. ^ a b c Caplan, Arthur; McCartney, James; Sisti, Dominic (2004). Health, disease, and illness: concepts in medicine . ... Reprinted in DeBow's Review XI (1851). Available at Google Books and excerpted at PBS.org . ... Reprinted in Arthur L. Caplan, James J. McCartney, Dominic A. Sisti, eds, Health, Disease, and Illness: Concepts in Medicine (Washington, D.C.: Georgetown University Press , 2004) ISBN 1-58901-014-0 External links [ edit ] Wikiquote has quotations related to: Drapetomania An Early History – African American Mental Health Findlay, James A. (2000).
The patients had onset of variable myoclonic seizures in the first decade of life (between 1 and 7 years). ... Molecular Genetics In 5 unrelated patients with DEDSM, Hamdan et al. (2017) identified 2 different de novo heterozygous missense mutations in the DHDDS gene (R37H, 608172.0002 and R211Q, 608172.0003). The mutations were found by whole-exome or whole-genome sequencing of several cohorts of patients with developmental delay and epilepsy. Studies of patient cells and functional studies of the variants were not performed, but Hamdan et al. (2017) postulated a gain-of-function or dominant-negative effect. INHERITANCE - Autosomal dominant GROWTH Height - Short stature (in some patients) MUSCLE, SOFT TISSUES - Hypotonia NEUROLOGIC Central Nervous System - Global developmental delay - Seizures, multiple types - Myoclonic seizures - EEG abnormalities - Intellectual disability - Speech delay - Involuntary movements - Ataxia - Tremor - Dystonia - Bradykinesia - Rigidity MISCELLANEOUS - Seizure onset in first decade - Variable severity - Seizures tend to be difficult to control - Mutations occur de novo - Five unrelated patients have been reported (last curated January 2018) MOLECULAR BASIS - Caused by mutation in the dehydrodolichyl diphosphate synthase gene (DHDDS, 608172.0002 ) ▲ Close
Inheritance The transmission pattern of FTDALS4 in the families reported by Freischmidt et al. (2015) was consistent with autosomal dominant inheritance and incomplete penetrance. Pottier et al. (2015) reported a deceased patient (case B) showing digenic inheritance of a neurodegenerative disorder: whole-genome sequencing identified heterozygous mutations in the OPTN (602432; G538EfsX27) and TBK1 (R117X) genes. ... Molecular Genetics Cirulli et al. (2015) performed whole-exome sequencing of 2,869 ALS patients and 6,405 controls. ... In affected members from 13 European Caucasian families with FTDALS4, Freischmidt et al. (2015) identified 8 different heterozygous loss-of-function mutations in the TBK1 gene (see, e.g., 604834.0001-604834.0005). Mutations in the first 9 families were found by whole-exome sequencing of 252 patients with ALS, and accounted for about 4% of cases overall. ... In 3 unrelated patients with FTDALS4, Pottier et al. (2015) identified heterozygous missense mutations in the TBK1 gene (see, e.g., 604834.0006 and 604834.0007). The mutations were found by whole-genome sequencing of 107 deceased patients with FTD.
Description Frontotemporal dementia (FTD) and/or amyotrophic lateral sclerosis (ALS) is an autosomal dominant neurodegenerative disorder characterized by adult onset of one or both of these features in an affected individual, with significant intrafamilial variation. ... Age of onset ranged from 40 to 72 years with a mean survival of 3 years. Inheritance was autosomal dominant with reduced penetrance. Three individuals who presented with motor symptoms of ALS subsequently developed personality and behavioral changes, including apathy, social isolation, emotional lability, and hallucinations. ... Molecular Genetics In affected members of large families with autosomal dominant frontotemporal dementia and/or amyotrophic lateral sclerosis (FTD/ALS) mapping to chromosome 9p21, DeJesus-Hernandez et al. (2011) and Renton et al. (2011) simultaneously and independently identified a heterozygous expanded hexanucleotide repeat (GGGGCC) located between the noncoding exons 1a and 1b of the C9ORF72 gene (614260.0001). ... These results revealed a pathogenic consequence of decreased C9orf72 levels, supporting a loss of function mechanism of disease. INHERITANCE - Autosomal dominant HEAD & NECK Eyes - Supranuclear gaze palsy (less common) MUSCLE, SOFT TISSUES - Muscle weakness - Muscle atrophy NEUROLOGIC Central Nervous System - Frontotemporal deme
Inheritance The transmission pattern of FTDALS2 in the families reported by Bannwarth et al. (2014) was consistent with autosomal dominant inheritance. Molecular Genetics In affected members of a family with FTDALS in whom mutations in C9ORF72 (614260) and other candidate genes were excluded, Bannwarth et al. (2014) identified a heterozygous missense mutation in the CHCHD10 gene (S59L; 615903.0001). The mutation was found by whole-exome sequencing and segregated with the disorder in the family. ... The findings implicated a role for dysfunctional mitochondria in the pathogenesis of late-onset frontotemporal dementia with motor neuron disease. INHERITANCE - Autosomal dominant HEAD & NECK Ears - Sensorineural deafness (in some patients) Eyes - Ptosis (uncommon) ABDOMEN Gastrointestinal - Dysphagia MUSCLE, SOFT TISSUES - Proximal muscle weakness (in some patients) - Ragged red fibers seen on biopsy - Cytochrome c oxidase deficiency - Lipid and glycogen accumulation - Mitochondrial DNA deletions - Abnormal mitochondria morphology - Fragmented mitochondrial network - Paracrystalline inclusions - Combined mitochondrial respiratory chain deficiency (in most patients) NEUROLOGIC Central Nervous System - Cerebellar ataxia - Dysarthria - Bulbar weakness - Frontal lobe dementia - Motor neuron disease - Extensor plantar responses - Parkinsonism (less common) - Cortical atrophy (in some patients) Peripheral Nervous System - Hyporeflexia - Areflexia MISCELLANEOUS - Two unrelated families have been reported (last curated July 2014) - Late-adult onset (usually after age 50 years) - Progressive disorder MOLECULAR BASIS - Caused by mutation in the coiled-coil-helix-coiled-coil helix domain-containing protein 10 gene (CHCHD10, 615903.0001 ) ▲ Close
Frontotemporal dementia with motor neuron disease (FTD-MND) is a type of frontotemporal lobar degeneration characterized by the insidious onset (between the ages of 38-78 years) of dementia-associated psychiatric symptoms (e.g. personality changes, uninhibited behavior, irritability, aggressiveness), memory difficulties, global intellectual impairment, emotional disorders and transcortical motor aphasia that eventually leads to mutism, in addition to the manifestations of motor neuron disease such as neurogenic muscular wasting (similar to what is seen in amyotrophic lateral sclerosis; see this term). The disease is progressive, with death occurring 2-5 years after onset.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( March 2017 ) Hypertension and brachydactyly syndrome Other names Brachydactyly-arterial hypertension syndrome This condition is inherited in an autosomal dominant manner. Hypertension and brachydactyly syndrome (HTNB) also known as Bilginturan syndrome and brachydactyly type E among others is a very rare genetic disorder. [1] [2] It was first reported in 1973 by N. Bilginturan et al. [3] [4] The estimated prevalence is less than 1 out of 1,000,000. [2] Contents 1 Symptoms 2 Genetics 3 Treatment 4 References 5 External links Symptoms [ edit ] The disorder is characterized by: severe salt-independent but age-dependent hypertension brachydactyly malformations of the hands and fingers increased fibroblast growth rate neurovascular contact at the rostral-ventrolateral medulla altered baroreflex blood pressure regulation death from stroke before age 50 years when untreated Genetics [ edit ] The disorder is thought to be related to mutations in the PDE3A gene. [1] [5] Treatment [ edit ] This section is empty. ... "PDE3A mutations cause autosomal dominant hypertension with brachydactyly" (PDF) .
