-
Telogen Effluvium
Wikipedia
Lookingbill and Marks' Principles of Dermatology (4th ed.). Elsevier Inc. Page 263. ISBN 1-4160-3185-5 . ^ James, William; Berger, Timothy; Elston, Dirk (2005).
-
Meningoencephalitis
Wikipedia
Please help improve it or discuss these issues on the talk page . ( Learn how and when to remove these template messages ) This article may require cleanup to meet Wikipedia's quality standards .PLAT, AIF1, LAMC2, CSF2, GFAP, AQP4, MOG, CARD9, IFNG, TNF, CXCL13, TLR8, SIRT1, DDX58, ROBO3, SLC9B1, IFNL3, PPIG, CYTB, PLAAT4, PTX3, MAPT, IL10, IL6, IGF2R, ENO2, CSNK2A1, CD40LG, CAT, OCLN

-
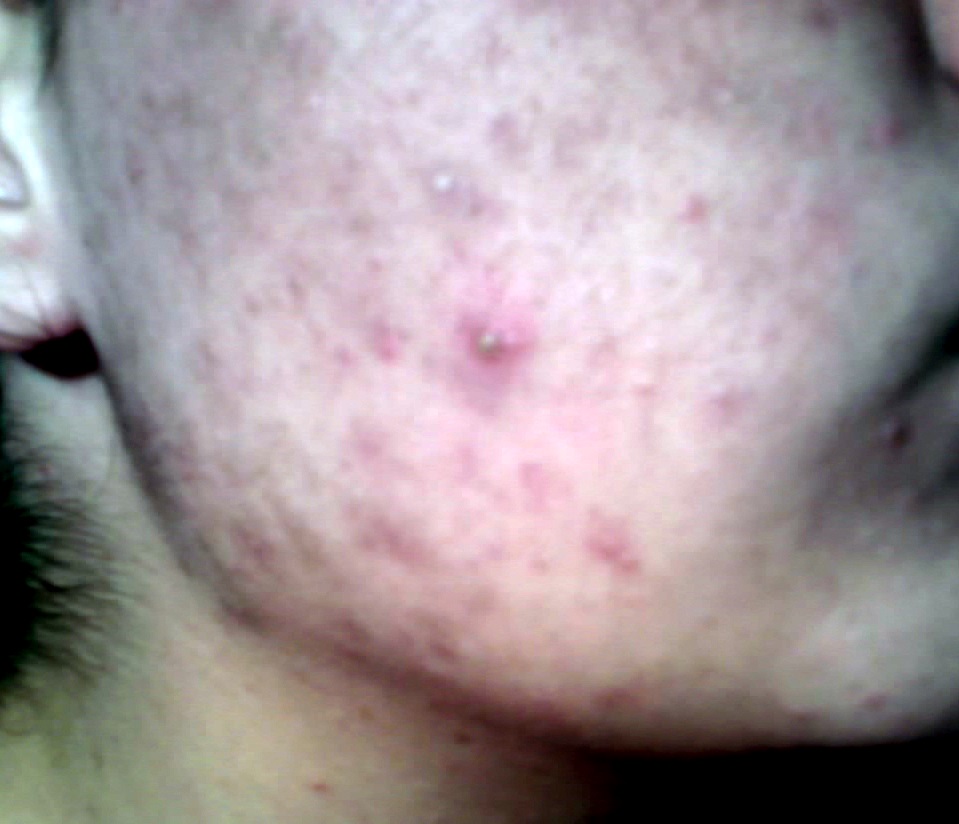
Pimple
Wikipedia
Retrieved 1 June 2017 . See FDA Index page for NDA 018662 for updates ^ Strauss JS, Krowchuk DP, Leyden JJ, Lucky AW, Shalita AR, Siegfried EC, Thiboutot DM, Van Voorhees AS, Beutner KA, Sieck CK, Bhushan R (April 2007).CTSC, CTSB, TMC8, CARD14, SLC39A4, IL36RN, IL17RA, TMC6, CIB1, LPIN2, PSTPIP1, ADAM17, JAK3, IL7, IL1RN, EGFR, ECM1, STING1
-
Pigmented Paravenous Chorioretinal Atrophy
Omim
Inheritance Father-to-son transmission of PPCRA in the family reported by Skalka (1979) indicated autosomal dominant inheritance. The pedigree described by Traboulsi and Maumenee (1986) was considered consistent with X-linked inheritance because the sons were more severely affected than the mother. ... Eyes - Pigmented paravenous chorioretinal atrophy - Bone corpuscle fundus pigmentation - Hyperopia - Esotropia - Vitreoretinal degeneration Misc - Usually asymptomatic Inheritance - Autosomal dominant vs. X-linked ▲ Close
-
Albinism, Ocular, With Sensorineural Deafness
Omim
Deafness, which was accompanied by vestibular hypofunction, lentigines even in unexposed areas, optic nerve dysplasia, and dominant inheritance distinguished this form of ocular albinism. ... Eyes - Reduced visual acuity - Photophobia - Nystagmus - Translucent irides - Strabismus - Hypermetropia - Albinotic fundus - Foveal hypoplasia - Optic nerve dysplasia Ears - Deafness - Vestibular hypofunction Inheritance - Autosomal dominant form - also X-linked Lab - Macromelanosomes Skin - Hypomelanosis - Lentigines ▲ Close
-
Wt Limb-Blood Syndrome
Omim
Important characteristics differentiating this condition from Fanconi anemia (227650) were autosomal dominant inheritance and absence of chromosome breakage. ... Shahidi (1987) gave comparisons with Fanconi anemia and dyskeratosis congenita (305000). INHERITANCE - Autosomal dominant HEAD & NECK Face - Micrognathia - Retrognathia Ears - Sensorineural hearing loss GENITOURINARY Internal Genitalia (Male) - Cryptorchidism SKELETAL Limbs - Radioulnar synostosis Hands - Fifth finger clinodactyly - Short fingers - Absent thumbs - Hypoplastic thumbs - Ulnar deviation of 1st, 3rd fingers - Fifth finger camptodactyly SKIN, NAILS, & HAIR Skin - Irregular hyperpigmentation HEMATOLOGY - Pancytopenia - Thrombocytopenia - Hypoplastic anemia NEOPLASIA - Leukemia LABORATORY ABNORMALITIES - No chromosome breakage ▲ Close
