Children's umbilical hernias often close on their own in the first two years of life, though some remain open into the fifth year or longer. ... For children, surgery is typically reserved for umbilical hernias that: Are painful Are slightly larger than 1/4 to 3/4 inch (1 to 2 centimeters) in diameter Are large and don't decrease in size over the first two years of life Don't disappear by age 5 Become trapped or block the intestine For adults, surgery is typically recommended to avoid possible complications, especially if the umbilical hernia gets bigger or becomes painful. ... What to expect from your doctor Your doctor is likely to ask you a number of questions, such as: When did you first notice the problem? Has it gotten worse over time?
Clinical Features Seven et al. (2002) described a Turkish brother and sister, offspring of a first-cousin marriage, who were affected with severe neurodegeneration beginning in infancy. ... Seven et al. (2002) suggested that facial dysmorphism could permit diagnosis in the first months of life without any clinical or neurologic signs. ... The patients with the frameshift mutation were more severely affected than those with the missense mutation. The mutations in the first family were found by linkage analysis and candidate gene sequencing.
Hypotonia-speech impairment-severe cognitive delay syndrome is a rare, genetic neurodegenerative disorder characterized by severe, persistent hypotonia (presenting at birth or in early infancy), severe global developmental delay (with poor or absent speech, difficulty or inability to roll, sit or walk), profound intellectual disability, and failure to thrive. Additional manifestations include microcephaly, progressive peripheral spasticity, bilateral strabismus and nystagmus, constipation, and variable dysmorphic facial features (including plagiocephaly, broad forehead, small nose, low-set ears, micrognathia and open mouth with tented upper lip).
The second duplication was 269.97 kb in length, was found to be 400 kb distal to the first duplication, and included the entire BIRC4 gene (XIAP; 300079) and the first 4 exons of the STAG2 gene (300826). RT-PCR studies of the first duplication identified 3 different transcripts, with duplication of exons 1 to 12, exons 2 to 12, and exons 3 to 12, all of which resulted in a frameshift and premature termination after exon 12, with lack of synthesis of a functional AMPA receptor.
A rare, genetic, X-linked syndromic intellectual disability disorder characterized by moderate to severe intellectual disability associated with epilepsy, short stature, autistic features and behavioral problems, such as self injury and aggressive outbursts. Observed facial dysmorphism includes brachycephaly, prominent supraorbital ridges, and deep set eyes. Additional variable manifestations include malposition of feet, asthenic habitus, hyporeflexia, bowel occlusions, hydronephrosis, ren arcuatus, delayed motor development and disturbed sleep-wake cycle.
Fraser et al. (1987) described 2 unrelated children with multiple malformations characteristic of the aminopterin syndrome, but without any evidence of exposure to aminopterin in the mothers. The first patient, a girl, had hydrocephaly, cleft lip/palate, and dysmorphic features, including hypertelorism, facial asymmetry, dolichocephaly, frontal bossing, unusual hair pattern, and hypoplastic right thumb. ... Krajewska-Walasek (1994) reported affected sibs. The first-born affected child, a girl, was delivered at 36 weeks' gestation with a weight at the 3rd percentile and a head circumference greater than the 97th percentile. ... Inheritance Krajewska-Walasek (1994) reported the first familial cases. Abnormalities in a brother and sister supported autosomal recessive inheritance, although parental consanguinity was excluded.
Pseudoaminopterin syndrome is a developmental anomalies syndrome that resembles the aminopterin embryopathy (see this term) without history of fetal exposure to aminopterin. It is characterized by skull (craniosynostosis and poorly mineralized cranial vault), dysmorphic (ocular hypertelorism, palpebral fissure anomalies, micrognathia cleft lip and/or high arched palate and small and low set/rotated ears) and limb (brachydactyly, syndactyly and clinodactyly) anomalies, associated with mild-to-moderate intellectual deficit and short stature.