Chitayat et al. (1997) reported 2 families with this disorder. All affected members of the first family had proportionate short stature, whereas the propositus and the affected relatives in the second family were only short compared to unaffected relatives. ... In a review of the clinical and genetic features of autosomal dominant hypertension and brachydactyly, Luft et al. (2003) noted that magnetic resonance angiographic studies in 15 hypertensive affected and 12 normotensive unaffected members of the Turkish family originally described by Bilginturan et al. (1973) demonstrated neurovascular contact in all 15 affected individuals and none of the unaffected individuals (Naraghi et al., 1997). ... By genomewide parametric linkage analysis, Gong et al. (2003) identified a locus for primary hypertension on chromosome 12p (HYT4; 608742) that overlaps the locus associated with severe autosomal dominant hypertension and brachydactyly. ... In the Turkish family with HTNB mapping to chromosome 12p12.2-p11.2, originally reported by Bilginturan et al. (1973), Maass et al. (2015) performed whole-exome sequencing and identified a heterozygous missense mutation in the PDE3A gene (T445N; 123805.0001). ... Maass et al. (2015) suggested that these mutations cause hypertension by contributing to a general increase in peripheral vascular resistance. INHERITANCE - Autosomal dominant GROWTH Height - Short stature CARDIOVASCULAR Vascular - Severe hypertension, salt-independent - Altered baroreflex blood pressure regulation SKELETAL Hands - Brachydactyly, type E - Thickening of metacarpal bones - Shortening of metacarpal bones - Thickening of phalangeal bones - Shortening of phalangeal bones - Cone-shaped epiphyses (in some patients) Feet - Thickening of metatarsal bones - Shortening of metatarsal bones - Thickening of phalangeal bones - Shortening of phalangeal bones - Fusion of middle and distal phalanges of fifth toes (in some patients) NEUROLOGIC Central Nervous System - Neurovascular contact at the rostral-ventrolateral medulla - Altered baroreflex blood pressure regulation MISCELLANEOUS - Severe hypertension develops in childhood - Increasing hypertension with increasing age - By age 50 years, affected family members have a 50mm Hg increase in mean arterial blood pressure compared to unaffected relatives - Death from stroke if untreated MOLECULAR BASIS - Caused by mutation in the cGMP-inhibited phosphodiesterase-3A gene (PDE3A, 123805.0001 ) ▲ Close
Brachydactyly - arterial hypertension is a rare genetic brachydactyly syndrome characterized by the association of brachydactyly type E (see this term) with hypertension (due to vascular or neurovascular anomalies) as well as the additional features of short stature and low birth weight (compared to non-affected family members), stocky build and a round face. The onset of hypertension is often in childhood and, if untreated, most patients will have had a stroke by the age of 50.
Genetically, myotubular myopathy can have two causes: autosomal dominant and autosomal recessive. When caused by a mutation in the DNM2 gene, the disorder is autosomal dominant, meaning it can be passed on by one mutated gene. ... Autosomal recessive onset is most common. [9] Central core disease [ edit ] Central core disease or central core myopathy was first described in 1956 [10] and usually presents in infancy or early childhood as non-progressive mild proximal weakness that persists throughout life. ... S2CID 5490760 . ^ "Congenital Myopathy Information Page" . National Institute of Neurological Disorders and Stroke . ... PMID 8198769 . ^ Chevessier, F; Marty, I; Paturneau-Jouas, M; Hantaı̈, D; Verdière-Sahuqué, M (2004). "Tubular aggregates are from whole sarcoplasmic reticulum origin: Alterations in calcium binding protein expression in mouse skeletal muscle during aging". ... PMID 7436360 . ^ Taratuto, AL; Matteucci, M; Barreiro, C; Saccolitti, M; Sevlever, G (1991). "Autosomal dominant neuromuscular disease with cylindrical spirals".
One such film, Terrifying Girls' High School: Lynch Law Classroom in 1973, would be the first to depict an omorashi scenario to a cinematic audience. ... However, these did not have the erotic context which characterizes modern omorashi media, since they predated the first full-blown anime pornography, which was not available until 1984, when the advent of the first H anime OVAs such as Wonder Kids' Lolita Anime were made possible by the widespread availability of home video . ... In New Zealand for example, creating, trading, distributing (e.g., making available on one's web page ) anything promoting or supporting "the use of urine or excrement in association with degrading or dehumanising conduct or sexual conduct" is a felony punishable by up to ten years in jail. [23] [24] Nonetheless, urination is a common fetish in pornography; the 1915 film A Free Ride , widely considered the first pornographic film made in the US, contains scenes of urination. ... Archived from the original on October 31, 2007 . Retrieved July 4, 2014 . ^ "Googlepage" . Retrieved September 16, 2014 . ^ "Information about: MAID iN HEAVEN SuperS / めいどいんへぶんすーぱーす" . ... New York: Atria. pp. 115–142 . ISBN 0-671-00957-5 . ^ "World's first incontinence lingerie collection hits the runway" .
