, NOS1, NAGLU, GATA6, OBP2A, HOXA6, HSD17B4, TMED7, PDE11A, NEUROG3, SLC40A1, HSPD1, HLA-G, ID1, IFI27, IFNB1, IGF2R, IL2, MAT2B, LEF1, HINT1, IL13, GRIN2B, GCG, GFAP, GHSR, GLP1R, ULK4, SFN, NR3C1, SIRT6, GSTP1, DCUN1D1, SULT2A1, HDC, HGF, NBAS, CXCL8, B3GAT1, NAGA, MPZ, SPEN, MGMT, MMRN1, KMT2A, HRH3, MMP9, COX2, TBC1D9, SDS, LILRB1, MUC1, DCTN6, MYC, ZNRD2, KIF1B, MEF2C, PELP1, KRT15, ITGAV, JUN, PRPF31, JUNB, JUND, KIF22, LY96, MCAM, LHCGR, LRP2, NNT, EPCAM, DICER1, MARCKS, H3P10
-
Bathophobia
Wikipedia
A Wikidata element is linked to this page: Bathophobia (Q6898244) .
-
Genital Leiomyoma
Wikipedia
Fitzpatrick's Dermatology in General Medicine . (6th ed.). Page 1033. McGraw-Hill. ISBN 0-07-138076-0 .
-
Spastic Paraplegia 74, Autosomal Recessive
OMIM
Description Spastic paraplegia-74 is an autosomal recessive neurologic disorder characterized by onset of slowly progressive lower limb spasticity, optic atrophy, and peripheral neuropathy in the first decade (summary by Lossos et al., 2015). ... Clinical Features Lossos et al. (2015) reported a multigenerational, highly consanguineous Arab family in which 11 individuals had slowly progressive spastic paraplegia with optic atrophy and peripheral neuropathy. Symptoms first appeared in the first decade, manifest as gait impairment associated with mild to moderate spasticity, hyperreflexia of the knee, and extensor plantar responses. ... The mutation, which was found by a combination of linkage analysis and whole-exome sequencing, segregated with the disorder in the family. ... INHERITANCE - Autosomal recessive HEAD & NECK Eyes - Optic atrophy - Visual impairment - Visual field defects MUSCLE, SOFT TISSUES - Distal leg muscle atrophy NEUROLOGIC Central Nervous System - Spastic paraplegia - Hyperreflexia of the knee - Extensor plantar responses Peripheral Nervous System - Axonal peripheral neuropathy - Distal sensory impairment - Areflexia of the ankles - Reduced compound muscle action potentials and velocities LABORATORY ABNORMALITIES - Patient lymphoblastoid cells showed decreased activity of mitochondrial complexes I and II MISCELLANEOUS - Onset in first decade - Slowly progressive - One consanguineous Arab family has been reported (last curated July 2015) MOLECULAR BASIS - Caused by mutation in the iron-sulfur cluster assembly factor IBA57 gene (IBA57, 615316.0002 ) ▲ Close
-
Col1a1/2 Osteogenesis Imperfecta
GeneReviews
Perform sequence analysis first. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... A small proportion of infants with OI type I have femoral bowing at birth. The first fractures may occur at birth or with diapering. More often, the first fractures occur when the infant begins to walk and, more importantly, to fall. ... More than 60% of affected infants die on the first day; 80% die within the first week; survival beyond one year is exceedingly rare and usually involves intensive support such as continuous assisted ventilation [Byers et al 1988].COL1A1, COL1A2, SERPINF1, CRTAP, P3H1, SPARC, WNT1, TMEM38B, PLOD2, P4HB, MESD, PPIB, SERPINH1, TENT5A, TAPT1, SMPD3, SMAD4, SUCO, CREB3L1, FKBP10, BMP1, IFITM5, SOST, PPP1R2C, BGLAP, DMD, BEST1, GH1, ACVR2B, SEC24D, PNPLA2, FGFR3, AGA2, PLS3, COX8A, DCN, LRP5, TLL1, PDIA2, GPATCH8, RER, WWTR1, MTCO2P12, PADI1, EFEMP2, PLA1A, MBTPS2, GPR180, NBAS, MED18, LINC01672, TRAP, MIR29B2, MIR29B1, MIR145, SP7, ACTB, EIF2AK3, TNC, ALPL, BAAT, BGN, TSPO, KRIT1, CD38, CD44, CHRM3, COL3A1, COL5A1, CSF2, DLX3, FN1, MSTN, IGF1, TNFSF11, LAMC2, LOX, LRP6, COX2, PPP1CB, PTH, PTGS2, RNASE1, SRSF2, SLC6A2, TNXB, TSC1, TSHR, CXCR4, LOC107984355
-
Apraxia
Wikipedia
This is one of the 3 major components of Balint's syndrome . [8] Causes [ edit ] Apraxia is most often due to a lesion located in the dominant (usually left) hemisphere of the brain, typically in the frontal and parietal lobes . ... It is also possible for apraxia to be caused by lesions in other areas of the brain. [11] Ideomotor apraxia is typically due to a decrease in blood flow to the dominant hemisphere of the brain and particularly the parietal and premotor areas. It is frequently seen in patients with corticobasal degeneration . [11] Ideational apraxia has been observed in patients with lesions in the dominant hemisphere near areas associated with aphasia; however, more research is needed on ideational apraxia due to brain lesions. ... Retrieved 2019-08-02 . ^ a b c "Apraxia Information Page | National Institute of Neurological Disorders and Stroke" . www.ninds.nih.gov . 2019 . ... PMID 19614961 . ^ (Manasco, 2014) ^ "NINDS Apraxia Information Page" . Retrieved 8 March 2012 . ^ Worthington, Andrew (2016).