-
Cortical Defects, Wormian Bones, And Dentinogenesis Imperfecta
Omim
Inheritance Moog et al. (1999) proposed either autosomal recessive inheritance, with dentinogenesis imperfecta in the mother and children representing a separate entity (dentinogenesis imperfecta type II; 125490), or, alternatively, autosomal dominant inheritance with dentinogenesis imperfecta forming part of this condition, with the lack of other features in the mother due to variable expression. INHERITANCE - Autosomal dominant GROWTH Height - Short stature (3rd percentile) HEAD & NECK Head - Dolichocephaly - Delayed closure of anterior fontanel Eyes - Hypertelorism - Periorbital fullness Teeth - Dentinogenesis imperfecta - Dental caries CHEST External Features - Asymmetric chest Ribs Sternum Clavicles & Scapulae - Slender ribs SKELETAL - Bone fragility Skull - Wormian bones Limbs - Restricted elbow extension - Wave-like defects of tibial corticalis (alternating hyperostosis and thinning) - Epiphyseal streaking Feet - 3rd-4th toe clinodactyly LABORATORY ABNORMALITIES - Normal collagen type I studies ▲ Close
-
Tibial Hemimelia
Omim
They suggested that there are 4 well-established and 2 other possible autosomal dominant tibial hemimelia syndromes in addition to 2 types with autosomal recessive inheritance. ... Limbs - Absent tibia Inheritance - Autosomal recessive - also other dominant and recessive tibial hemimelia syndromes ▲ Close
-
Nievergelt Syndrome
Omim
Nakamura et al. (2007) suggested that the Savarirayan and Nievergelt types may be allelic autosomal dominant disorders. History Urban and Kruger (1998) suggested that Nievergelt syndrome was the correct diagnosis in 24-year-old Alice Vance from Mount Pleasant, Texas, who was among the malformed individuals presented at the world exhibition in Antwerp in 1894. ... Her mother was known to have similar malformations. INHERITANCE - Autosomal dominant GROWTH Height - Dwarfism, short limb mesomelic HEAD & NECK Face - Normal face SKELETAL Limbs - Lower leg mesomelia - Genua valga - Rhomboidal shape of tibiae and fibulae - Relative overgrowth of fibula - Radioulnar synostosis - Radial head subluxation Feet - Clubfoot - Tarsal bone synostosis - Metatarsal synostosis SKIN, NAILS, & HAIR Skin - Dimples (medial and lateral aspect of lower leg) NEUROLOGIC Central Nervous System - Normal intelligence ▲ Close