Ears and leaves dry up and sometimes look like a mature plant. The whole plant dies and maize cobs remain without kernels. ... To learn how to add open license text to Wikipedia articles, please see this how-to page . For information on reusing text from Wikipedia , please see the terms of use . ... To learn how to add open license text to Wikipedia articles, please see this how-to page . For information on reusing text from Wikipedia , please see the terms of use . ... To learn how to add open license text to Wikipedia articles, please see this how-to page . For information on reusing text from Wikipedia , please see the terms of use . ... To learn how to add open license text to Wikipedia articles, please see this how-to page . For information on reusing text from Wikipedia , please see the terms of use .
Because the maternal allele is not normally expressed, the findings were consistent with a loss of MAGEL2 function. The mutation in the first patient was found by clinical whole-exome sequencing. Based on these results, a research database of 1,248 whole-exome sequencing cases were reviewed, and the 3 remaining cases were identified. In 2 sisters with Schaaf-Yang syndrome, Soden et al. (2014) identified a heterozygous truncating mutation in the MAGEL2 gene (605283.0005). The mutation was found by whole-genome sequencing and apparently resulted from gonadal mosaicism; the mutation was missed by initial whole-exome sequencing. The patients were part of a larger cohort of 100 families with neurodevelopmental disorders who underwent whole-exome or whole-genome sequencing. ... The patients were ascertained based on genotype from whole-exome or direct Sanger sequencing through multiple research-based centers or laboratories.
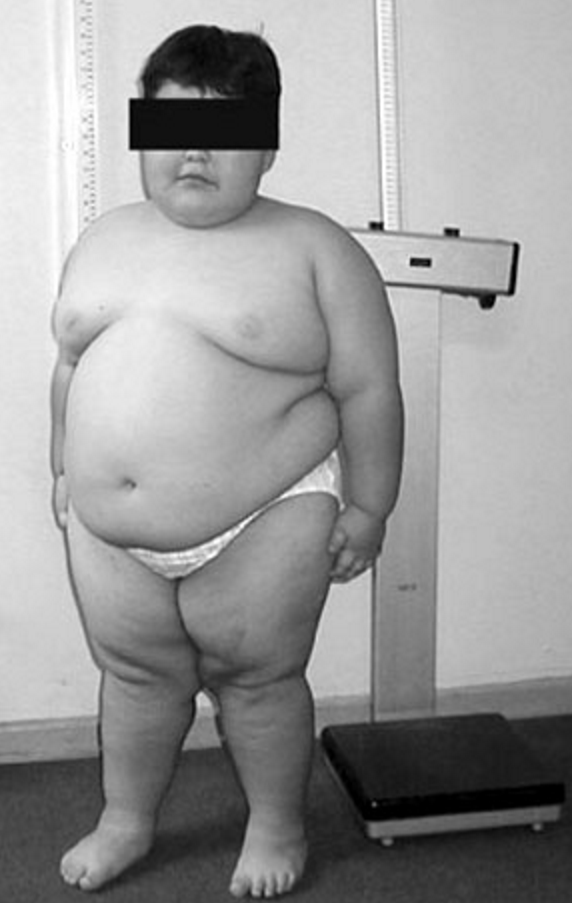
., SNRPN ). FISH does not query the whole AS/PWS region and will miss small deletions. ... Same as CMA; in addition, use of small nucleotide polymorphisms (SNP) will allow detection of UPD in cases with long stretches of homozygosity. 7. Not a first line test; performed after DNA methylation analysis diagnoses PWS, but FISH or CMA analysis indicates disomy. 8. ... Cohen syndrome, characterized by failure to thrive in infancy and childhood; truncal obesity in the teen years; early-onset hypotonia and developmental delays; microcephaly developing during the first year of life; moderate to profound psychomotor retardation; progressive retinochoroidal dystrophy and high myopia; neutropenia in many with recurrent infections and aphthous ulcers in some; a cheerful disposition; joint hypermobility; and characteristic facial features which are different from PWS. ... Special feeding techniques, including special nipples or gavage feeding, may be necessary for the first weeks to months of life to assure adequate nutrition and avoid failure to thrive. ... To assure appropriateness of exercise program and diet, including adequacy of vitamin and mineral intake and monitor height, weight, and BMI (weight in kg/height in m 2 ): Every month in infancy; Every six months in the first decade of life; At least annually thereafter.
Managing eating problems, behavior and medical issues can impact the whole family. Some options for coping and support can include: Talking to a mental health professional.
Prader-Willi syndrome (PWS) is a genetic condition that affects many parts of the body. Infants with PWS have severe hypotonia (low muscle tone), feeding difficulties, and slow growth. In later infancy or early childhood, affected children typically begin to eat excessively and become obese . Other signs and symptoms often include short stature, hypogonadism, developmental delays, cognitive impairment, and distinctive behavioral characteristics such as temper tantrums, stubbornness, and obsessive-compulsive tendencies. PWS is caused by missing or non-working genes on chromosome 15 . Most cases are not inherited and occur randomly.
While PWS results from the loss of PW genes within this region on the paternal chromosome, loss of a different gene ( UBE3A ) within the same region on the maternal chromosome causes AS. [32] PWS and AS represent the first reported instances of disorders related to imprinting in humans. ... Retrieved August 20, 2016 . ^ Teitelbaum, Jonathan E. (2007). In a Page: Pediatrics . Lippincott Williams & Wilkins. p. 330.
Genetic counseling Most cases are sporadic; however, in rare cases dominant transmission may occur with 50% risk where the father carries the imprinting defect.
Prader-Willi syndrome is a complex genetic condition that affects many parts of the body. In infancy, this condition is characterized by weak muscle tone (hypotonia), feeding difficulties, poor growth, and delayed development. Beginning in childhood, affected individuals develop an insatiable appetite, which leads to chronic overeating (hyperphagia) and obesity. Some people with Prader-Willi syndrome, particularly those with obesity, also develop type 2 diabetes (the most common form of diabetes). People with Prader-Willi syndrome typically have mild to moderate intellectual impairment and learning disabilities.