Clinical Features Nieminen et al. (2011) studied 3 consanguineous families of Pakistani origin and 2 families of northern European origin in which affected children had craniosynostosis involving the metopic, coronal, sagittal, and/or lambdoid sutures, maxillary hypoplasia, and variable dental anomalies. In the first Pakistani family ('family 1'), 4 of 6 children were affected, whereas the parents and 2 daughters had normal craniofacial features with no history or signs of supernumerary tooth formation or delayed tooth eruption. ... Dental anomalies in this pedigree consisted of large lateral incisors seen in 2 affected children; additional anomalies included broad first toes with hallux valgus and mild syndactyly of the second and third toes. ... INHERITANCE - Autosomal recessive HEAD & NECK Head - Brachycephaly (in some patients) - Trigonocephaly (in some patients) - Turricephaly (in some patients) - Scaphocephaly (in some patients) - Sloping or flat forehead Face - Maxillary hypoplasia Eyes - Exorbitism - Papilledema - Hypertelorism (in some patients) Teeth - Malocclusion, class III - Delayed eruption - Ectopic eruption - Supernumerary teeth (in some patients) SKELETAL Skull - Metopic suture synostosis - Coronal suture synostosis - Sagittal suture synostosis - Lambdoid suture synostosis (in some patients) Hands - Syndactyly (in some patients) - Short phalanges (rare) Feet - Broad first toe (in some patients) - Hallux valgus (in some patients) - Syndactyly of second and third toes, mild (in some patients) - Clinodactyly (rare) SKIN, NAILS, & HAIR Nails - Dysplastic fingernails (rare) MISCELLANEOUS - Some patients have a Crouzon-like appearance MOLECULAR BASIS - Caused by mutation in the interleukin 11 receptor alpha gene (IL11RA, 600939.0001 ) ▲ Close
Craniosynostosis-dental anomalies is a rare, genetic, cranial malformation syndrome characterized by premature fusion of multiple or all calvarial sutures (resulting in variable abnormal shape of the head), midface hypoplasia, delayed and ectopic tooth eruption and supernumerary teeth. Associated facial dysmorphism includes proptosis, hypertelorism, beaked nose, and relative prognathism. Variable digital anomalies (e.g. finger and/or toe syndactyly, clinodactyly), short stature, cognitive and/or motor delay, high palate, ear deformity and conductive hearing loss have also been reported.
The disease typically affects boys , but it can also affect girls . It was first described in 1908 by Alban Köhler (1874–1947), a German radiologist . [2] [3] Dr. ... This means that the affected bone and the Tarsal Navicular begin to regain their normal size, density and structure. [6] Cause [ edit ] The cause of Köhler's disease, has thus far, been declared unknown by scientists. [6] However, it is suspected that possible causes may be caused by strain on the foot and the blood vessels associated with it, before the bone is fully able to grow into its adult form (ossification). [6] This bone ossification usually begins within the first 18 to 24 months of a female's life and the first 24 to 30 months of a male's life.
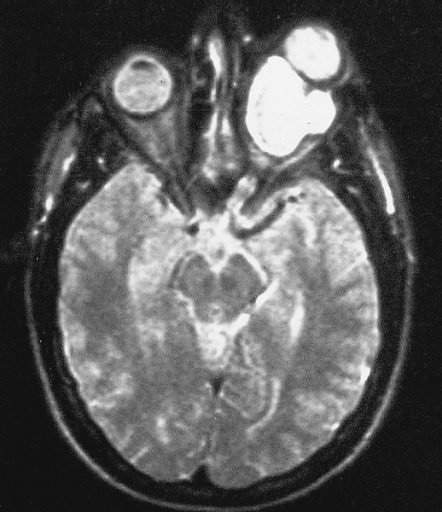
Optic nerve glioma Magnetic resonance image of a large retrobulbar optic nerve tumor causing massive proptosis Specialty Oncology Optic nerve glioma (or optic glioma ), a form of glioma which affects the optic nerve , is often one of the central nervous system manifestations of neurofibromatosis 1 . [1] [2] Optic gliomas are usually pilocytic tumors, and can involve the optic nerve or optic chiasm . [3] Optic gliomas are usually associated with neurofibromatosis type 1 in 30% of patients. [3] Optic nerve gliomas have low mortality but extremely high prevalence of vision loss & eye proptosis (exophthalmos) in children. [4] As of 2014, approximately 1000 cases have been reported so far. [4] Contents 1 Diagnosis 2 Treatment 3 Prognosis 4 References 5 External links Diagnosis [ edit ] Optic nerve gliomas are diagnosed using magnetic resonance imaging (MRI) and computed tomography (CT) scans. [5] The tumor adopts a fusiform appearance, appearing wider in the middle and tapered at the ends. [5] Enlargement of the optic nerve along with a downward kink in the mid-orbit is usually observed. [5] While CT scans allow for optic nerve evaluation, MRI allows for intracranial evaluation to observe if the tumor has extended to other regions such as the optic chiasm & hypothalamus. [6] Treatment [ edit ] Currently, the main goal of treating optic gliomas is to preserve the patient’s visions for as long as possible. [5] The tumor’s indolent & self-limiting growth indicates that it is not immediately problematic in most benign cases, with long-term studies showing that optic glioma patients may still have stable functional vision without intervention. [4] [6] [7] As a result, the first & preferred course of action is usually observation of the patient’s optic glioma over time. [5] [6] Once the first signs of visual deterioration and/or tumor progression are observed, interventional treatment will then commence. [6] These include radiation therapy, chemotherapy and/or surgical excision. ... ISBN 0-412-38920-7 . ^ Skelley, Tao Le, Vikas Bhushan, Nathan William (2012-03-12). First aid for the USMLE step 2 CK (8th ed.).