-
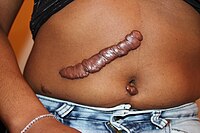
Keloid Formation
Omim
The pattern of inheritance observed in these families was consistent with an autosomal dominant mode with incomplete clinical penetrance and variable expression. ... In addition, association with keloid was found with rs8032158 in the NEDD4 gene (602278) on chromosome 15q21.3 (p = 5.96 x 10(-13); OR, 1.51). Inheritance - Autosomal dominant Skin - Keloids ▲ Close
-
Congenital Dyserythropoietic Anemia Type Iii
Orphanet
Three families have been reported with autosomal dominant CDA III in Sweden, America and Argentina. ... It is inherited in an autosomal dominant mode. Other sporadic CDA III-like cases have been reported with an autosomal recessive pattern of inheritance, suggesting a different genetic alteration than KIF23 associated CDA III and possibly another subtype of CDA.
-
Lutheran Null
Omim
Since it is inherited as an autosomal dominant trait, it was initially postulated to result from an inhibitor of the Lu antigen. ... INHERITANCE - Autosomal recessive HEMATOLOGY - Absence of Lutheran antigen on red blood cells - Lu(a-b-) phenotype LABORATORY ABNORMALITIES - Presence of serum anti-Lu3 antibodies MISCELLANEOUS - No phenotypic manifestations - See also autosomal dominant Lutheran-null phenotype ( 111150 ) MOLECULAR BASIS - Caused by mutation in the B-cell adhesion molecule gene (BCAM, 612773.0001 ) ▲ Close
-
Fibular Aplasia, Tibial Campomelia, And Oligosyndactyly Syndrome
Omim
The authors suggested recessive inheritance or gonadal mosaicism for a dominant mutation. Courtens et al. (2005) reported an infant born of nonconsanguineous Moroccan parents with oligosyndactyly of the left hand, complete absence of the right fibula, bowing of the right tibia, and absence of the right fifth metatarsal and phalanges. ... Genetic analysis showed no mutation in the WNT7A gene (601570). INHERITANCE - Autosomal dominant - Isolated cases SKELETAL Limbs - Absence of the fibula - Hypoplasia of the fibula - Bowing of the tibia - Shortening of the tibia Hands - Oligosyndactyly Feet - Oligosyndactyly MISCELLANEOUS - Present at birth - Variable phenotype ▲ Close
-
Deafness, Autosomal Recessive 26
Omim
Riazuddin et al. (2000) also mapped a dominant deafness modifier, designated DFNM1 (605429), that suppressed deafness in the 7 nonpenetrant individuals to a 5.6-cM region on chromosome 1q24, with a lod score of 4.31 at theta = 0.0 for D1S2815. ... Reviews Nadeau (2001) reviewed modifier genes in mice and humans, dividing the types of modification as follows: reduced penetrance, dominance modification, expressivity, and pleiotropy.