Hall and Smith (1972) reported 2 affected male maternal first cousins. One was of normal stature and intelligence. ... Clarren and Smith (1977) reported affected sibs and affected first cousins. They found a recurrence risk of 1.6% in sibs of probands. ... Clarren and Smith (1977) reported affected sibs and first cousins. They found a recurrence risk of 1.6% in sibs of probands. ... Although there were no major clinical differences between the 2 classes of patients analyzed as a whole, mean maternal and paternal age were significantly higher in the UPD patients. ... Multiple Affected Relatives There are several mechanisms that explain the simultaneous occurrence of affected first- and second-degree relatives in PWS families.
This article is an orphan , as no other articles link to it . Please introduce links to this page from related articles ; try the Find link tool for suggestions. ( September 2015 ) Tuberculous dactylitis Specialty Infectious disease Tuberculous dactylitis is a skeletal manifestation of tuberculosis , one of the commonest bacterial osteitis . It affects children more often than adults. [1] The first radiological description of the condition is credited to Feilchenfeld in 1896; however, the first histological description was given by Rankin in 1886. [2] The Swedish botanist and fysician Carl von Linne was the first to mention the condition by the name "spina ventosa" 1746 in his "Västgöta resa". [3] Multiple bones are involved in children and usually only a single bone is involved in adults suffering from tuberculous dactylitis. [1] Tuberculous dactylitis affects the short tubular bones of the hands and feet in children. ... Proximal phalanx of the index and middle fingers are the commonest sites of involvement.Up to nearly 7% of children with pulmonary tuberculosis may develop this condition. [8] Spread to the skeletal system is believed to occur via blood and lymphatics. [9] Contents 1 Pathogenesis 2 Diagnosis 3 References 4 External links Pathogenesis [ edit ] In the pediatric age group, the marrow in the phalangeal bones are still active, a conducive place for the tuberculous bacilli to multiply. Slowly, the whole marrow space gets involved and this underlying granulomatous disease leads to expansion of the overlying soft cortex.
A number sign (#) is used with this entry because of evidence that autosomal dominant dyskeratosis congenita-6 (DKCA6) and autosomal recessive dyskeratosis congenita-7 (DKCB7) are caused by heterozygous and compound heterozygous mutation, respectively, in the ACD gene (609377) on chromosome 16q22. ... He was one of identical twin boys; the other twin died at age 4 months from complications of pertussis infection. During the first year of life, the proband was noted to have developmental delay, microcephaly, oral leukoplakia, nail dystrophy, esophageal stenosis, and cerebellar hypoplasia. ... Laboratory studies showed extremely shortened telomeres (much less than first percentile in length). The proband's older sister had no clinical features of DKC, but had very short telomeres. ... Inheritance The transmission pattern of DKCA6 in the family reported by Guo et al. (2014) was consistent with autosomal dominant inheritance. The transmission pattern of DKCB7 in the family reported by Kocak et al. (2014) was consistent with autosomal recessive inheritance. ... Dyskeratosis Congenita 7, Autosomal Recessive In a boy with DKCB7, Kocak et al. (2014) identified compound heterozygous mutations in the ACD gene: K170del and P491T (609377.0002). The mutations were found by whole-exome sequencing and confirmed by Sanger sequencing.
Hoyeraal–Hreidarsson syndrome Other names Progressive pancytopenia-immunodeficiency-cerebellar hypoplasia syndrome [1] This condition is inherited in an X-linked recessive manner. Specialty Medical genetics Hoyeraal–Hreidasson syndrome [2] ) is a very rare multisystem X-linked recessive disorder characterized by excessively short telomeres and is considered a severe form of dyskeratosis congenita . [2] [3] Being an X-linked disorder, Hoyeraal–Hreidasson syndrome primarily affects males. Patients typically present in early childhood with cerebellar hypoplasia , immunodeficiency , progressive bone marrow failure , and intrauterine growth restriction . [2] The primary cause of death in Hoyeraal–Hreidasson syndrome is bone marrow failure, but mortality from cancer and pulmonary fibrosis is also significant. [4] [5] [6] Contents 1 Presentation 1.1 Overlap with dyskeratosis congenita 2 Pathogenesis 3 Diagnosis 4 Treatment 5 See also 6 References 7 External links Presentation [ edit ] The currently recognized features are cerebellar hypoplasia , immunodeficiency , progressive bone marrow failure , and intrauterine growth restriction . Patients also commonly exhibit symptoms such as microcephaly , aplastic anemia , and intellectual disability . [3] Overlap with dyskeratosis congenita [ edit ] Patients with Hoyeraal–Hreidasson syndrome frequently present with the mucocutaneous triad of nail dysplasia , lacy skin pigmentation , and oral leukoplakia . [ citation needed ] Pathogenesis [ edit ] Although the pathogenesis remains unknown, it is strongly suspected that the clinical sequelae of Hoyeraal–Hreidasson syndrome arise from the accelerated telomere shortening. [2] It has been associated with mutations in the poly(A)-specific ribonuclease poly(A)-specific ribonuclease gene. [7] Diagnosis [ edit ] Neuroimaging – cerebellar hypoplasia /atrophy, small brainstem, thin corpus callosum and cerebral calcifications Molecular genetic testing – for confirmation Treatment [ edit ] Current treatment is supportive: [ citation needed ] The aplastic anemia and immunodeficiency can be treated by bone marrow transplantation. Supportive treatment for gastrointestinal complications and infections.
Patient cells showed deficiencies in trimming of specific small nucleolar RNAs (snoRNAs), abnormal ribosomal assembly, abnormally adenylated TERC (602322), and short telomeres (less than the first percentile of controls). The patient had severe bone marrow failure with decreased numbers of CD34+ hematopoietic progenitor cells; patient fibroblasts also showed growth defects. ... The mutations, which were found by whole-exome sequencing, segregated with the disorder in the families.
