-
Nosebleed
Wikipedia
Pronunciation Epistaxis / ˌ ɛ p ɪ ˈ s t æ k s ɪ s / EP -ih- STAK -sis Specialty Otorhinolaryngology Symptoms Bleeding from the nose [1] Usual onset Less than 10 and over 50 years old [2] Risk factors Trauma, Excessive Nose-picking , Certain infections , blood thinners , high blood pressure , alcoholism , seasonal allergies , dry weather [3] Diagnostic method Direct observation [1] Differential diagnosis Bleeding from the lungs , esophageal varices [1] Prevention Petroleum jelly in the nose [4] Treatment Pressure over the lower half of the nose, nasal packing , endoscopy [5] Medication Tranexamic acid [6] Frequency 60% at some point in time [7] Deaths Rare [3] A nosebleed , also known as epistaxis , is bleeding from the nose . [1] Blood can also flow down into the stomach and cause nausea and vomiting . [8] In more severe cases blood may come out of both nostrils . [9] Rarely bleeding may be so significant low blood pressure occurs. [1] Rarely the blood can come up the nasolacrimal duct and out from the eye. [10] Risk factors include trauma including putting the finger in the nose, blood thinners , high blood pressure , alcoholism , seasonal allergies , dry weather, and inhaled corticosteroids . [3] There are two types: anterior, which is more common; and posterior, which is less common but more serious. [3] Anterior nosebleeds generally occur from Kiesselbach's plexus while posterior bleeds generally occur from the sphenopalatine artery . [3] The diagnosis is by direct observation. [1] Prevention may include the use of petroleum jelly in the nose. [4] Initially treatment is generally by applying pressure for at least five minutes over the lower half of the nose. [5] If this is not sufficient nasal packing may be used. [5] Tranexamic acid may also be helpful. [6] If bleeding episodes continue endoscopy is recommended. [5] About 60% of people have a nosebleed at some point in their life. [7] About 10% of nosebleeds are serious. [7] Nosebleeds are rarely fatal, accounting for only 4 of the 2.4 million deaths in the U.S. in 1999. [11] Nosebleeds most commonly affect those younger than 10 and older than 50. [2] Contents 1 Cause 2 Pathophysiology 3 Prevention 4 Treatment 4.1 Nasal packing 4.2 Tranexamic acid 4.3 Cauterization 4.4 Surgery 4.5 Other 5 Society and culture 5.1 Etymology 6 References 7 External links Cause [ edit ] Two children boxing , the one on the right having a nosebleed due to a punch to the face. ... As a result, many forms of nasal packing involve use of topical antistaphylococcal antibiotic ointment. [2] Tranexamic acid [ edit ] Tranexamic acid helps promote blood clotting. [6] For nosebleeds it can be applied to the site of bleeding, taken by mouth, or injected into a vein. [6] Cauterization [ edit ] This method involves applying a chemical such as silver nitrate to the nasal mucosa, which burns and seals off the bleeding. [12] Eventually the nasal tissue to which the chemical is applied will undergo necrosis . [12] This form of treatment is best for mild bleeds, especially in children, that are clearly visible. [12] A topical anesthetic (such as lidocaine ) is usually applied prior to cauterization. ... PMID 24439881 . ^ a b c d Tunkel, David E.; Anne, Samantha; Payne, Spencer C.; Ishman, Stacey L.; Rosenfeld, Richard M.; Abramson, Peter J.; Alikhaani, Jacqueline D.; Benoit, Margo McKenna; Bercovitz, Rachel S.; Brown, Michael D.; Chernobilsky, Boris; Feldstein, David A.; Hackell, Jesse M.; Holbrook, Eric H.; Holdsworth, Sarah M.; Lin, Kenneth W.; Lind, Meredith Merz; Poetker, David M.; Riley, Charles A.; Schneider, John S.; Seidman, Michael D.; Vadlamudi, Venu; Valdez, Tulio A.; Nnacheta, Lorraine C.; Monjur, Taskin M. (7 January 2020). ... Retrieved January 31, 2010 . ^ J. F. Lubianca Neto; F. D. Fuchs; S. R. Facco; M. Gus; L. Fasolo; R. Mafessoni; A. ... E.; Williams, R. J.; Kuhn, I.; Carrie, S. (December 2017). "Intranasal packs and haemostatic agents for the management of adult epistaxis: systematic review".ACVRL1, DNAH11, ZBTB16, WIPF1, WAS, VWF, TERT, TERC, TBXAS1, TBXA2R, STIM1, STAT5B, STAT3, RARA, PRTN3, PRKAR1A, PRKACG, PRF1, PML, GFI1B, FCGR2C, CACNA1D, RASGRP2, IRF2BP2, SLFN14, MCFD2, HPS4, DTNBP1, FIP1L1, HPS6, TBL1XR1, NABP1, P2RY12, BCOR, TET2, GP6, SBDS, PTPN22, NBEAL2, HPS5, PLAU, NUMA1, NPM1, MYH9, FGB, FGA, F13B, F13A1, F11, F10, F7, F5, F2, ETV6, EPOR, ENG, CYP11B2, CYP11B1, CTLA4, LYST, RUNX1, FGG, FYB1, GATA1, IFNG, MYD88, MPL, SMAD4, KCNJ5, JAK2, ITGB3, ITGA2B, HPS1, GBA, HLA-DPB1, HLA-DPA1, GP9, GP1BB, GP1BA, GGCX, GDF2, BLOC1S3

-
Latent Hypoxia
Wikipedia
Retrieved 24 January 2017 . ^ Brubakk, A. O.; Neuman, T. S. (2003). Bennett and Elliott's physiology and medicine of diving, 5th Rev ed . ... Navy diver U.S.Navy master diver United States Navy SEALs Underwater Demolition Team Underwater work Commercial offshore diving Dive leader Diver training Recreational diver training Hyperbaric welding Media diving Nondestructive testing Pearl hunting Police diving Potable water diving Public safety diving Scientific diving Ships husbandry Sponge diving Submarine pipeline Underwater archaeology Archaeology of shipwrecks Underwater construction Offshore construction Underwater demolition Underwater photography Underwater search and recovery Underwater videography Salvage diving SS Egypt Kronan La Belle SS Laurentic RMS Lusitania Mars Mary Rose USS Monitor HMS Royal George Vasa Diving contractors COMEX Helix Energy Solutions Group Tools & equipment Abrasive waterjet Airlift Baited remote underwater video In-water surface cleaning Brush cart Cavitation cleaning Pressure washing Pigging Lifting bag Remotely operated underwater vehicle Thermal lance Tremie Water jetting Underwater weapons Limpet mine Speargun Hawaiian sling Polespear Underwater firearm Gyrojet Mk 1 Underwater Defense Gun Powerhead Underwater pistols Heckler & Koch P11 SPP-1 underwater pistol Underwater revolvers AAI underwater revolver Underwater rifles ADS amphibious rifle APS underwater rifle ASM-DT amphibious rifle Recreational diving Specialties Altitude diving Cave diving Deep diving Ice diving Muck diving Open-water diving Rebreather diving Sidemount diving Solo diving Technical diving Underwater photography Wreck diving Diver organisations British Sub-Aqua Club (BSAC) Cave Divers Association of Australia (CDAA) Cave Diving Group (CDG) Comhairle Fo-Thuinn (CFT) Confédération Mondiale des Activités Subaquatiques (CMAS) Federación Española de Actividades Subacuáticas (FEDAS) Fédération Française d'Études et de Sports Sous-Marins (FFESSM) International Association for Handicapped Divers (IAHD) National Association for Cave Diving (NACD) Woodville Karst Plain Project (WKPP) Diving tourism industry Dive center Environmental impact of recreational diving Scuba diving tourism Shark tourism Sinking ships for wreck diving sites Diving events and festivals Diversnight Underwater Bike Race Recreational dive sites Reef diving regions Aliwal Shoal Marine Protected Area Arrecifes de Cozumel National Park Edmonds Underwater Park Great Barrier Reef iSimangaliso Marine Protected Area Poor Knights Islands Table Mountain National Park Marine Protected Area Reef dive sites Artificial reef Gibraltar Artificial Reef Shark River Reef Osborne Reef Fanadir Gamul Kebir Palancar Reef Underwater artworks Cancún Underwater Museum Christ of the Abyss Molinere Underwater Sculpture Park Wreck diving regions Chuuk Lagoon