-
Aortic Valve Disease 3
OMIM
In addition, the authors studied a mother and son (family 2) with aortic valve stenosis and atrial septal defect; the son also exhibited BAV with a left-to-right fusion pattern. Molecular Genetics By whole-exome sequencing in a cohort of 9 families with bicuspid aortic valve and/or thoracic aortic aneurysm (AAT), who were negative for mutation in AAT-associated genes, Gould et al. (2019) identified 2 families with heterozygous mutations in the ROBO4 gene: in family 1, a splicing mutation (607528.0001) was present in 8 affected individuals as well as in 2 unaffected family members; in family 2, an affected mother and son were heterozygous for a missense mutation (R64C; 607528.0002). ... One of the independent probands carried the same R64C missense variant that was identified in family 2; clinical details were not reported for that patient. INHERITANCE - Autosomal dominant CARDIOVASCULAR Heart - Aortic valve stenosis - Bicuspid aortic valve - Atrial septal defect (in some patients) Vascular - Aortic root aneurysm - Aneurysm of ascending aorta MISCELLANEOUS - Reduced penetrance has been observed MOLECULAR BASIS - Caused by mutation in the roundabout guidance receptor 4 gene (ROBO4, 607528.0001 ) ▲ Close
-
Autosomal Recessive Cutis Laxa Type 1
Orphanet
Clinical description The skin manifestations affect the whole body and are usually recognizable from birth. ... Differential diagnosis The differential diagnosis should include other forms of CL (autosomal recessive type 2, autosomal dominant and X-lined CL) and related syndromes (gerodermia osteodysplastica, Cantu syndrome, wrinkly skin syndrome and De Barsy syndrome), together with the Ehlers-Danlos syndromes and Costello syndrome (see these terms).
-
Progressive Deafness With Stapes Fixation
Orphanet
Otosclerosis appears to be a multifactorial disease. Autosomal dominant and a low penetrance (40%) transmission is observed in familial cases. ... In osteogenesis imperfecta and Paget's disease, lesions involve the whole temporal bone and the skull, and result in thickening of the ossicles.
-
Histiocytoid Cardiomyopathy
Orphanet
Mutation in cytochrome b, as well as A8344G mitochondrial DNA mutation have been described but seem to be sporadic variants. Whole Genome Expression Analysis support cluster of candidate gene at 1q21.3c, 2q12.1a and a decrease in copy number of the genes encoding S100A calcium binding protein, along with a strong decrease in interleukin 33 expression. ... Genetic counseling There is a familial tendency of 5%; however, mode of inheritance (X-linked dominant inheritance and autosomal recessive) is still debated.
-
Spondyloepimetaphyseal Dysplasia, Di Rocco Type
OMIM
X-rays at age 38 showed osteoarthropathy in the wrists and shoulders, as well as progressive spine involvement, whereas the metaphyseal lesions of the knees were no longer detectable. Molecular Genetics By whole-exome sequencing in an Italian family with spondyloepimetaphyseal dysplasia, Di Rocco et al. (2018) identified heterozygosity for a missense mutation in the UFSP2 gene (D426A; 611482.0002) that segregated with disease and was not found in controls or in public variant databases. INHERITANCE - Autosomal dominant GROWTH Height - Short stature SKELETAL Spine - Hypoplasia of anterior vertebral bodies - Slight platyspondyly Pelvis - Irregular acetabular roof Limbs - Waddling gait - Joint pain - Restricted mobility - Genua vara - Absent ossification nucleus of proximal femoral epiphysis - Irregular profile of femoral neck - Irregular acetabular roof - Metaphyseal dysplasia of distal femur - Metaphyseal dysplasia of proximal tibia - Osteoarthropathy of shoulders - Osteoarthropathy of wrists Hands - Delayed carpal bone age MISCELLANEOUS - Based on report of 1 family (last curated May 2018) MOLECULAR BASIS - Caused by mutation in the UFM1-specific peptidase-2 gene (UFSP2, 611482.0002 ) ▲ Close
-
Arthrogryposis, Cleft Palate, Craniosynostosis, And Impaired Intellectual Development
OMIM
Molecular Genetics In 2 patients with ACCIID, Mizuguchi et al. (2018) identified de novo heterozygous mutations in the autoinhibitory domain of the PPP3CA gene (114105.0007-114105.0008). The mutations were found by whole-exome sequencing and confirmed by Sanger sequencing. Using a yeast model, the mutations were found to be constitutively activating. INHERITANCE - Autosomal dominant GROWTH Height - Short stature (-4.4 to -5 SD) Weight - Low weight (-2.9 to -4 SD) HEAD & NECK Head - Plagiocephaly - Trigonocephaly Face - Micrognathia Mouth - Cleft palate CHEST Ribs Sternum Clavicles & Scapulae - Slender ribs GENITOURINARY Kidneys - Hydronephrosis Ureters - Vesicoureteral reflux SKELETAL - Gracile bones - Perinatal fractures - Arthrogryposis - Tubular bones Skull - Craniosynostosis Hands - Brachydactyly NEUROLOGIC Central Nervous System - Intellectual disability, moderate to severe (DQ