A number sign (#) is used with this entry because autosomal dominant dyskeratosis congenita-3 (DKCA3) is caused by heterozygous mutation in the TINF2 gene (604319) on chromosome 14q12. ... Clinical Features Savage et al. (2008) reported a family with autosomal dominant DKC affecting 6 individuals, including 2 monozygotic male twins. ... Telomere flow-FISH analysis showed telomere lengths below the first percentile in all white blood cell subsets tested. ... Molecular Genetics Using a candidate gene approach with evidence for linkage to chromosome 14q11.2, Savage et al. (2008) identified a heterozygous missense mutation in the TINF2 gene (K280Q; 604319.0001) as the cause of dyskeratosis congenita in a family with autosomal dominant inheritance. The mutation was present only in individuals with telomere lengths below the first percentile for age. ... These findings indicated that disrupted TERF1 binding is not the main factor driving disease pathogenesis, but may contribute to a more severe phenotype. INHERITANCE - Autosomal dominant GROWTH Height - Short stature Other - Intrauterine growth retardation - Poor growth HEAD & NECK Head - Microcephaly Ears - Deafness Eyes - Blockage of the lacrimal ducts - Epiphora - Retinopathy Mouth - Oral leukoplakia Teeth - Tooth loss RESPIRATORY Lung - Pulmonary fibrosis - Pulmonary failure GENITOURINARY Internal Genitalia (Male) - Cryptorchidism SKELETAL - Osteoporosis Limbs - Avascular necrosis of the hip (2 patients) SKIN, NAILS, & HAIR Skin - Reticular pigmentation pattern - Leukoplakia - Dry skin Nails - Dysplastic nails Hair - Premature greying - Short, fine hair - Alopecia NEUROLOGIC Central Nervous System - Speech delay - Learning difficulties - Intracranial calcifications - Cerebellar hypoplasia - Cerebellar ataxia HEMATOLOGY - Bone marrow failure - Pancytopenia - Aplastic anemia - Thrombocytopenia - Leukopenia - Increased fetal hemoglobin NEOPLASIA - Increased risk of malignancy LABORATORY ABNORMALITIES - Shortened telomeres - Decreased telomerase activity MISCELLANEOUS - Highly variable phenotype and severity, even within families - Age at onset ranges from childhood to adulthood - Phenotypic overlap with Revesz syndrome ( 268130 ) MOLECULAR BASIS - Caused by mutation in the TRF1-interacting nuclear factor 2 gene (TINF2, 604319.0001 ) ▲ Close
An X-linked syndromic intellectual disability considered to be a severe variant of dyskeratosis congenita characterized by intrauterine growth retardation, microcephaly, cerebellar hypoplasia, progressive combined immune deficiency and aplastic anemia. Epidemiology Hoyeraal-Hreidarsson syndrome (HHS) prevalence is unknown. The syndrome may be underdiagnosed due to high mortality rates. Clinical description The disease generally presents in early childhood and primarily affects males. Growth retardation is usually of prenatal onset. Other clinical manifestations include microcephaly, mucocutaneous lesions (hyperpigmentation, nail dystrophy, premalignant leukoplakia affecting oral and gastrointestinal mucosa), early onset bone marrow failure, immunodeficiency and pancytopenia. Cancer predisposition is also reported. Etiology HSS is caused by mutations in the DKC1 gene (Xq28), encoding the nucleolar protein dyskerin which interacts with the human telomerase RNA complex.
Heterozygous mutation in the RTEL1 gene can cause autosomal dominant dyskeratosis congenita-4 (DKCA4). ... Ballew et al. (2013) reported a family in which DKCB5/HHS was transmitted in an autosomal dominant pattern with evidence of genetic anticipation. ... The mutations, which were found by whole-genome linkage analysis combined with whole-exome sequencing and confirmed by Sanger sequencing, segregated with the disorder in the families. ... Autosomal Dominant Dyskeratosis Congenita 4 In 2 brothers with severe autosomal dominant dyskeratosis congenita manifest as Hoyeraal-Hreidarsson syndrome, Ballew et al. (2013) identified a heterozygous mutation in the RTEL1 gene (R1010X; 608833.0012). The mutation, which was identified by whole-exome sequencing, was also present in the clinically unaffected mother, who had shortened telomeres.
A number sign (#) is used with this entry because autosomal dominant dyskeratosis congenita-2 (DKCA2) and autosomal recessive dyskeratosis congenita-4 (DKCB4) are caused by heterozygous and homozygous or compound heterozygous mutation, respectively, in the TERT gene (187270) on chromosome 5p15. ... Clinical Features Armanios et al. (2005) reported a 3-generation family in which at least 6 members had autosomal dominant dyskeratosis congenita without skin manifestations. ... Basel-Vanagaite et al. (2008) reported an Iraqi Jewish family with autosomal dominant dyskeratosis congenita-2. Affected males presented with thrombocytopenia, and later developed aplastic anemia, premature graying of the hair, and pulmonary and hepatic fibrosis. ... Molecular Genetics In all 6 affected members of a family with autosomal dominant dyskeratosis congenita, Armanios et al. (2005) identified a heterozygous mutation in the TERT gene (K902N; 187270.0007). ... Autosomal Recessive Dyskeratosis Congenita 4 Marrone et al. (2007) identified homozygous TERT mutations (187270.0012 and 187270.0013) in patients with a severe form of autosomal recessive dyskeratosis congenita-4. INHERITANCE - Autosomal dominant - Autosomal recessive GROWTH Height - Short stature Other - Failure to thrive (seen in recessive form) HEAD & NECK Head - Microcephaly (seen in recessive form) Mouth - Leukoplakia (seen in recessive form) - Bluish discoloration