Edmonds Underwater Park Finger Lakes Underwater Preserve Association Maritime Heritage Trail – Battle of Saipan Michigan Underwater Preserves Robben Island Marine Protected Area Table Mountain National Park Marine Protected Area Tulagi Tulamben Whitefish Point Underwater Preserve Wreck Alley, San Diego Wreck dive sites HMS A1 HMS A3 USS Aaron Ward Abessinia Aeolian Sky Albert C. ... Bradley Carnatic SMS Dresden Dunraven Eastfield HMT Elk Ellengowan RMS Empress of Ireland HMS Falmouth Fifi SS Francisco Morazan Fujikawa Maru Fumizuki SATS General Botha USNS General Hoyt S. Vandenberg HMS Ghurka Glen Strathallan SAS Good Hope Gothenburg Herzogin Cecilie Hilma Hooker Hispania HMS Hood HMAS Hobart Igara James Eagan Layne Captain Keith Tibbetts King Cruiser SMS Kronprinz Kyarra HMS Laforey USAT Liberty Louis Sheid USS LST-507 SMS Markgraf Mikhail Lermontov HMS M2 Maine Maloja HMS Maori Marguerite SS Mauna Loa USAT Meigs Mendi USCGC Mohawk Mohegan RMS Moldavia HMS Montagu MV RMS Mulheim Nagato Oceana USS Oriskany Oslofjord P29 P31 Pedernales Persier HMAS Perth SAS Pietermaritzburg Piłsudski Pool Fisher HMS Port Napier Preußen President Coolidge PS Queen Victoria Radaas Rainbow Warrior RMS Rhone Rondo Rosehill Rotorua Royal Adelaide Royal Charter Rozi HMS Safari Salem Express USS Saratoga USS Scuffle HMS Scylla HMS Sidon USS Spiegel Grove Stanegarth Stanwood Stella HMAS Swan USS Tarpon Thesis Thistlegorm Toa Maru Torrey Canyon SAS Transvaal U-40 U-352 U-1195 Um El Faroud Varvassi Walter L M Russ Washingtonian (1913) HMNZS Wellington USS Yancey Yongala Zenobia Zealandia Zingara Cave diving sites Blauhöhle Chinhoyi Caves Devil's Throat at Punta Sur Engelbrecht Cave Fossil Cave Jordbrugrotta Piccaninnie Ponds Pluragrotta Pollatoomary Sistema Ox Bel Ha Sistema Sac Actun Sistema Dos Ojos Sistema Nohoch Nah Chich Freshwater dives Dutch Springs Ewens Ponds Little Blue Lake Training sites Capernwray Dive Centre Deepspot National Diving and Activity Centre Stoney Cove Open ocean diving Blue-water diving Black-water diving Diving safety Human factors in diving equipment design Human factors in diving safety Life-support system Safety-critical system Scuba diving fatalities Diving hazards List of diving hazards and precautions Environmental Current Delta-P Entanglement hazard Overhead Silt out Wave action Equipment Freeflow Use of breathing equipment in an underwater environment Failure of diving equipment other than breathing apparatus Single point of failure Physiological Cold shock response Decompression Nitrogen narcosis Oxygen toxicity Seasickness Uncontrolled decompression Diver behaviour and competence Lack of competence Overconfidence effect Panic Task loading Trait anxiety Willful violation Consequences Barotrauma Decompression sickness Drowning Hypothermia Hypoxia Hypercapnia Hyperthermia Diving procedures Ascending and descending Emergency ascent Boat diving Canoe and kayak diving Buddy diving buddy check Decompression Decompression practice Pyle stop Ratio decompression Dive briefing Dive log Dive planning Scuba gas planning Diver communications Diving hand signals Diving line signals Diver voice communications Diver rescue Diver training Doing It Right Drift diving Gas blending for scuba diving Night diving Solo diving Water safety Risk management Checklist Hazard identification and risk assessment Hazard analysis Job safety analysis Risk assessment Risk control Hierarchy of hazard controls Incident pit Lockout–tagout Permit To Work Redundancy Safety data sheet Situation awareness Diving team Bellman Chamber operator Diver medical technician Diver's attendant Diving supervisor Diving systems technician Gas man Life support technician Stand-by diver Equipment safety Breathing gas quality Testing and inspection of diving cylinders Hydrostatic test Sustained load cracking Diving regulator Breathing performance of regulators Occupational safety and health Approaches to safety Job safety analysis Risk assessment Toolbox talk Housekeeping Association of Diving Contractors International Code of practice Contingency plan Diving regulations Emergency procedure Emergency response plan Evacuation plan Hazardous Materials Identification System Hierarchy of hazard controls Administrative controls Engineering controls Hazard elimination Hazard substitution Personal protective equipment International Marine Contractors Association Occupational hazard Biological hazard Chemical hazard Physical hazard Psychosocial hazard Occupational hygiene Exposure assessment Occupational exposure limit Workplace health surveillance Safety culture Code of practice Diving safety officer Diving superintendent Health and safety representative Operations manual Safety meeting Standard operating procedure Diving medicine Diving disorders List of signs and symptoms of diving disorders Cramp Motion sickness Surfer's ear Pressure related Alternobaric vertigo Barostriction Barotrauma Air embolism Aerosinusitis Barodontalgia Dental barotrauma Pulmonary barotrauma Compression arthralgia Decompression illness Dysbarism Oxygen Freediving blackout Hyperoxia Hypoxia Oxygen toxicity Inert gases Avascular necrosis Decompression sickness Isobaric counterdiffusion Taravana Dysbaric osteonecrosis High-pressure nervous syndrome Hydrogen narcosis Nitrogen narcosis Carbon dioxide Hypercapnia Hypocapnia Breathing gas contaminants Carbon monoxide poisoning Immersion related Asphyxia Drowning Hypothermia Immersion diuresis Instinctive drowning response Laryngospasm Salt water aspiration syndrome Swimming-induced pulmonary edema Treatment Demand valve oxygen therapy First aid Hyperbaric medicine Hyperbaric treatment schedules In-water recompression Oxygen therapy Therapeutic recompression Personnel Diving Medical Examiner Diving Medical Practitioner Diving Medical Technician Hyperbaric nursing Screening Atrial septal defect Effects of drugs on fitness to dive Fitness to dive Psychological fitness to dive Research Researchers in diving physiology and medicine Arthur J. ... Thalmann Jacques Triger Diving medical research organisations Aerospace Medical Association Divers Alert Network (DAN) Diving Diseases Research Centre (DDRC) Diving Medical Advisory Council (DMAC) European Diving Technology Committee (EDTC) European Underwater and Baromedical Society (EUBS) National Board of Diving and Hyperbaric Medical Technology Naval Submarine Medical Research Laboratory Royal Australian Navy School of Underwater Medicine Rubicon Foundation South Pacific Underwater Medicine Society (SPUMS) Southern African Underwater and Hyperbaric Medical Association (SAUHMA) Undersea and Hyperbaric Medical Society (UHMS) United States Navy Experimental Diving Unit (NEDU) Law Civil liability in recreational diving Diving regulations Duty of care List of legislation regulating underwater diving Investigation of diving accidents UNESCO Convention on the Protection of the Underwater Cultural Heritage History of underwater diving History of decompression research and development History of scuba diving List of researchers in underwater diving Timeline of diving technology Underwater diving in popular culture Archeological sites SS Commodore USS Monitor Queen Anne's Revenge Whydah Gally Underwater art and artists The Diver Jason deCaires Taylor Engineers and inventors William Beebe Georges Beuchat John R. ... Barnette Victor Berge Philippe Diolé Gary Gentile Bret Gilliam Bob Halstead Trevor Jackson