Odditorium" in Times Square in New York City . This is the first time they were billed as Lori and George Schappell. [3] Personal lives [ edit ] Born as Lori and Dori Schappell, they are craniopagus conjoined twins joined at the head, but having very different personalities and living—insofar as possible—individual lives. As a mark of individuality, and disliking the fact that their names rhymed, George first chose to go by the name Reba, after his favorite singer Reba McEntire . By 2007 he preferred to be publicly known as George. Lori and George spent the first 24 years of their lives living in an institution in Hamburg, Pennsylvania , in which the majority of patients were suffering from severe intellectual disabilities.
The injury can be sustained through either blunt forces , such as a motor vehicle accident , or penetrative forces , such as that of a gunshot wound . [1] The pancreas is one of the least commonly injured organs in abdominal trauma . [2] Contents 1 Management 1.1 Diagnosis 1.2 Classification 1.3 Surgical treatment 2 History 3 References 4 External links 5 External links Management [ edit ] Diagram of the pancreas, showing its relation to the duodenum and the mesenteric veins and arteries Diagnosis [ edit ] The diagnosis of this form of injury can be challenging because of the pancreas' location inside the abdomen. [3] The use of ultrasound can reveal fluid around the site of injury. [1] Computed tomography (CT) can also be utilized as a non-invasive diagnostic tool, [3] but its reliability is low; one retrospective case review found that computed tomography had either failed to find injuries or had underestimated the severity of injury in more than half of 17 pancreatic injury patients. [4] Serum amylase has also been shown to be of limited diagnostic utility within the first three hours following injury. [4] Management of a pancreatic injury can be difficult because other abdominal organs , such as the liver , usually have sustained trauma as well. [3] [5] Several common symptoms manifest hours after the injury such as tachycardia , abdominal distension , and midepigastric tenderness . [5] Indications for surgical intervention include: peritonitis based on physical examination ; hypotension in combination with a positive focussed assessment with sonography (ultrasound) for trauma (FAST); and pancreatic duct disruption based on the results of thin-cut computed tomography or endoscopic retrograde cholangiopancreatography (ERCP). [3] Commonly, a laparotomy is done in order to directly visualize the injury, and generally this approach is the most accurate diagnostic method. [1] [5] Classification [ edit ] 1: Head of pancreas 2: Uncinate process of pancreas 3: Pancreatic notch 4: Body of pancreas 5: Anterior surface of pancreas 6: Inferior surface of pancreas 7: Superior margin of pancreas 8: Anterior margin of pancreas 9: Inferior margin of pancreas 10: Omental tuber 11: Tail of pancreas 12: Duodenum Pancreatic injuries are classified according to the criteria of the American Association for the Surgery of Trauma (AAST). ... When injuries are not close to the mesenteric vessels, a distal pancreatectomy may be done; this procedure preserves much of the pancreas and usually avoids loss of its endocrine and exocrine functions. [4] In severe cases of pancreaticoduodenal injury, a pancreaticoduodenectomy can be used. [4] [7] Common complications after surgery include pancreatitis , pancreatic fistula , abscess , and pseudocyst formation. [2] Initial management of hemorrhage includes controlling it by packing the wound. [5] [7] History [ edit ] The first recorded case of pancreatic injury was published in The Lancet in 1827. [8] At the time, death as a result of injury was deemed to be "universal". [8] The first successful surgery to repair a transected pancreas was performed in 1904 by Garré, who reported the case the following year. [5] [9] [10] References [ edit ] ^ a b c d Degiannis E, Glapa M, Loukogeorgakis SP, Smith MD (January 2008).