of the tongue (seen in recessive form) Teeth - Abnormal dentition - Tooth loss (seen in recessive form) CARDIOVASCULAR Heart - Cardiac fibrosis - Dilated cardiomyopathy RESPIRATORY Lung - Pulmonary fibrosis ABDOMEN Liver - Liver fibrosis Gastrointestinal - Esophageal stricture (seen in recessive form) - Chronic diarrhea (seen in recessive form) SKELETAL - Osteoporosis Limbs - Avascular necrosis of the hip (seen in recessive form) SKIN, NAILS, & HAIR Skin - Reticulated pigmentation (seen in recessive form) - Leukoplakia (seen in recessive form) - Hyperkeratosis of the palms (seen in recessive form) Nails - Nail dystrophy (seen in recessive form) Hair - Gray forelock - Premature graying NEUROLOGIC Central Nervous System - Learning difficulties (seen in recessive form) - Developmental delay (seen in recessive form) - Cerebellar hypoplasia (seen in recessive form) HEMATOLOGY - Bone marrow failure - Pancytopenia - Aplastic anemia - Thrombocytopenia - Leukopenia LABORATORY ABNORMALITIES - Shortened telomeres - Decreased telomerase activity MISCELLANEOUS - Two autosomal dominant families have been reported (as of May 2011) - Highly variable phenotype and severity, even within families - Variable penetrance and expressivity - Age at onset ranges from childhood to adulthood - Genetic anticipation - Patients with the autosomal recessive disorder have a more severe phenotype MOLECULAR BASIS - Caused by mutation in the telomerase reverse transcriptase gene (TERT, 187270.0007 ) ▲ Close
Merchant et al. (1998) described the changes of chronic keratoconjunctivitis in a case of congenital dyskeratosis thought to be autosomal dominant with 'variable penetration.' In general, the disorder is more likely to be X-linked, and the family history suggested that this was the case with partial expression in heterozygous females. ... Patients with Hoyeraal-Hreidarsson syndrome show only growth retardation and microcephaly in the first months of life, whereas those with the pseudo-TORCH syndrome have symptoms resembling TORCH infection shortly after birth, including hepatosplenomegaly. Furthermore, in the intrauterine infection-like syndrome, the neonatally present thrombocytopenia resolves within a year if the child survives, whereas in the Hoyeraal-Hreidarsson syndrome the first symptoms of pancytopenia do not occur before the age of 5 months and continue to increase for years. ... The pattern of methylation of HpaII and HhaI sites with a highly polymorphic CAG repeat in the coding region of the first exon of the androgen receptor gene (AR; 313700) (Allen et al., 1992) was used in these studies. Ferraris et al. (1997) studied 2 families and found that indeed carrier females showed nonrandom X inactivation in whole blood leukocytes, granulocytes, and mononuclear cells.
Description Hypomyelinating leukodystrophy-16 is an autosomal dominant neurologic disorder characterized by onset of hypotonia, nystagmus, and mildly delayed motor development in infancy. ... The patients had onset of nystagmus and hypotonia in the first days or weeks of life, but were alive at ages 19, 5, 38, and 26 years, respectively. ... The mutation, which was identified by trio-based whole-exome or whole-genome sequencing, was not found in the dbSNP, ExAC, or gnomAD databases. ... Yan et al. (2018) identified a de novo heterozygous D252N mutation in the TMEM106B gene in a 3-year-old girl of Chinese descent with HLD16. The mutation was found by trio whole-exome sequencing and confirmed by Sanger sequencing. Functional studies of the variant and studies of patient cells were not performed, but Yan et al. (2018) noted that the mutation occurred at a CpG dinucleotide, suggesting that it is a recurrent mutation due to a hotspot within the gene. INHERITANCE - Autosomal dominant HEAD & NECK Eyes - Nystagmus (horizontal, vertical, rotary) - Saccadic pursuit ABDOMEN Gastrointestinal - Poor feeding (in some patients) MUSCLE, SOFT TISSUES - Hypotonia - Hypertonia NEUROLOGIC Central Nervous System - Delayed development, primarily motor - Intellectual disability (in some patients) - Learning disabilities - Speech delay - Seizures (rare) - Gait ataxia - Wide-based gait - Intention tremor - Dysarthria - Dystonia - Dysmetria - Hyperreflexia - Pyramidal signs, mild - Hypomyelinating leukodystrophy - Thin corpus callosum MISCELLANEOUS - Onset in the first days or weeks of life - Variable severity - Some patients may have normal cognitive development - De novo mutation (in most patients) MOLECULAR BASIS - Caused by mutation in the transmembrane protein 106B gene (TMEM106B, 613413.0001 ) ▲ Close
Description Dystonia-28 is an autosomal dominant neurologic disorder characterized by onset of progressive dystonia in the first decade of life. ... All patients had onset of symptoms in the first decade, except for a 46-year-old affected mother who reportedly had onset at age 23 years; her son had onset at age 8 years. ... Inheritance The transmission pattern of DYT28 in 1 of the families reported by Zech et al. (2016) was consistent with autosomal dominant inheritance. Molecular Genetics In 4 unrelated probands with DYT28, Zech et al. (2016) identified 4 different heterozygous loss-of-function mutations in the KMT2B gene (606834.0001-606834.0004). The mutations, which were found by whole-exome sequencing, were confirmed by Sanger sequencing. ... There were 7 frameshift mutations, 2 nonsense mutations, 1 splice site mutation, and 7 missense mutations. The mutations were found by whole-exome or whole-genome sequencing and confirmed by Sanger sequencing.