Steve Lewis John Mattera Rescuers Craig Challen Richard Harris Rick Stanton John Volanthen Frogmen Lionel Crabb Commercial salvors Keith Jessop Science of underwater diving Diving physics Breathing performance of regulators Buoyancy Archimedes' principle Neutral buoyancy Concentration Diffusion Molecular diffusion Force Oxygen fraction Permeation Psychrometric constant Solubility Henry's law Saturation Solution Supersaturation Surface tension Hydrophobe Surfactant Temperature Torricellian chamber Underwater acoustics Modulated ultrasound Underwater vision Snell's law Underwater computer vision Weight Apparent weight Gas laws Amontons's law Boyle's law Charles's law Combined gas law Dalton's law Gay-Lussac's law Ideal gas law Pressure Absolute pressure Ambient pressure Atmospheric pressure Gauge pressure Hydrostatic pressure Metre sea water Partial pressure Diving physiology Artificial gills Cold shock response Diving reflex Equivalent narcotic depth Lipid Maximum operating depth Metabolism Physiological response to water immersion Tissue Underwater vision Circulatory system Blood shift Patent foramen ovale Perfusion Pulmonary circulation Systemic circulation Decompression theory Decompression models: Bühlmann decompression algorithm Haldane's decompression model Reduced gradient bubble model Thalmann algorithm Thermodynamic model of decompression Varying Permeability Model Equivalent air depth Equivalent narcotic depth Oxygen window in diving decompression Physiology of decompression Respiration Blood–air barrier Breathing CO₂ retention Dead space Gas exchange Hypocapnia Respiratory exchange ratio Respiratory quotient Respiratory system Work of breathing Diving environment Classification List of diving environments by type Altitude diving Benign water diving Confined water diving Deep diving Inland diving Inshore diving Muck diving Night diving Open-water diving Black-water diving Blue-water diving Penetration diving Cave diving Ice diving Wreck diving Recreational dive sites Underwater environment Impact Environmental impact of recreational diving Low impact diving Environmental factors Algal bloom Currents: Current Longshore drift Ocean current Rip current Tidal race Undertow Upwelling Ekman transport Halocline Reef Coral reef Stratification Thermocline Tides Turbidity Wind wave Breaking wave Surf Surge Wave shoaling Other Bathysphere Defense against swimmer incursions Diver detection sonar Offshore survey Underwater domain awareness Awards and events Hans Hass Award International Scuba Diving Hall of Fame London Diving Chamber Dive Lectures NOGI Awards Deep-submergence vehicle Aluminaut DSV Alvin American submarine NR-1 Bathyscaphe Archimède FNRS-2 FNRS-3 FNRS-4 Harmony class bathyscaphe Sea Pole -class bathyscaphe Trieste II Deepsea Challenger Ictineu 3 JAGO Jiaolong Konsul -class submersible DSV Limiting Factor Russian submarine Losharik Mir Nautile Pisces -class deep submergence vehicle DSV Sea Cliff DSV Shinkai DSV Shinkai 2000 DSV Shinkai 6500 DSV Turtle DSV-5 Nemo Deep-submergence rescue vehicle LR5 LR7 MSM-1 Mystic -class deep-submergence rescue vehicle DSRV-1 Mystic DSRV-2 Avalon NATO Submarine Rescue System Priz -class deep-submergence rescue vehicle Russian deep submergence rescue vehicle AS-28 Russian submarine AS-34 ASRV Remora SRV-300 Submarine Rescue Diving Recompression System Type 7103 DSRV URF (Swedish Navy) Special interest groups Artificial Reef Society of British Columbia CMAS Europe Coral Reef Alliance Diving Equipment and Marketing Association Divers Alert Network Green Fins Historical Diving Society Karst Underwater Research Nautical Archaeology Program Nautical Archaeology Society Naval Air Command Sub Aqua Club Project AWARE Reef Check Reef Life Survey Rubicon Foundation Save Ontario Shipwrecks SeaKeys Sea Research Society Society for Underwater Historical Research Society for Underwater Technology Underwater Archaeology Branch, Naval History & Heritage Command Submarine escape and rescue Escape trunk International Submarine Escape and Rescue Liaison Office McCann Rescue Chamber Submarine Escape and Rescue system (Royal Swedish Navy) Submarine escape training facility Submarine Escape Training Facility (Australia) Submarine rescue ship Neutral buoyancy facilities for Astronaut training Neutral Buoyancy Laboratory Neutral buoyancy pool Neutral buoyancy simulation as a training aid Neutral Buoyancy Simulator Space Systems Laboratory Yuri Gagarin Cosmonaut Training Center Other Nautilus Productions Category Commons Glossary Indexes: dive sites divers diving Outline Portal v t e Freediving Activities Aquathlon (underwater wrestling) Apnoea finswimming Freediving Pearl hunting Spearfishing Underwater football Underwater hockey Underwater ice hockey Underwater rugby Underwater target shooting Competitions Nordic Deep Vertical Blue Disciplines Constant weight (CWT) Constant weight without fins (CNF) Dynamic apnea (DYN) Dynamic apnea without fins (DNF) Free immersion (FIM) No-limits apnea (NLT) Static apnea (STA) Skandalopetra diving Variable weight apnea (VWT) Variable weight apnea without fins Equipment Diving mask Diving suit Fins Hawaiian sling Monofin Polespear Snorkel Speargun Water polo cap Freedivers Deborah Andollo Peppo Biscarini Sara Campbell Derya Can Göçen Goran Čolak Carlos Coste Robert Croft Mandy-Rae Cruickshank Yasemin Dalkılıç Leonardo D'Imporzano Flavia Eberhard Şahika Ercümen Emma Farrell Francisco Ferreras Pierre Frolla Flavia Eberhard Mehgan Heaney-Grier Elisabeth Kristoffersen Loïc Leferme Enzo Maiorca Jacques Mayol Audrey Mestre Karol Meyer Stéphane Mifsud Alexey Molchanov Natalia Molchanova Dave Mullins Patrick Musimu Guillaume Néry Herbert Nitsch Umberto Pelizzari Annelie Pompe Michal Risian Stig Severinsen Tom Sietas Aharon Solomons Martin Štěpánek Walter Steyn Tanya Streeter William Trubridge Devrim Cenk Ulusoy Danai Varveri Alessia Zecchini Hazards Barotrauma Drowning Freediving blackout (Deep-water blackout and Shallow-water blackout) Hypercapnia Hypothermia Historical Ama (diving) Octopus wrestling Swimming at the 1900 Summer Olympics – Men's underwater swimming Organisations AIDA International SSI Australian Underwater Federation British Freediving Association CMAS FFESSM Performance Freediving International Related Ama (diving) Haenyeo Snorkeling Submarine escape training facility Underwater diving Underwater sports Category: Free-diving Commons: Category:Freediving

-
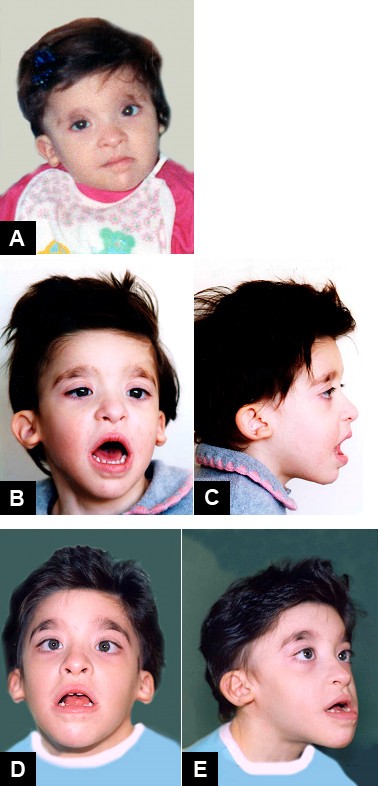
Mowat-Wilson Syndrome
Gene_reviews
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. ... Sequence analysis of ZEB2 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Option 2 When the diagnosis of MWS is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... A person with mild facial features (atypical but reminiscent of the MWS gestalt) had only mild speech delay and a novel splice site variant in the 5'UTR [Zweier et al 2006].
-
Tracheobronchial Injury
Wikipedia
PMID 17767714 . ^ a b c d e f g h i j k l m n o p q r s t u v w x Chu CP, Chen PP (2002). ... PMID 3283046 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y Karmy-Jones R, Wood DE (2007). ... ISBN 0-7234-2595-7 . Retrieved 2008-06-12 . ^ Gabor S, Renner H, Pinter H, et al. (2001). ... PMID 11584802 . ^ a b c d e f g h i j k l m n o p q r s Kiser AC, O'Brien SM, Detterbeck FC (2001). ... PMID 11288843 . ^ a b Scaglione M, Romano S, Pinto A, Sparano A, Scialpi M, Rotondo A (2006).