Faioni et al. (2014) reported 27-year-old twins, a brother and sister, from a large Sardinian family, who had lifelong mucosal bleeding. The patients presented in the first years of life with epistaxis, gum bleeding, gastrointestinal bleeding, hematuria, and recurrent duodenal ulcers. ... Brooke et al. (2014) reported a brother and sister from a small region in Serbia with onset of severe gastrointestinal disease before 4 years of age. They first presented with peptic ulceration, bleeding, and pyloric stenosis, and both underwent vagotomy and eventually gastroenterostomy or gastrojejunostomy for disease recurrence. ... INHERITANCE - Autosomal recessive ABDOMEN Gastrointestinal - Mucosal ulceration, recurrent - Duodenal ulcers - Small bowel ulcers - Esophageal ulcers - Fistulas HEMATOLOGY - Impaired platelet aggregation due to impaired thromboxane-A2 production - Iron deficiency anemia, secondary LABORATORY ABNORMALITIES - Decreased serum eicosanoids - Decreased thromboxane-B2 (TxB2) - Decreased 12-HETE MISCELLANEOUS - Onset in first years of life MOLECULAR BASIS - Caused by mutation in the phospholipase A2, group IVA gene (PLA2G4A, 600522.0001 ) ▲ Close
A rare genetic hematologic and intestinal disease characterized by childhood onset of bleeding tendency with epistaxis, gum bleeding, gastrointestinal bleeding, hematuria, and menorrhagia due to impaired platelet aggregation and secretion, as well as recurrent gastrointestinal ulcera. Mildly reduced levels of coagulation factor XI have been reported in addition.
They performed nonparametric linkage analyses in 152 families segregating autism, focusing on 3 traits derived from the Autism Diagnostic Interview: 'age at first word,' 'age at first phrase,' and a composite measure of 'repetitive and stereotyped behaviors.' Using nonparametric multipoint linkage analysis, they found the strongest QTL evidence for the age at first word on 7q35-q36, and subsequent linkage analyses of additional markers and association analyses of the same region supported the initial result.
Tubulinopathies caused by mutation of TUBA1A , TUBB2A, TUBB2B , TUBB3 , TUBB ( TUBB5 ), or TUBG1 are inherited in an autosomal dominant manner. Most often a de novo heterozygous pathogenic variant is causative; however, in some families the pathogenic variant is inherited from an affected parent.
Mutations in connexin-26 (121011) provide another example of syndrome deafness and nonsyndromic deafness being caused by different alleles of the same gene: isolated autosomal recessive deafness (220290), autosomal dominant isolated deafness (601544), and keratoderma with deafness (148350).
Van Laer et al. (2008) noted that a frameshift mutation in the GRHL2 gene (608576.0001) had been associated with an autosomal dominant form of progressive nonsyndromic sensorineural hearing loss (DFNA28; 608641), and suggested that variation in the GRHL2 gene may change gene expression and influence susceptibility to age-related hearing impairment.
It can begin as early as a person's thirties or forties and worsens gradually over time. Age-related hearing loss first affects the ability to hear high-frequency sounds, such as speech.
Learn more about the gene associated with Central precocious puberty MKRN3 Additional Information from NCBI Gene: KISS1 KISS1R Inheritance Pattern Central precocious puberty follows an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder.
Precocious puberty is when a person's sexual and physical traits develop and mature earlier than normal. Normal puberty typically begins between ages 10 and 14 for girls, and ages 12 and 16 for boys. The start of puberty depends on various factors such as family history, nutrition and gender. The cause of precocious puberty is not always known. Some cases of precocious puberty are due to conditions that cause changes in the body's release of hormones. Treatment involves medications that can stop the release of sexual hormones.