DYT- KMT2B is inherited in an autosomal dominant manner. To date, ~84% of individuals have the disorder as the result of a de novo KMT2B pathogenic variant and ~16% have inherited the KMT2B variant (10% from an affected parent; 6% from a clinically asymptomatic parent). ... Sequence analysis of KMT2B detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications. ... Exome array (when clinically available) may be considered if exome sequencing is not diagnostic, particularly when evidence supports autosomal dominant inheritance. For an introduction to comprehensive genomic testing click here. ... Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
Clinical Features Wagnon et al. (2018) reported a 3-generation family in which 5 individuals had onset of isolated action-induced nonepileptic myoclonus affecting the upper limb in the first decade. Two patients were examined and the other 3 were reportedly affected with a similar disorder. ... Electrophysiologic studies of 1 patient in the first decade showed evidence of subcortical origin, but not cortical origin. ... Inheritance The transmission pattern of MYOCL2 in the family reported by Wagnon et al. (2018) was consistent with autosomal dominant inheritance. Molecular Genetics In 3 affected members of a family with MYOCL2, Wagnon et al. (2018) identified a heterozygous missense mutation in the SCN8A gene (P1719R; 600702.0012). The mutation, which was found by whole-exome sequencing and confirmed by Sanger sequencing, segregated with the disorder in the family in those who agreed to testing. In vitro functional expression studies in transfected neuron-derived cells showed that the mutation caused a partial loss of function, manifest as decreased inward sodium current compared to controls. INHERITANCE - Autosomal dominant NEUROLOGIC Central Nervous System - Myoclonic seizures, isolated - Possible subcortical origin - No dystonia - No seizures MISCELLANEOUS - Onset in the first decade - Nonprogressive disorder - Alcohol may alleviate the symptoms - One family has been reported (last curated March 2019) MOLECULAR BASIS - Caused by mutation in the sodium channel, voltage gated, type VIII, alpha polypeptide gene (SCN8A, 600702.0012 ) ▲ Close
Description Leber congenital amaurosis with early-onset deafness is an autosomal dominant syndrome manifesting as early-onset and severe photoreceptor and cochlear cell loss. ... Hearing loss in the 5 patients was diagnosed within the first decade of life, and ranged from mild to severe. ... The authors designated the severe and early-onset retinal degeneration seen in these patients to be consistent with advanced Leber congenital amaurosis type 2 (see LCA2, 204100), although they noted that all 5 had high hypermetropia, which was atypical for LCA2. Molecular Genetics By whole-exome sequencing in 3 affected members of a family (family 1) with early-onset retinal degeneration and hearing loss, Luscan et al. (2017) identified heterozygosity for a missense mutation in the TUBB4B gene (R391H; 602660.0001) in the affected mother and son. The maternal grandmother, who had milder symptoms, was found to be mosaic for the mutation, which was present in 29% of reads and had arisen de novo. Analysis of an in-house whole-exome sequencing database identified 3 more probands with LCAEOD and mutations in TUBB4B, 2 with the same R391H mutation, and 1 with an R391C substitution (602660.0002). In 2 of the families, the mutation was shown to have arisen de novo, but in the third family (family 2), the mutation was inherited from an unaffected mosaic father, in whom the variant was present in only 13% of reads. INHERITANCE - Autosomal dominant HEAD & NECK Ears - Sensorineural hearing loss, early-onset - Endocochlear deafness Eyes - Retinal degeneration, early-onset - Reduced visual acuity - High hypermetropia - Retinal vessel attenuation - Generalized choroid atrophy - Macular rearrangements - Peripheral pigmentary deposits - Severely reduced to extinguished responses on electroretinography MISCELLANEOUS - Symptoms occur within the first decade of life - Onset at birth in some patients MOLECULAR BASIS - Caused by mutation in the beta-4B tubulin gene (TUBB4B, 602660.0001 ) ▲ Close
A number sign (#) is used with this entry because of evidence that autosomal dominant mental retardation-55 with seizures (MRD55) is caused by heterozygous mutation in the NUS1 gene (610463) on chromosome 6q22. Clinical Features Hamdan et al. (2017) reported 3 unrelated patients with onset of myoclonic seizures in the first years of life. The most severely affected patient (indvKW) was an 8-year-old boy with global developmental delay, moderate intellectual disability, speech delay, and ataxic gait. ... She had mild motor delay and could jump and run, but was unable to ride a bicycle. She could speak and read at a first grade level and had mild dysarthria and tremor, but no ataxia. ... Two of the mutations were truncating, and 1 was an intragenic deletion encompassing all of exon 2. Two patients were found by whole-genome sequencing of 197 individuals with developmental epileptic encephalopathy; the third patient was found by clinical exome sequencing from another patient cohort. Functional studies of the variants and studies of patients cells were not performed, but Hamdan et al. (2017) postulated haploinsufficiency as the pathogenic mechanism. INHERITANCE - Autosomal dominant NEUROLOGIC Central Nervous System - Delayed development - Intellectual disability, moderate to severe - Language delay - Dysarthria - Ataxic gait - Clumsy - Poor fine motor skills - Tremor - Myoclonic seizures - Seizures, multiple types - EEG abnormalities Behavioral Psychiatric Manifestations - Autism spectrum disorder MISCELLANEOUS - Onset of seizures in first years of life - Seizures tend to be controlled by multiple medications - De novo mutation - Three unrelated patients have been reported (last curated January 2018) MOLECULAR BASIS - Caused by mutation in the nuclear undecaprenyl pyrophosphate synthase 1, S. cerevisiae, homolog of, gene (NUS1, 610463.0002 ) ▲ Close
The patients had hypotonia and variably delayed motor development: 2 patients were nonambulatory at ages 12 and 20 years, whereas the others learned to walk in the first years of life but had an ataxic gait. ... Molecular Genetics In 4 unrelated patients with HADDTS, Beck et al. (2016) identified a de novo heterozygous missense mutation in the CTBP1 gene (R331W; 602618.0001). The mutations were found by whole-exome sequencing and confirmed by Sanger sequencing. ... Functional studies of the variant and studies of patient cells were not performed, but the authors postulated a dominant-negative effect. The patients were part of a cohort of 5,471 trios containing probands with neurodevelopmental disorders who underwent whole-exome sequencing. INHERITANCE - Autosomal dominant HEAD & NECK Face - Dysmorphic features (in some patients) - Frontal bossing - Retrognathia Eyes - Deep-set eyes Mouth - High-arched palate Teeth - Enamel defects - Soft enamel - Discolored teeth ABDOMEN Gastrointestinal - Feeding difficulties MUSCLE, SOFT TISSUES - Hypotonia - Nonspecific dystrophic changes NEUROLOGIC Central Nervous System - Delayed motor development - Ataxic gait - Inability to walk - Intellectual disability, variable (in some patients) - Speech delay - Cerebellar atrophy (in some patients) MISCELLANEOUS - Variable phenotype - Four unrelated patients have been reported (last curated March 2018) MOLECULAR BASIS - Caused by mutation in the C-terminal-binding protein 1 gene (CTBP1, 602618.0001 ) ▲ Close