-
Squamous Cell Skin Cancer
Wikipedia
The antimetabolites azathioprine and mycophenolic acid have an intermediate risk profile. [37] Diagnosis [ edit ] Diagnosis is confirmed via biopsy of the tissue(s) suspected to be affected by SCC. ... This way the radiation source can be aplied to complexe locations and minimize radiation to healthy tissue. [49] After removal of the cancer, closure of the skin for patients with a decreased amount of skin laxity involves a split-thickness skin graft. A donor site is chosen and enough skin is removed so that the donor site can heal on its own. Only the epidermis and a partial amount of dermis is taken from the donor site which allows the donor site to heal. ... Archived from the original on 2008-12-21. ^ Kuschal, C.; Thoms, K. M.; Schubert, S.; et al. (2012). "Skin cancer in organ transplant recipients: effects of immunosuppressive medications on DNA repair" . ... M.; Patrizi, A.; Baraldi, C.; Zagni, F.; Vichi, S.; Pettinato, C.; Morganti, A. G.; Strigari, L. (2020-11-02).BRAF, FYN, CDKN2A, TP53, HRAS, KNSTRN, COL7A1, TERT, OCA2, AHR, MC1R, RSPO1, ERBB2, IRF4, TERC, ERCC4, LAMB3, LAMA3, KRT14, KRT5, ERCC3, RALY, BCL2L12, IRF3, CYP21A2, SEC16A, DDB2, LAMC2, GJB6, ERCC2, PIK3CA, GJB2, EGFR, TRPS1, ERCC5, TYR, MMP1, XPC, LMNA, CD274, TINF2, STK19, SLC45A2, RAC1, LPP, CRNKL1, PIK3CG, PIK3CD, PIK3CB, FUT1, PTGS2, CD44, KRT7, EPHB2, MYC, TAL1, STAT3, SCLY, MTCO2P12, BCL2, CCND1, NOTCH1, COX2, VEGFA, GRHL3, SOX2, YAP1, MAPK1, INPP5A, PTEN, RARRES2, H3P10, TMC8, TSPAN1, AKT1, MIR20A, TNF, MIR21, RELA, CDH13, ACKR3, KLF4, SIRT1, MIR497, NET1, COL17A1, SLC6A5, KRT16, MIR135B, TMC6, S100A8, CTNNB1, MIR361, CABIN1, IL10, PRPF38B, LGR5, ATF3, POLH, YBX1, PPARG, KRT20, VIM, CD68, MTHFR, CAMP, CD200, MMP13, MMP9, MS4A1, MIR31, IL22, SRC, CTTN, EGF, MIR186, FOXE1, SLC6A2, EIF4E, MIR205, TGFB1, FGFR2, HOXA9, HOXB7, LINC00319, MIR203A, IGFBP3, MIR30A, EPHA1, PICSAR, HPLH1, DLC1, BECN1, CADM1, SIN3A, CLDN1, ZNRD2, G0S2, SLCO1B3, CFDP1, KYNU, TNFSF12, ARTN, HDAC3, HERC2, CCRL2, CXCL13, CLEC2D, HPGDS, WWP1, SMPX, DAPK2, PPARGC1A, RHOBTB1, LZTS1, PRAF2, PPP1R13L, NLRP1, ZHX2, FAM114A2, HDAC9, TRIM32, EFS, DNM1L, CYFIP1, AIM2, POLQ, DCTN6, CXCR6, SLIT2, SF3B1, LPAR2, BRD4, IGF2BP1, PDPN, TRIM16, USP8, SMUG1, CKAP4, ISG15, ZMIZ1, GEMIN4, MIR143, TNFSF12-TNFSF13, MIR17HG, MIR34A, MIR221, MIR217, MIR204, MIR181A2, MIR154, MIR148A, MIR142, FOPNL, MALAT1, TICAM2, DPH3, CAVIN1, TINCR, PRSS55, RMDN2, KRT72, CD200R1, MIR326, MIR346, MIR424, MIR490, H3P23, LOC110806263, LINC01048, OCLN, LINC00963, MIR3619, MIR1193, TMED7-TICAM2, MIR664A, MIR1238, MIR1207, MIR1247, DEFB4B, HOTAIR, MIR766, LINC00520, POU5F1P4, POU5F1P3, MIR506, RBM45, NLRP3, TMED7, MRGBP, SPHK2, METTL3, PARD3, ERBIN, PAG1, POLR3E, NBPF1, SYBU, ZNF654, RMDN3, TP53RK, DEF8, RETREG1, RIPK4, WWOX, RAB23, SIRT6, TNFRSF12A, RMDN1, NDUFA13, PCBP4, RAB25, TP63, AICDA, LMLN, SPZ1, CARD11, TMPRSS13, ARHGAP24, TCF7L1, TXNDC5, SRCIN1, CD276, PDCD1LG2, UBA5, TNFAIP8L2, MAPKAP1, CARD14, WNK1, GORASP1, LGR6, OVOL2, ANKRD36B, AKR1C3, ADRA1A, DENR, HSD11B2, HLA-DRB1, HLA-DQA1, HIF1A, NR3C1, GRN, GPR42, GPER1, GPR1, GNA12, GLB1, GABPA, FN1, FOXC2, FOXG1, FHL1, FGF10, EFEMP1, FAP, PTK2B, HMGB1, HSF1, ESRRA, TNC, MAGEA12, SMAD4, SMAD2, LIMK1, LAMC1, KTN1, KRT17, KRT8, KRAS, KPNA4, KLKB1, JAK1, IVL, STT3A, ITGB1, IL6, IL1B, RBPJ, IFI27, EZH2, ESR2, MCL1, CKS1B, CDKN2B, CDK9, CDK6, CDH1, CDC25A, CDC20, RUNX1, C3, BRS3, CFB, BAX, AR, AQP3, FAS, APRT, XIAP, APAF1, AGRP, AGER, CHI3L1, CCR5, ESR1, CLDN7, ERCC1, EIF4G2, EGR3, EDNRA, HBEGF, DLX3, TIMM8A, DEFB4A, GADD45A, DAPK1, CD55, CYP3A4, CTNNA1, CTLA4, CCN2, CSHL1, CRMP1, CRIP2, CREBBP, MAL, MCM2, MLRL, ZEB1, SYT1, STAT2, STAT1, SSTR4, SPRR1A, ADRA2B, SOAT1, SLC2A1, ST3GAL1, SFRP5, SFRP1, CXCL12, SDC2, SDC1, CCL8, SERPINB4, SERPINB3, S100A9, S100A7, TCF7L2, PPP1R11, RPE65, TGFBR1, TAF15, SEM1, TFEB, XRCC1, WNT1, NSD2, VSNL1, EZR, VEGFC, VCP, UVRAG, UTRN, USF1, TRAF6, CRISP2, TP73, TLR4, THBS1, TGFBR2, S100A4, RNF2, MAP3K4, PCBP1, PCSK6, PEBP1, OVOL1, CLDN11, OSM, ODC1, NOTCH2, NOS2, NFE2L2, NEDD8, NCAM1, MMUT, MST1, MSMB, MSH2, MMP7, MAP3K9, MLH1, MFAP1, PRKN, CDK16, RET, PDCD1, REN, OPN1LW, RASA1, RAF1, PTPRD, PTGDS, PTCH1, PSMD9, PSMD2, MAP2K7, MAPK8, MAPK3, PPARD, POU5F1, PLK1, PLAT, PIN1, SERPINB9, PDK1, SOX4

-
Lynch Syndrome I
Omim
Genetic analysis suggested that Saccharomyces cerevisiae has a mismatch repair system similar to the bacterial MutHLS system. The S. cerevisiae pathway has a MutS homolog, MSH2. In both bacteria and S. cerevisiae, mismatch repair plays a role in maintaining the genetic stability of DNA. In S. cerevisiae, Msh2 mutants exhibit increased rates of expansion and contraction of dinucleotide repeat sequences. ... They identified a T-to-C transition in the -6 position of a splice acceptor site in sporadic colon tumors and as a constitutional change in affected members of 2 small families with HNPCC. ... In most families there was no definite evidence that the missense mutations or splice site mutation were causally associated with an increased risk of developing colorectal carcinoma.MSH2, MLH1, PMS2, MSH6, MLH3, EPCAM, TGFBR2, FAN1, APC, KRAS, PMS1, MSH3, EXO1, PALB2, CHEK2, ATM, PIK3CA, CDH1, PTPRJ, CTNNA1, CD44, EPHX1, CDKN1B, NFKBIZ, SMARCA4, BARD1, SEMA4A, XRCC4, RPS20, MUTYH, FBXO11, CTNNB1, BRCA2, MRC1, BRCA1, BRAF, TP53, PTEN, FAP, CDKN2A, CCND1, RINT1, PTGS2, H3P10, CD274, REEP5, NHS, BAX, BAAT, SMAD4, NAT2, STK11, COX2, IGF1, MTHFR, HRAS, POLE, HDAC2, ARID1A, MTCO2P12, XRCC6P5, PMS2CL, ARSA, CEACAM5, RAD51C, GSTT1, CYP1A1, MYC, BRIP1, EGFR, POLD1, GSTM1, GSTP1, LRRFIP2, XPA, ANXA10, VHL, RASSF1, TIMP2, AIP, TGFB1, SOCS1, CTCF, RNF43, SEC63, EMB, MIR18B, MIR31, MIR23B, MIR223, MIR152, MIR148A, LINC01194, GSTK1, ARSI, CISD3, HEPACAM, SLCO6A1, MUC16, ZHX2, ATAD1, STN1, MYH14, CPAT1, NLRP2, CDHR2, CDHR5, F11R, SGSM3, NPTN, GREM1, SPEN, ZEB1, NAT1, ST8, SPTBN1, KAT2A, FMR1, ETS1, ERCC2, ERBB2, EPHB1, ENG, ELK3, DNMT3B, DCC, CYP17A1, CYP1B1, CTLA4, KLF6, COMT, COL11A2, CEACAM7, CEACAM3, CDX2, CASP2, BLM, APEX1, APBA1, ALK, AKT1, PARP1, ACVR2A, GJA8, HFE, IGF2, PCNA, SPRR2A, SLC6A2, RNASEL, RAF1, PTPRG, PSG2, MAP2K7, PRB1, PIK3CG, PIK3CD, PIK3CB, SERPINA1, NDUFAB1, IGF2R, NBN, MMP7, MMP1, MGMT, MEN1, MDM2, MCC, MAX, MAT2A, SMAD7, SMAD2, ITGA9, PDCD1

-
Chagas Disease
Wikipedia
Human infectious disease Chagas disease Other names American trypanosomiasis Photomicrograph of Giemsa -stained Trypanosoma cruzi Pronunciation / ˈ tʃ ɑː ɡ ə s / , Portuguese pronunciation: [ˈʃaɡɐs] Specialty Infectious disease Symptoms Fever, large lymph nodes, headache [1] Complications Heart failure , enlarged esophagus , enlarged colon [1] Causes Trypanosoma cruzi spread by kissing bugs [1] Diagnostic method Finding the parasite, its DNA, or antibodies in the blood [2] Prevention Eliminating kissing bugs and avoiding their bites [1] Medication Benznidazole , nifurtimox [1] Frequency 6.2 million (2017) [3] Deaths 7,900 (2017) [4] Chagas disease , also known as American trypanosomiasis , is a tropical parasitic disease caused by Trypanosoma cruzi . [1] It is spread mostly by insects known as Triatominae , or "kissing bugs". [1] The symptoms change over the course of the infection. ... People infected through ingestion of parasites tend to develop severe disease within three weeks of consumption, with symptoms including fever, vomiting , shortness of breath , cough , and pain in the chest, abdomen , and muscles . [2] Those infected congenitally typically have few to no symptoms, but can have mild non-specific symptoms, or severe symptoms such as jaundice , respiratory distress , and heart problems. [2] People infected through organ transplant or blood transfusion tend to have symptoms similar to those of vector-borne disease, but the symptoms may not manifest for anywhere from a week to five months. [2] Chronically infected individuals who become immunosuppressed due to HIV infection can suffer particularly severe and distinct disease, most commonly characterized by inflammation in the brain and surrounding tissue or brain abscesses . [5] Symptoms vary widely based on the size and location of brain abscesses, but typically include fever, headaches, seizures, loss of sensation, or other neurological issues that indicate particular sites of nervous system damage. [18] Occasionally, these individuals also experience acute heart inflammation, skin lesions, and disease of the stomach, intestine, or peritoneum . [5] Cause [ edit ] Life cycle and transmission of T. cruzi Chagas disease is caused by infection with the protozoan parasite T. cruzi , which is typically introduced into humans through the bite of triatomine bugs, also called "kissing bugs". [5] At the bite site, motile T. cruzi forms called trypomastigotes invade various host cells. [6] Inside a host cell, the parasite transforms into a replicative form called an amastigote, which undergoes several rounds of replication. [6] The replicated amastigotes transform back into trypomastigotes, which burst the host cell and are released into the bloodstream. [2] Trypomastigotes then disseminate throughout the body to various tissues, where they invade cells and replicate. [2] Over many years, cycles of parasite replication and immune response can severely damage these tissues, particularly the heart and digestive tract. [2] Transmission [ edit ] Triatoma infestans , a common vector of T. cruzi [19] T. cruzi can be transmitted by various triatomine bugs in the genera Triatoma , Panstrongylus , and Rhodnius . [2] The primary vectors for human infection are the species of triatomine bugs that inhabit human dwellings, namely Triatoma infestans , Rhodnius prolixus , Triatoma dimidiata and Panstrongylus megistus . [19] These insects are known by a number of local names, including vinchuca in Argentina, Bolivia, Chile and Paraguay, barbeiro (the barber ) in Brazil, pito in Colombia, chinche in Central America, and chipo in Venezuela. [20] The bugs tend to feed at night, preferring moist surfaces near the eyes or mouth. [15] [19] A triatomine bug can become infected with T. cruzi when it feeds on an infected host. [15] T. cruzi replicates in the insect's intestinal tract and is shed in the bug's feces. [15] When an infected triatomine feeds, it pierces the skin and takes in a blood meal , defecating at the same time to make room for the new meal. [15] The bite is typically painless, but causes itching. [15] Scratching at the bite introduces the T. cruzi -laden feces into the bite wound, initiating infection. [15] In addition to classical vector spread, Chagas disease can be transmitted through food or drink contaminated with triatomine insects or their feces. [21] Since heating or drying kills the parasites, drinks and especially fruit juices are the most frequent source of infection. [21] This route of transmission has been implicated in several outbreaks, where it led to unusually severe symptoms, likely due to infection with a higher parasite load than from the bite of a triatomine bug. [7] [21] T. cruzi can also be transmitted independent of the triatomine bug during blood transfusion, following organ transplantation, or across the placenta during pregnancy. [2] Transfusion with the blood of an infected donor infects the recipient 10–25% of the time. [2] To prevent this, blood donations are screened for T. cruzi in many countries with endemic Chagas disease, as well as the United States. [7] Similarly, transplantation of solid organs from an infected donor can transmit T. cruzi to the recipient. [2] This is especially true for heart transplant, which transmits T. cruzi 75–100% of the time, and less so for transplantation of the liver (0–29%) or a kidney (0–19%). [2] An infected mother can also pass T. cruzi to her child through the placenta; this occurs in up to 15% of births by infected mothers. [22] As of 2019, 22.5% of new infections occurred through congenital transmission. [23] Pathophysiology [ edit ] Large scale anatomy of a heart damaged by chronic Chagas disease In the acute phase of the disease, signs and symptoms are caused directly by the replication of T. cruzi and the immune system 's response to it. [2] During this phase, T. cruzi can be found in various tissues throughout the body and circulating in the blood. [2] During the initial weeks of infection, parasite replication is brought under control by production of antibodies and activation of the host's inflammatory response , particularly cells that target intracellular pathogens such as NK cells and macrophages , driven by inflammation-signaling molecules like TNF-α and IFN-γ . [2] During chronic Chagas disease, long-term organ damage develops over years due to continued replication of the parasite and damage from the immune system. ... Techniques such as microhematocrit centrifugation can be used to concentrate the blood, which makes the test more sensitive . [2] On microscopic examination, T. cruzi trypomastigotes have a slender body, often in the shape of an S or U, with a flagellum connected to the body by an undulating membrane. [25] Alternatively, T. cruzi DNA can be detected by polymerase chain reaction (PCR). ... Endothelin-1 has been studied as a prognostic marker in animal models. [61] T. cruzi shed acute-phase antigen (SAPA), which can be detected in blood using ELISA or Western blot, [22] has been used as an indicator of early acute and congenital infection. [61] A novel assay for T. cruzi antigens in urine has been developed to diagnose congenital disease. [22] See also [ edit ] Drugs for Neglected Diseases Initiative Chagas: Time to Treat campaign Association for the Promotion of Independent Disease Control in Developing Countries References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w x y "Chagas disease (American trypanosomiasis)" . ... Retrieved 9 March 2020 . ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am an ao ap aq ar as at au av aw ax ay az ba bb bc bd Pérez-Molina JA, Molina I (2018).CYP51A1, TNF, IL10, IFNG, MMP2, LGALS3, CCR5, PPARG, MMP9, IL2, HLA-A, HLA-DRB1, HSPA4, MTCO2P12, IL6, IL17A, MBL2, TLR4, COX2, IL18, IL1B, PTGS2, RBM45, CALR, PPP1R2C, TLR2, NFE2L2, NDUFA5, TGFB1, MYD88, CYTB, REN, IL1RN, NLRP3, ACHE, APOA1, ATM, PRL, VIP, TP53, VDR, KNG1, HP, TPI1, FN1, TLR9, CTSL, TRIM33, CYP2B6, ACE, BACE1, EPGN, NPNT, CCL4, CCL3, SPN, AMZ1, CCL2, THBS1, RRAD, TNFRSF1B, MIR208A, CCR2, SPINK1, CX3CL1, SDC4, SKIL, SLC11A1, SMPD1, SOD2, TRBV20OR9-2, KIR2DS2, TCN2, TWIST1, RO60, TRIM21, SRM, ROS1, COLEC11, PPARGC1B, PANX1, MASP2, PPARGC1A, SBNO2, SIRT2, RPIA, PLCB1, SIRT1, PTPN22, EBI3, PYCARD, HSPA14, UBAP1, GGNBP2, SBNO1, OTUB1, RABEP2, HEXIM1, KNTC1, TYK2, TNFSF11, VIM, VIPR1, VPS11, FOSL1, CDR3, DYSF, DBA2, TP63, ARHGEF10, MCU, ADAM7, EIF2S2, LMLN, MBD2, SLC25A31, TMPRSS11D, VIPR2, ABO, RAD51, CSF3, CYBB, DECR1, DHODH, DMD, EIF2S1, EIF2S3, FBN1, FCN2, FDPS, FHIT, FOXO3, GAST, G6PD, GABPA, GCK, GEM, GLUL, CTSB, CR1, HLA-G, CDKN1A, ACTB, ADA, PARP1, AGT, ALB, ALOX5, ARG1, STS, ATP2A3, ATP2B1, BCHE, BDNF, C3, CACNA1C, CAMP, CD80, CD86, HLA-DPB1, HSPD1, PPBP, COX1, NGF, NHS, NOS2, PNP, NRF1, OCA2, P2RX7, PAEP, PCNA, PDE2A, PIK3CA, PIK3CB, PIK3CD, PIK3CG, PLCB4, POLG, POMC, PPP1R12A, MIF, IGF1, MEF2D, IGHG3, IL2RA, IL4R, IL7, IL12B, IL13, IL15, ISG20, ITPR1, KIR2DL2, KIR3DL1, KRT8, LGALS9, LTA, LY6E, MAP6, MEF2A, NT5E