-
Deafness, Autosomal Recessive 26, Modifier Of
Omim
Riazuddin et al. (2000) mapped a dominant deafness modifier, which they designated DFNM1, that suppressed deafness in the 7 nonpenetrant individuals to a 5.6-cM region on chromosome 1q24, with a lod score of 4.31 at theta = 0.0 for D1S2815. ... The authors suggested that differential regulation of SPRY2 might be the mechanism by which the METTL13 variant functions as a modifier to prevent deafness caused by mutation in the GAB1 gene. INHERITANCE - Autosomal dominant HEAD & NECK Ears - Normal hearing despite being homozygous for a deafness-causing mutation in the GAB1 gene ( 604439.0001 ) MISCELLANEOUS - Based on report of 1 large consanguineous Pakistani family (last curated June 2018) MOLECULAR BASIS - Protection conferred by mutation in the methyltransferase-like 13 gene (METTL13, 617987.0001 ) ▲ Close
-
Fatal Insomnia
Wikipedia
Similar to other prion diseases, the diagnosis can only be confirmed by a brain autopsy at post-mortem. [1] Fatal insomnia has no known cure and involves progressively worsening insomnia, which leads to hallucinations, delirium, confusional states like that of dementia, and eventually death. [6] The average survival time from onset of symptoms is 18 months. [6] The first recorded case was an Italian man, who died in Venice in 1765. [7] Contents 1 Signs and symptoms 2 Cause 3 Pathophysiology 4 Diagnosis 4.1 Differential diagnosis 5 Treatments 6 Prognosis 7 Epidemiology 7.1 Silvano, 1983, Bologna, Italy 7.2 Unnamed patient of Schenkein and Montagna, 2001 7.3 Egyptian man, 2011, Netherlands 8 Research 9 Popular culture 10 References 11 External links Signs and symptoms [ edit ] The disease has four stages: [8] The person has increasing insomnia , resulting in panic attacks , paranoia , and phobias . ... In 1998, 40 families were known to carry the gene for FFI globally: eight German, five Italian, four American, two French, two Australian, two British, one Japanese, and one Austrian. [18] In the Basque Country , Spain, 16 family cases of the 178N mutation were seen between 1993 and 2005 related to two families with a common ancestor in the 18th century. [19] In 2011, another family was added to the list when researchers found the first man in the Netherlands with FFI. ... He managed to write a book and drive hundreds of miles in this time, but nonetheless, over the course of his trials, the person succumbed to the classic four-stage progression of the illness. [24] [23] Egyptian man, 2011, Netherlands [ edit ] Timeline of an FFI patient (same as the one above this one) In 2011, the first reported case in the Netherlands was of a 57 year-old man of Egyptian descent. ... The 2019 movie, Awoken , uses FFI as a major plot element. [ citation needed ] FFI is a major plot element and is described in detail in the Lewis episode "Falling Darkness". In the first episode of the 2020 TV series Next , one of the main characters confesses to have the disease and says "It's a real thing, look it up." ... W.; Capellari, S.; Rozemuller, A. J. M. (13 July 2011). "The first case of fatal familial insomnia (FFI) in the Netherlands: a patient from Egyptian descent with concurrent four repeat tau deposits".PRNP, C4BPA, CARD14, GLS, ABCB6, PRDX2, RYR2, ZBTB38, MLKL, MPRIP, RIPK3, UQCRC2, SDHB, AQP1, RASA2, TSPO, PRL, NDUFB8, COX2, MDH1, GH1, GFAP, CSNK2A2, CHI3L1, MIR146A

-
Aniridia And Absent Patella
Omim
The patella was either hypoplastic or aplastic. INHERITANCE - Autosomal dominant HEAD & NECK Eyes - Aniridia - Cataracts - Glaucoma SKELETAL Limbs - Hypoplastic patella ▲ Close
-
Ear Antitragus, Tag At Base Of
Omim
One of the families contained an instance of male-to-male transmission. Inheritance - Autosomal dominant Ears - Tag, nodule or localized elevation at base of antitragus ▲ Close
-
Deafness-Ear Malformation-Facial Palsy Syndrome
Orphanet
It has been described in three sibs and their mother. Inheritance is autosomal dominant.
-
Pseudoatrophoderma Colli
Omim
Frost and Epstein (1939) described affected sisters. Inheritance - Autosomal dominant Skin - Pseudoatrophic papillary and pigmentary dermatosis of neck ▲ Close