As opposed to a forward viewing angle, an extreme downward angle such as this has been shown to markedly increase sickness in virtual environments. [16] Time spent immersed in a virtual environment contributes to sickness symptom presence due to the increasing effects of fatigue on the user. [16] Oculomotor symptoms are the most common to occur due to immersion time, but the nature of the user's movements (e.g., whole-body vs. head-only) is suggested to be the primary cause of nausea or physical sickness. [16] Techniques for reducing VR sickness [ edit ] According to several studies, introducing a static frame of reference ( independent visual background ) may reduce simulation sickness. [17] [18] [19] A technique called Nasum Virtualis shows a virtual nose as a fixed frame of reference for VR headsets. [20] [21] Other techniques for reducing nausea involve simulating ways of displacement that don't create or reduce discrepancies between the visual aspects and body movement, such as reducing rotational motions during navigation, [22] dynamically reducing the field of view , [23] teleportation, [24] and movement in zero gravity . [25] In January 2020, the French start-up Boarding Ring , known for their glasses against motion sickness, [26] released an add-on device against virtual reality sickness. [27] Using two small screens in the user's peripheral field of view, the device displays visual information consistent with vestibular inputs, avoiding the sensory conflict. ... Retrieved 2019-03-08 . ^ a b Lang, B. (January 16, 2014). "First impressions of Valve's VR head mounted display prototype" . ... PMID 11538019 . v t e Mixed and virtual reality Concepts Applications of virtual reality Artificial reality Augmented reality Degrees of freedom Immersion Projection augmented model Real life Reality–virtuality continuum Room-scale Telepresence Virtual reality sickness Virtual world persistent Technology Omnidirectional treadmill Wearable computer Haptic suit Display Head-mounted display optical Head-up display Virtual retinal display Virtual reality headset ( comparison ) 3D interaction Positional tracking Eye tracking Finger tracking Simultaneous localization and mapping Software Asynchronous reprojection Foveated rendering Image-based modeling and rendering Photography Free viewpoint television 360-degree video VR photography Omnidirectional camera Peripherals Cyberith Virtualizer Oculus Touch Leap Motion PlayStation Move Razer Hydra Virtuix Omni Wired glove Wizdish ROVR Immersive devices Current Google Cardboard HTC Vive Nintendo Labo VR Kit Oculus Go Oculus Quest 2 Oculus Rift CV1 Rift S OSVR Magic Leap Samsung Gear VR PlayStation VR Pimax Valve Index Vuzix Blade Windows Mixed Reality Microsoft HoloLens 2 Google Glass Legacy Famicom 3D System Google Daydream Liquid Image Sensorama Sega VR Sword of Damocles Vuzix VFX1 Headgear Virtual Boy Virtuality Applications ARToolKit ARCore Interactive art Virtual graffiti OpenVR OpenXR Pervasive game WebVR Games List of HTC Vive games List of Oculus Quest games List of Oculus Rift games List of PlayStation VR games v t e Motion sickness Types Airsickness Seasickness Simulator sickness Ski sickness Space adaptation syndrome Virtual reality sickness Medicine treatment Bonine Cinnarizine Dramamine Marezine Promethazine Transdermscop Related Bárány chair Sickness bag
To identify additional susceptibility loci, Miceli-Richard et al. (2004) genotyped 120 multiplex SpA families who were included in a genomewide scan. Linkage analyses on the first 65 families identified 4 candidate non-MHC regions on chromosomes 5q, 9q, 13q, and 17q, which were further explored in the remaining 55 multiplex families. ... Nonparametric multipoint linkage analyses of the whole data set yielded evidence of significant linkage to 9q31-q34, in the vicinity of marker D9S1776 (NPL = 4.87, lod = 5.15, p = 0.00002). By examining cosegregation of this haplotype with the disease status within the family, and by taking into account the HLA-B27 carrier status, Miceli-Richard et al. (2004) inferred a possible dominant model of inheritance with incomplete penetrance. Joints - Spondyloarthropathy Lab - Rheumatoid serologic tests negative Inheritance - Autosomal dominant ▲ Close
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article needs additional citations for verification . ... Check that Wiktionary does not have an article on this word or phrase, as verified using the search page . If Wiktionary has a definition already, change this tag to {{ TWCleanup2 }} or else consider a soft redirect to Wiktionary by replacing the text on this page with {{ Wi }}. If Wiktionary does not have the definition yet, consider moving the whole article to Wiktionary by replacing this tag with the template {{ Copy to Wiktionary }}.
Auer-Grumbach et al. (2016) reported families with autosomal dominant transmission of susceptibility to late-onset CMT2T. ... The mutations, which were found by whole-exome sequencing, segregated with the disorder in the families in which segregation was analyzed. ... MME mutations accounted for 13% of patients with a diagnosis of autosomal recessive CMT who underwent whole-exome sequencing, and Higuchi et al. (2016) concluded that mutations in the MME gene are the most frequent cause of autosomal recessive axonal CMT in the Japanese population. Susceptibility to Late-Onset Charcot-Marie-Tooth Disease Type 2T In 19 probands of European descent with late-onset axonal CMT, Auer-Grumbach et al. (2016) identified 11 different heterozygous variants in the MME gene (see, e.g., 120520.0007-120520.0009), including 7 loss-of-function alleles and 4 missense alleles. The variants in the first 3 families were found by whole-exome sequencing; subsequent MME variants were found in 6 of 45 additional probands with a similar disorder who underwent direct sequencing of the MME gene. Select patient samples and in vitro cellular studies were consistent with decreased tissue availability of neprilysin and impaired enzymatic activity. Examination of repositories of whole-exome data from more than 10,000 individuals with neurologic and other disorders found 12 more individuals with heterozygous truncating variants, 10 of whom had polyneuropathy, motor neuron disorder, or sensory ataxia.
A rare autosomal dominant hereditary axonal motor and sensory neuropathy characterized by adult onset of slowly progressive distal muscle weakness and atrophy, sensory impairment, and hyporeflexia beginning in the lower limbs.
A rare autosomal recessive axonal hereditary motor and sensory neuropathy characterized by adult onset of slowly progressive distal muscle weakness and atrophy, sensory impairment, and decreased or absent deep tendon reflexes predominantly in the lower extremities. Patients present gait disturbances but remain ambulatory. Mild involvement of the upper limbs may be seen.
Spinocerebellar ataxia type 43 is a rare autosomal dominant cerebellar ataxia type I disorder characterized by late adult-onset of slowly progressive cerebellar ataxia, typically presenting with balance and gait disturbances, in association with axonal peripheral neuropathy resulting in reduced/absent deep tendon reflexes and sensory impairment.