-
Endometrial Intraepithelial Neoplasia
Wikipedia
Progression of EIN to carcinoma, effectively a conversion from a benign neoplasm to a malignant neoplasm, is accomplished through acquisition of additional mutations and accompanied by a change in behavior characterized by the ability to invade local tissues and metastasize to regional and distant sites. Table I : Precancer Characteristics of EIN Precancer Characteristics EIN Evidence Precancers differ from normal tissue Monoclonal EIN [3] [4] [5] [6] arise from polyclonal normal field. [7] Mutations are acquired in EIN. [6] [8] [9] [10] [11] [12] [13] Precancers share some, but not all, features of cancer EIN-cancer lineage hierarchy [14] EIN may share PTEN, [11] [13] [15] K-ras, [9] [10] [12] [16] MLH1 [8] [17] changes with cancer. ... PMID 8150459 . ^ a b Baak JP, Mutter GL, Robboy S, et al. (June 2005). "The molecular genetics and morphometry-based endometrial intraepithelial neoplasia classification system predicts disease progression in endometrial hyperplasia more accurately than the 1994 World Health Organization classification system" . ... External links [ edit ] Patient Resources American Cancer Society Women's Cancer Network an educational and research organization Gynecologic Oncology Group an NIH-Funded research group that runs clinical trials OncoLink an excellent educational site from the U. of Pennsylvania Find a board certified specialist at the Society of Gynecologic Oncologists Pathology US and Canadian Academy of Pathology www.endometrium.org a pathology site focusing on endometrial disease PTEN Gene Cancer Genetics Web PTEN entry PTEN and the Endometrium at PubMed Entrez Gene PTEN entry Other CancerNet an NIH database with clinical and scientific information PubMed a search engine and database for Medical Literature v t e Tumors of the female urogenital system Adnexa Ovaries Glandular and epithelial / surface epithelial- stromal tumor CMS: Ovarian serous cystadenoma Mucinous cystadenoma Cystadenocarcinoma Papillary serous cystadenocarcinoma Krukenberg tumor Endometrioid tumor Clear-cell ovarian carcinoma Brenner tumour Sex cord–gonadal stromal Leydig cell tumour Sertoli cell tumour Sertoli–Leydig cell tumour Thecoma Granulosa cell tumour Luteoma Sex cord tumour with annular tubules Germ cell Dysgerminoma Nongerminomatous Embryonal carcinoma Endodermal sinus tumor Gonadoblastoma Teratoma / Struma ovarii Choriocarcinoma Fibroma Meigs' syndrome Fallopian tube Adenomatoid tumor Uterus Myometrium Uterine fibroids/leiomyoma Leiomyosarcoma Adenomyoma Endometrium Endometrioid tumor Uterine papillary serous carcinoma Endometrial intraepithelial neoplasia Uterine clear-cell carcinoma Cervix Cervical intraepithelial neoplasia Clear-cell carcinoma SCC Glassy cell carcinoma Villoglandular adenocarcinoma Placenta Choriocarcinoma Gestational trophoblastic disease General Uterine sarcoma Mixed Müllerian tumor Vagina Squamous-cell carcinoma of the vagina Botryoid rhabdomyosarcoma Clear-cell adenocarcinoma of the vagina Vaginal intraepithelial neoplasia Vaginal cysts Vulva SCC Melanoma Papillary hidradenoma Extramammary Paget's disease Vulvar intraepithelial neoplasia Bartholin gland carcinoma
-
Verrucous Carcinoma
Wikipedia
Contents 1 Signs and symptoms 2 Cause 3 Risk factors 4 Locations 5 Diagnosis 5.1 Subtypes 6 Treatment 7 Prognosis 8 See also 9 References 10 Further reading 11 External links Signs and symptoms [ edit ] Age - usually over 60 years old Sex - males are more prone Site - gingiva, buccal mucosa, alveolar mucosa , hard palate, floor of the mouth, larynx, oesophagus, penis, vagina, scrotum. ... Risk factors [ edit ] The major risk factors are cigarette smoking and alcohol consumption, while betel nut is an additional factor in Taiwan. Different gene mutation sites in head and neck cancer between western countries and Taiwan have been reported. [2] [3] [4] [5] The presentation of VC originated from exposure to different carcinogens may not be the same. ... The oral cavity is the most common site of this tumor. [6] The ages range from 50 to 80 years with a male predominance and a median age of 67 years. [7] VC may grow large in size, resulting in the destruction of adjacent tissue, such as bone and cartilage. [8] Diagnosis [ edit ] Surgeons must provide adequate specimens including the full thickness of the tumors and adjacent uninvolved mucosa for correct histopathology diagnosis. [9] Low-magnification micrograph of penile verrucous carcinoma. ... ISBN 978-1-4160-2999-1 . ^ a b Suen, K.; Wijeratne, S.; Patrikios, J. (2012). "An unusual case of bilateral verrucous carcinoma of the foot (epithelioma cuniculatum)" .TP53, CDKN2A, H3P10, MMP26, TP63, SOCS1, ZNRD2, DCTN6, HPSE, CKAP4, RASSF1, LRIG1, TMED7, HCCAT5, MIB1, TICAM2, MIR125A, MIR195, MIR19A, MIR203A, MIR21, MIR31, TMED7-TICAM2, H3P23, UVRAG, CCND1, BCL2, MGMT, CDH1, CDK6, EGFR, ERBB2, IFI27, KRT5, MCM2, MCM7, MDM2, MKI67, SPRR3, MMP1, NOTCH4, MAPK1, PSMD9, PTEN, RAF1, RNASE3, RPE65, SLC2A1, TGFA

-
Bovine Viral Diarrhea
Wikipedia
Chronic infections [ edit ] BVD virus can be maintained as a chronic infection within some immunoprivileged sites following transient infection. These sites include ovarian follicles, testicular tissues, central nervous system and white blood cells. ... PMID 28190502 . ^ Pinior, B; Firth, C; Richter, V; Lebl, K; Trauffler, M; Dzieciol, M; Hutter, S; Burgstaller, J; Obritzhauser, W; Winter, P; Käsbohrer, A (2017). ... Factors regulating template switch in vitro by viral RNA-dependent RNA polymerases: implications for RNA-RNA recombination. Proc Natl Acad Sci U S A. 2001 Apr 24;98(9):4972-7. doi: 10.1073/pnas.081077198. ... Bovine Viral Diarrhoea Virus, expert reviewed and published by Wikivet at http://en.wikivet.net/Bovine_Viral_Diarrhoea_Virus , accessed 21/07/2011 External links [ edit ] New York State Cattle Health Assurance Program BVD Module Description of the entity on the Merck Veterinary Manual Animal viruses Bovine Viral Diarrhea Resource Page Specialist BVD site, Royal Veterinary College, London Taxon identifiers Bovine viral diarrhea virus 1 Wikidata : Q18968322 Wikispecies : Pestivirus A EoL : 541189 IRMNG : 11459300 NCBI : 11099 Bovine viral diarrhea virus 2 Wikidata : Q18968331 Wikispecies : Pestivirus B EoL : 541186 IRMNG : 11459301 NCBI : 54315 Pestivirus A Wikidata : Q51930329 NCBI : 2170080 Pestivirus B Wikidata : Q51930337 NCBI : 2170081
-
Kearns-Sayre Syndrome
Omim
Two of the 3 deletions may have included nucleotide 8993 which has been demonstrated to be the site of mutation in NARP syndrome (551500; 516060.0001). ... The deletion was bracketed by direct repeats that were unusual in that one of them was located 11-13 nucleotides from the deletion site and both were conserved, which should not occur in slip replication or illegitimate elongation. ... To test this concept, Shoubridge et al. (1997) rebiopsied a patient with the KSS phenotype and an mtDNA point mutation in the MTTL2 gene (590055.0001) and analyzed muscle fibers regenerating at the site of the original muscle biopsy. Regenerating fibers were identified by morphologic criteria and by expression of neural cell adhesion molecule (116930). ... As with several other reported deletions, the defect in this case preserved the promoters of transcription of heavy and light strands, the 12-S and 16-S ribosomal RNA genes, and the origin of heavy strand replication.