Functional studies suggested that the failure of testis development in the French and Moroccan patients could be explained by the impaired ability of the mutant ZFPM2 proteins to interact with GATA4 (600576), a regulator of early testis development. INHERITANCE - Autosomal dominant GENITOURINARY External Genitalia (Male) - Ambiguous external genitalia - Genital tubercle present at birth - Striated genital folds External Genitalia (Female) - Hypertrophic labia majora - Fused labia minora Internal Genitalia (Male) - Partial or complete gonadal dysgenesis - Absence of germ cells on histology Internal Genitalia (Female) - Rudimentary vaginal cavity may be present - Rudimentary uterus may be present SKELETAL Hands - Bilateral fifth-finger clinodactyly (in some patients) NEUROLOGIC Central Nervous System - Learning and language difficulties (in some patients) - Subtentorial ventricular dilation (in some patients) Behavioral Psychiatric Manifestations - Autism spectrum disorder (in some patients) MISCELLANEOUS - Based on report of 2 probands (last curated October 2014) - Only 46,XY individuals are affected MOLECULAR BASIS - Caused by mutation in the zinc finger protein, multitype-2 gene (ZFPM2, 603693.0007 ) ▲ Close
Overall, the findings suggested that the mutations increased NFE2L2 levels in the absence of stress and caused constitutive chronic activation of stress response genes, consistent with a gain-of-function effect. INHERITANCE - Autosomal dominant GROWTH Height - Short stature Other - Failure to thrive - Poor growth CARDIOVASCULAR Heart - Congenital heart defects (in some patients) - Atrial septal defect (in some patients) - Cardiomyopathy, (in 1 patient) - Thickened bicuspid aortic valve (in 1 patient) RESPIRATORY - Recurrent respiratory infections SKIN, NAILS, & HAIR Skin - Recurrent skin infections NEUROLOGIC Central Nervous System - Delayed development, mild - Intellectual disability, mild - Speech delay - Learning disabilities - Periventricular and subcortical T2-weighted white matter abnormalities seen on brain imaging - Leukoencephalopathy IMMUNOLOGY - Immunodeficiency - Recurrent infections - Hypogammaglobulinemia - Decreased memory switched B cells LABORATORY ABNORMALITIES - Hypohomocysteinemia - Decreased creatine - Decreased plasma cysteine MISCELLANEOUS - Onset in infancy - De novo mutation - Four unrelated patients have been reported (last curated October 2017) MOLECULAR BASIS - Caused by mutation in the nuclear factor erythroid 2-like 2 gene (NFE2L2, 600492.0001 ) ▲ Close
The parents were nonconsanguineous, and the mother and father were 23 and 27 years old, respectively, at the birth of their first child. Buntincx et al. (1993) reported a female child with the same syndrome.
A very rare syndrome of congenital hypothyroidism characterized by thyroid dysgenesis (in most cases athyreosis), cleft palate and spiky hair, with or without choanal atresia, and bifid epiglottis. Facial dysmorphism and porencephaly have been reported in isolated cases. Epidemiology Only 8 patients from 6 families have been reported to date. Clinical description The syndrome is typically observed at birth with cleft palate, spiky hair and thyroid dysgenesis (in most cases athyreosis) leading to congenital hypothyroidism that manifests with lethargy, poor feeding, macroglossia, cold or mottled skin, persistent jaundice, and umbilical hernia. Neonatal hyperbilirubinemia is also common. Some may also present with choanal atresia and bifid epiglottis.
Inheritance Baughman (1971) suggested that this disorder is autosomal dominant; however, based on additional pedigree data obtained after the original report, Valdmanis et al. (1977) and Toriello et al. (1979) concluded that autosomal recessive inheritance is more likely, with an instance of pseudodominance as a result of multiple consanguineous matings in the family.
A rare ectodermal dysplasia syndrome characterized by the association of sparse, woolly, curly hair, ankyloblepharon, and nail dysplasia. Additional reported features include abnormal oral frenula, bifid tongue, lip pits, adhesions between upper and lower lips, hypertelorism and flat nasal bridge, alveolar synechia, and imperforate vagina.
Antenatal diagnosis Prenatal diagnosis can be made in first-trimester ultrasonography by detection of multiple thoraco-abdominal defects, including EC and CNS anomalies.
Epidemiology [ edit ] Pentalogy of Cantrell occurs in 1/65,000 to 1/200,000 live births. [2] History [ edit ] It was first characterized in 1958. [4] A 2010 study concluded that the 13th-century Christian saint Rose of Viterbo died of complications of Pentalogy of Cantrell. [5] References [ edit ] ^ Katranci AO, Görk AS, Rizalar R, et al. (1998).
Pentalogy of Cantrell is a condition characterized by a combination of midline birth defects that can potentially involve the breastbone (sternum); the muscle that separates the chest cavity from the abdomen and aids in breathing ( diaphragm ); the thin membrane that lines the heart (pericardium); the abdominal wall; and the heart. It can have varying degrees of severity, and can be lethal. Most affected infants do not have all potential defects (incomplete pentalogy of Cantrell). The exact cause of the condition is not known. Most cases occur sporadically, but familial cases have been reported. Treatment is based on the symptoms present in each affected person.