-
Shox Deficiency Disorders
Gene_reviews
The phenotypic spectrum of SHOX deficiency disorders, caused by haploinsufficiency of the s hort stature ho meobo x- containing gene ( SHOX ), ranges from Leri-Weill dyschondrosteosis (LWD) at the severe end of the spectrum to nonspecific short stature at the mild end of the spectrum. ... Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. ... Ideally, determining heterozygosity for (a) SNP(s) involves analysis of DNA from the proband and both parents. ... Option 2 When SHOX-deficient short stature is indistinguishable from many other inherited disorders characterized by short stature, comprehensive genomic testing –which does not require the clinician to determine which gene(s) are likely involved – is the best option. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.
-
Kat6b Disorders
Gene_reviews
Diagnosis KAT6B disorders include genitopatellar syndrome (GPS) and Say-Barber-Biesecker variant of Ohdo s yndrome ( S ay- B arber- B iesecker- Y oung- S impson s yndrome; SBYSS). ... Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. ... Sequence analysis of KAT6B is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Genes of Interest in the Differential Diagnosis of KAT6B Disorders View in own window Gene(s) Differential Diagnosis Disorder MOI Clinical Features of Differential Diagnosis Disorder Overlapping w/ KAT6B Disorders Not Observed in KAT6B Disorders CDC45 CDC6 CDT1 GMNN ORC1 ORC4 ORC6 Meier-Gorlin syndrome (OMIM PS224690) AR (AD) Patellar aplasia or hypoplasia; microcephaly; genital anomalies; contractures Severe intrauterine & postnatal growth restriction; bilateral microtia ERCC6 ERCC8 Cerebrooculofacioskeletal syndrome (severe fetal form of Cockayne syndrome) AR Arthrogryposis; microcephaly; severe ID Progressive neurodegenerative disorder; congenital cataracts & facial dysmorphism FBN2 Congenital contractural arachnodactyly AD Congenital contractures Marfanoid habitus; aortic root dilatation; arachnodactyly; hypoplastic calf muscles FOXL2 Blepharophimosis, ptosis, epicanthus inversus syndrome 1 AD 2 Blepharophimosis & ptosis Epicanthus inversus (a fold of skin that runs from the lower lids inwards & upwards) LMX1B Nail-patella syndrome AD Absent patella; renal anomalies; flexion deformities of knees & hips, clubfoot Proteinuria; open-angle glaucoma; nail changes RECQL4 RAPADILINO syndrome (OMIM 266280) AR Patellar hypoplasia; hearing loss; cleft palate Irregular pigmentation w/café au lait spots; normal intelligence; palate defects; radial ray defects; GI abnormalities TBX4 Ischiocoxopodopatellar syndrome (OMIM 147891) AD Patellar aplasia or hypoplasia Absent, delayed, or irregular ossification of the ischiopubic junctions or infraacetabular axe-cut notches ZEB2 Mowat-Wilson syndrome AD 3 Agenesis of the corpus callosum; genital anomalies; ID; congenital heart disease; microcephaly Seizures; Hirschsprung disease; short stature AD = autosomal dominant; AR = autosomal recessive; GI = gastrointestinal; ID = intellectual disability; MOI = mode of inheritance; XL = X-linked 1.
-
Fibrous Dysplasia/mccune-Albright Syndrome
Gene_reviews
Fibrous dysplasia/McCune-Albright syndrome (FD/MAS), the result of an early embryonic postzygotic somatic activating pathogenic variant in GNAS (encoding the cAMP pathway-associated G-protein, G s α), is characterized by involvement of the skin, skeleton, and certain endocrine organs. However, because G s α signaling is ubiquitous, additional tissues may be affected. ... GNAS variants other than those previously reported to be associated with FD/MAS would likely be interpreted as variants of unknown significance. (3) G s α is expressed in nearly all tissues from both maternal and paternal GNAS alleles. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... However, because G s α signaling is present in virtually every tissue, additional sites may be affected.
-
Arsacs
Gene_reviews
Summary Clinical characteristics. A utosomal r ecessive s pastic a taxia of C harlevoix- S aguenay (ARSACS) is clinically characterized by a progressive cerebellar ataxia, peripheral neuropathy, and spasticity. ... Population screening for the most common SACS pathogenic variants is an option for individuals with a genealogic link to regions of high ARSACS prevalence. Diagnosis A utosomal r ecessive s pastic a taxia of C harlevoix- S aguenay (ARSACS) is clinically characterized by a progressive cerebellar ataxia, peripheral neuropathy, and spasticity. ... Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. ... Sequence analysis of SACS detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.SACS, ANKFY1, CRMP1, UTRN, DAPK2, DENR, DNM1L, SGCG, TRPS1, SPG7, VIM, LAMP2, AFG3L2, SETX, RIEG2, SUN2, POLG, DCPS, P4HB, APTX, PACC1, COQ8A, SLURP1

-
Nail-Patella Syndrome
Omim
Abnormalities of collagen at this site have also been demonstrated in Alport syndrome (104200). ... For the adults with NPS, BMD was 11 to 20% lower at the hip sites (P less than or equal to 0.001) and 8% lower at the spine (P less than 0.05) than that of controls. ... The results were as expected if the modifier(s) is an allele at the NPS locus, and inherited from the unaffected parent, i.e., as an isoallele. ... Animal Model Dunston et al. (2005) studied the expression of Lmx1b during development by inserting an internal ribosomal entry site-LacZ reporter into the 3-prime untranslated region (UTR) of the endogenous murine gene. The pattern of Lmx1b expression during the development of the limb, eye, and kidney correlated with the NPS phenotype. Additional sites of expression were observed in the central nervous system.LMX1B, LDB1, SMOC1, LAMC2, COL5A1, CSF2, ADAMTS2, PDLIM5, APOE, PAX2, MCIDAS, AK1, PTLAH, ABL1, EBI3, NXF1, USO1, SEC14L2, CD2AP, REM1, NPSR1, IL18R1, PAEP, TNF, MAPK8, MTTP, MEFV, MAPT, IL18, IL15, HCRT, GSN, GHRHR, NR5A1, FLNB, CASP3, ASS1, NPS
-
Short Syndrome
Gene_reviews
Summary Clinical characteristics. SHORT syndrome is a mnemonic for s hort stature, h yperextensibility, o cular depression (deeply set eyes), R ieger anomaly, and t eething delay. ... Diagnosis The designation SHORT syndrome was coined by Gorlin et al [1975] to reflect several of the most striking clinical features of the original reported cases: s hort stature, h yperextensibility, o cular depression (deeply set eyes), R ieger anomaly, and t eething delay. ... Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. ... Sequence analysis of PIK3R1 is performed first to detect missense, frameshift, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.

-
Pik3ca-Related Segmental Overgrowth
Gene_reviews
., CLOVES [ c ongenital l ipomatous asymmetric o vergrowth of the trunk, lymphatic, capillary, venous, and combined-type v ascular malformations, e pidermal nevi, s keletal and s pinal anomalies] syndrome, fibroadipose hyperplasia [FH]). ... CLOVES syndrome is characterized by c ongenital l ipomatous asymmetric o vergrowth of the trunk, lymphatic, capillary, venous, and combined-type v ascular malformations, e pidermal nevi, s keletal and s pinal anomalies. CLOVES syndrome differs from MCAP syndrome by more striking growth dysregulation with complex congenital overgrowth of lipomatous tissues (typically manifest as a truncal lipomatous mass) and combined lymphatic and vascular malformations (Figure 4A). ... Pathogenic variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... Most identified PIK3CA pathogenic variants to date are missense, with only a single in-frame 3-bp deletion identified in MCAP syndrome [Rivière et al 2012]. No nonsense or splice site variants, large deletions, or duplications involving PIK3CA have been identified to date. 6. ... Referral to the appropriate specialist(s) is recommended for the following findings: Significant or lipomatous segmental overgrowth: referral to a surgeon and/or thoracic surgeon (when lipomatous overgrowth involves the trunk) Leg-length discrepancy secondary to segmental somatic overgrowth Cardiac abnormalities (i.e., structural cardiovascular disease and arrhythmias) Renal abnormalities Intellectual disability and/or difficulties with learning, behavior, or speech, or motor difficulties Speech, swallowing, and feeding difficulties Neurologic and neurosurgical manifestations MCAP syndrome.
-
Morgellons
Wikipedia
"They said they were not interested in seeing him because I had Munchausen Syndrome by Proxy". [14] Leitao says that her son developed more sores, and more fibers continued to poke out of them. [5] [12] She and her husband, Edward Leitao, an internist with South Allegheny Internal Medicine in Pennsylvania , felt their son had "something unknown". [4] She chose the name Morgellons disease (with a hard g ) from a description of an illness in the medical case-history essay, A Letter to a Friend (c. 1656, pub. 1690) by Sir Thomas Browne , where the physician describes several medical conditions in his experience, including "that endemial distemper of children in Languedoc , called the morgellons , wherein they critically break out with harsh hairs on their backs". [5] [6] Leitao started the Morgellons Research Foundation (MRF) informally in 2002 and as an official non-profit in 2004. [5] [15] The MRF website states that its purpose is to raise awareness and funding for research into the proposed condition, described by the organization as a "poorly understood illness, which can be disfiguring and disabling". [16] Leitao stated that she initially hoped to receive information from scientists or physicians who might understand the problem, but instead, thousands of others contacted her describing their sores and fibers, as well as neurological symptoms, fatigue, muscle and joint pain, and other symptoms. [5] The MRF claimed to have received self-identified reports of Morgellons from all 50 US states and 15 other countries, including Canada, the UK, Australia, and the Netherlands, and states that it has been contacted by over 12,000 families. [16] In 2012 the Morgellons Research Foundation closed down and directed future inquiries to Oklahoma State University. [17] Media coverage In May 2006, a CBS news segment on Morgellons aired in Southern California. [18] The same day, the Los Angeles County Department of Health services issued a statement saying, "No credible medical or public health association has verified the existence or diagnosis of 'Morgellons Disease ' ", and "at this time there is no reason for individuals to panic over unsubstantiated reports of this disease". [19] In June and July 2006 there were segments on CNN , [20] ABC 's Good Morning America , [21] and NBC 's The Today Show . In August 2006, a segment of the ABC show Medical Mysteries [12] was devoted to the subject. Morgellons was featured on ABC 's Nightline on January 16, 2008, [22] and as the cover story of the January 20, 2008, issue of the Washington Post . [7] The first article to propose Morgellons as a new disease in a scientific journal was a review article co-authored by members of the MRF and published in 2006 by the American Journal of Clinical Dermatology . [23] A 2006 article in the San Francisco Chronicle reported, "There have been no clinical studies" of Morgellons disease. [23] A New Scientist article in 2007 also covered the phenomenon, noting that people are reporting similar symptoms in Europe and Australia. [24] In an article published in the Los Angeles Times on April 22, 2010, singer-songwriter Joni Mitchell claimed to have the condition. [25] On June 13, 2011, the Australian Broadcasting Corporation 's Radio National broadcast The Mystery of Morgellons with guests including Mayo Clinic Professor Mark Davis. [26] CDC investigation The Morgellons Research Foundation coordinated a mailing campaign via their website, in which thousands of people sent form letters to a Centers for Disease Control and Prevention (CDC) task force, which first met in June 2006. [7] [27] [28] By August 2006, the task force consisted of 12 people, including two pathologists, a toxicologist, an ethicist, a mental health expert, and specialists in infectious, parasitic, environmental and chronic diseases. [29] In June 2007, the CDC opened a website relating to Morgellons, CDC Study of an Unexplained Dermopathy , and by November 2007, the CDC opened an investigation into the condition. [8] Kaiser Permanente , a health-care consortium in Northern California, was chosen to assist with the investigation, which involved skin biopsies from affected people and characterization of foreign material such as fibers or threads obtained from people to determine their potential source. [8] [30] The U.S. ... In many cases, these attempts are well-intentioned, yet wrong, and a person's belief in some of these oftentimes unscientific sites online may preclude their trust in the evidence-based approaches and treatment recommendations of their physician." [35] Vila-Rodriguez states that the Internet promotes the spreading and supporting of "bizarre" disease beliefs, because "a belief is not considered delusional if it is accepted by other members of an individual's culture or subculture". [33] Robert Bartholomew , a sociologist who has studied the Morgellons phenomenon, states that the "World Wide Web has become the incubator for mass delusion and it (Morgellons) seems to be a socially transmitted disease over the Internet." ... Dermatologist Caroline Koblenzer specifically faults the Morgellons Research Foundation (MRF) website for misleading people: "Clearly, as more and more of our patients discover this site (MRF), there will be an ever greater waste of valuable time and resources on fruitless research into fibers, fluffs, irrelevant bacteria, and innocuous worms and insects." [37] A 2005 Popular Mechanics article stated that Morgellons symptoms are well known and characterized in the context of other disorders, and that "widespread reports of the strange fibers date back" only a few years to when the MRF first described them on the Internet. [38] The Los Angeles Times , in an article on Morgellons, notes that "[t]he recent upsurge in symptoms can be traced directly to the Internet, following the naming of the disease by Mary Leitao, a Pennsylvania mother". [34] In 2008, The Washington Post reported that Internet discussions about Morgellons include many conspiracy theories about the cause, including biological warfare , nanotechnology , chemtrails and extraterrestrial life . [7] The Atlantic says it "even received pop-culture attention" when it was featured on Criminal Minds , adding that "Morgellons patients have further alienated themselves from the mainstream medical community" by "linking Morgellons to another illness viewed skeptically by most doctors, chronic Lyme disease , and by attacking those who doubt their condition". [39] Jay Traver Jay Traver (1894–1974), a University of Massachusetts entomologist , was known for "one of the most remarkable mistakes ever published in a scientific entomological journal", [40] after publishing a 1951 account of what she called a mite infestation [41] which was later shown to be incorrect, [40] and that has been described by others as a classic case of delusional parasitosis as evidenced by her own detailed description. [42] [43] [44] [45] Matan Shelomi argues that the historical paper should be retracted because it has misled people about their delusion. [44] He says the paper has done "permanent and lasting damage" to people with delusional parasitosis, "who widely circulate and cite articles such as Traver's and other pseudoscientific or false reports" via the internet, making treatment and cure more difficult. [44] See also Culture-bound syndrome Formication Fringe medicine List of topics characterized as pseudoscience Mass psychogenic illness Matchbox sign Medicalization Münchausen syndrome Quaternary prevention Somatic symptom disorder References ^ a b Vulink, NC (August 23, 2016). ... Archived from the original on July 9, 2011. ^ Koblenzer, Caroline S. (2006). "The challenge of Morgellons disease".
-
L1 Syndrome
Gene_reviews
L1 syndrome involves a phenotypic spectrum ranging from severe to mild and includes three clinical phenotypes: X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (HSAS) MASA ( m ental retardation [intellectual disability], a phasia [delayed speech], s pastic paraplegia [shuffling gait], a dducted thumbs) syndrome including X-linked complicated hereditary spastic paraplegia type 1 X-linked complicated corpus callosum agenesis Males with HSAS are born with severe hydrocephalus, adducted thumbs, and spasticity; intellectual disability is severe. ... Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. ... Sequence analysis of L1CAM is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. ... Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. ... In MASA ( m ental retardation [intellectual disability], a phasia [delayed speech], s pastic paraplegia [shuffling gait], a dducted thumbs) syndrome, intellectual disability ranges from mild (IQ: 50-70) to moderate (IQ: 30-50).L1CAM, PLP1, PTHLH, PRDX5, PDXP, ENOPH1, CALM1, CALM2, CALM3, CANX, KRIT1, F7, F8, RNF6, CAMKMT, ASRGL1













