-
Osteosclerosis With Ichthyosis And Fractures
Omim
Koller et al. (1979) described an apparently 'new' dominant disorder characterized by cortical thickening of the diaphyses of long bones and bowing of femurs and tibias in a family from northern Norway. ... Skel - Osteosclerosis - Increased fractures Limbs - Bowed femurs and tibias Radiology - Cortical thickening of long bone diaphyses Inheritance - Autosomal dominant Skin - Ichthyosis ▲ Close
-
Dimples, Facial
Omim
Cheek dimples may be inherited as an irregular dominant. Wiedemann (1990) described a unilateral cheek dimple in a 5-year-old girl whose mother had a similar dimple when she was a child, also in her left cheek, and only when she smiled. ... Facies - Cheek dimples Inheritance - Autosomal dominant ▲ Close
-
Hypertelorism
Omim
Inheritance Bojlen and Brems (1938) traced hypertelorism through 5 generations in a pattern consistent with autosomal dominant inheritance. Friede (1954) described affected mother and daughter. Eyes - Hypertelorism Inheritance - Autosomal dominant ▲ CloseEFNB1, ACOX1, KAT6B, SPECC1L, SIK3, MED13L, DICER1, GRIP1, TGDS, SUZ12, ATP6V0A2, RPGRIP1L, PIGN, LEMD3, POLR1A, SIN3A, SH2B1, PARS2, SMCHD1, CAMTA1, WDR4, MRAS, TXNL4A, CIT, POLR3A, IL1RAPL1, AP4S1, MAN1B1, CHSY1, KIAA0556, IQSEC2, SPART, NFASC, POGZ, MAPK8IP3, PACS2, NSMF, SETBP1, AUTS2, TCTN3, PSAT1, EFEMP2, VSX1, SLC45A1, SOST, TPRKB, WDPCP, POLR1D, RLIM, TMEM216, RSRC1, WAC, ACTL6B, PTRH2, SUFU, SLC25A24, DSE, BLNK, FOXP1, KIFBP, PHGDH, B3GAT3, FGF20, ELP4, B9D1, AFF4, ANKRD11, INTU, AHDC1, PGAP2, CCDC22, SETD2, UBE2T, CPLX1, ZMYND11, RIPK4, ADAMTS3, DPM1, FGF17, CACNA1G, HERC1, SEMA5A, FIBP, TRIP12, TRIP4, SNAP29, COG1, HS6ST1, RECQL4, EIF2AK3, CHST3, PIGL, CCNK, GPAA1, EED, OFD1, RBM10, SMC1A, LAGE3, TRRAP, LTBP4, OGT, PEX3, PTCH2, CNTNAP1, ITGA8, CDK10, CASK, CDK13, DCHS1, TBX4, POLR1C, RAI1, SEC24C, IRX5, ZMPSTE24, APC2, KLHL41, MAD2L2, ZBTB18, SEC23A, FBLN5, DEAF1, COLEC10, STAMBP, SPINT2, EBP, TBR1, SIX2, LRPPRC, ABCC9, GNE, ZBTB24, TTC37, SEMA3E, TMEM94, KIAA0586, PTDSS1, ZEB2, KIAA0753, AKT3, RUSC2, SEC24D, FIG4, WASHC5, MED12, SLC12A6, FGFRL1, TMCO1, LZTR1, TP53RK, TMEM107, SLX4, KISS1R, MYPN, PIGO, TMEM87B, WDR73, PIGY, COL27A1, UBE3B, RSPRY1, TICRR, TMEM67, STRADA, PGAP3, ANTXR1, KIAA1109, BRIP1, FBXO11, CSPP1, TCTN2, ALG13, GREB1L, FRAS1, CEP290, B9D2, CDCA7, ASXL3, SLC2A10, SPRY4, MED25, DDX59, TRAPPC9, IFT43, CHST14, BNC2, C12orf57, PHACTR1, EBF3, NALCN, KANSL1, PIGW, DOK7, SH3PXD2B, FAM149B1, FREM2, CTU2, KIF7, KBTBD13, GTF2H5, RNU4ATAC, TTN-AS1, JMJD1C, TUBB, TAPT1, B3GLCT, TWIST2, PROKR2, DNAJC21, MPLKIP, AMER1, A2ML1, CCBE1, FLCN, CEP120, ESCO2, FREM1, SPRED1, BMPER, ASXL1, EHMT1, ALG9, PALB2, TBL1XR1, NUP133, NGLY1, ERMARD, MCTP2, CENPJ, HDAC8, KLHL7, SMG9, ALG1, ANKH, LRRC8A, FAM20C, KIF15, CCDC47, KNL1, WDR11, TENM3, VAC14, CEP55, QRICH1, NSUN2, MKS1, PHIP, FANCL, RFWD3, FANCI, PACS1, ASXL2, SLC29A3, CHD7, OSGEP, PIGV, PEX26, RPGRIP1, NUP107, SALL4, BCL11B, FAM111A, MRPS14, XYLT2, NSD1, LMBR1, NXN, TMEM237, BCORL1, CPLANE1, COLEC11, ALG8, TMEM231, FAT4, SRD5A3, PIEZO2, PROK2, THOC2, WDR35, ADGRG6, GATAD2B, TBC1D24, SHROOM4, ARID1B, HACE1, CC2D2A, ALX4, DOCK6, CHD8, ZSWIM6, FANCM, EPG5, KMT2C, USP9X, ALX1, ACTA1, GNRH1, FOXE3, FLI1, FLII, FLNA, FLNB, MTOR, FUCA1, FZD2, GATA6, GBA, GJA1, GK, GPC3, GLB1, GLE1, FOXC1, FH, FGFR2, FBN1, FANCD2, FANCE, BPTF, FANCB, FANCF, FANCG, GPC4, FGFR3, FGD1, FGF3, FGF8, FGF10, FGF14, FGFR1, GLI3, GNRHR, FANCA, GP1BB, KISS1, KRAS, LBR, LETM1, LIG4, LMNA, LOX, LRP2, LRP4, LRP5, SMAD3, SMAD4, MAF, MAT2A, MEF2C, KCNJ2, KCNH1, ANOS1, HNRNPU, GTF2E2, H3-3A, HBA1, HBA2, HELLS, HNRNPH2, HRAS, ITGA3, HSD17B4, HSPG2, IGHM, IGLL1, INPPL1, INSR, FANCC, EZH2, HMGA2, CENPF, BGN, BMP2, BMPR1A, BRCA1, BRAF, BRCA2, BUB1B, CAMK2A, RUNX2, CBL, CCND2, CD79A, CD79B, CDC42, CDH1, NKX3-2, AVP, ATRX, JAG1, ACTA2, ACTB, ACTG1, ACY1, ADK, AGA, AKT1, ATP6V1E1, ALX3, ANK1, APC, ARVCF, ATP6V1A, ATP6V1B2, CDH11, CHD3, EXT2, CHD4, DLX4, DNMT3A, DNMT3B, DPH1, SLC26A2, DUSP6, DVL1, DVL3, DYRK1A, MEGF8, ELN, ERCC2, ERCC3, ERCC4, ERF, DHCR7, DDX3X, CTNND2, COL2A1, CHRNA1, CHRND, CHRNG, CNTN1, COL1A1, COL1A2, COL3A1, CTNND1, COL11A1, COL11A2, COMT, COX7B, CSNK2A1, CTBP1, MID1, KMT2A, MLLT1, TAZ, SET, SIM1, SKI, SKIV2L, SMO, SMS, SOS1, SOS2, SOX9, SOX10, STAT3, STIM1, TAC3, TACR3, MAP3K7, SCN1A, RYR1, RREB1, RET, RAD51C, RAF1, RAPSN, RASA2, RB1, DPF2, RIT1, RRAS, RMRP, RPL5, RPL35A, RPS6KA3, RPS7, RPS19, TBX1, TBCD, MMP2, TBCE, UFD1, UMPS, WHCR, NSD2, NELFA, WNT5A, XRCC2, YWHAE, ZIC1, ZIC3, RNF113A, BRPF1, SHOC2, PDHX, MFAP5, UBE2A, TWIST1, HIRA, TFAP2B, TBX2, TBX15, TCF3, TCF12, TCOF1, TFAP2A, TGFB2, TTN, TGFB3, TGFBR1, TGFBR2, THRA, TPM2, TPM3, RAD51, ALDH18A1, PYCR1, PEX5, TRIM37, MUSK, MYH3, MYH11, MYLK, MYOD1, NEB, NF1, NFIX, TONSL, NONO, NOTCH2, NOTCH3, NRAS, ROR2, TRNW, TRNS2, TRNS1, COX3, ALDH6A1, MOCS1, MOCS2, ATP6, COX1, COX2, ND1, TRNQ, ND4, ND5, ND6, TRNF, TRNH, TRNL1, DDR2, NUP88, PAFAH1B1, PRPS1, PPP2R5D, PPP3CA, PRKAR1A, PRKG1, MAP2K1, MAP2K2, MASP1, PPP2CA, PSMD12, PTCH1, PTEN, PTH1R, PTPN11, PEX2, PPP2R1A, PPP1CB, PAX3, PIGA, PAX6, PDE4D, PDE6D, PEPD, PEX1, PEX6, PIK3CA, POR, PIK3R1, PIK3R2, PITX2, PMM2, EXOSC9, POLR2F, SCN1A-AS1

-
Fibromatosis, Gingival, With Progressive Deafness
Omim
Inheritance The transmission pattern of gingival fibromatosis with progressive deafness in the families reported by Jones et al. (1977) and Hartsfield et al. (1985) was consistent with autosomal dominant inheritance. INHERITANCE - Autosomal dominant HEAD & NECK Ears - Hearing loss, progressive sensorineural Mouth - Gingival fibromatosis ▲ Close
-
Phenytoin Toxicity
Omim
The infant was born of a nonepileptic mother who had a history of first trimester prophylactic anticonvulsant therapy after surgical excision of a meningioma. ... De Smet and Debeer (2002) described 2 children whose mother had been treated with phenylhydantoin for epilepsy that developed after surgery for a brain tumor. The first son had hypoplasia of the terminal phalanx of the fifth finger of the left hand. ... He had facial dysmorphism with ocular hypertelorism, a small triangular shaped skull, and a depressed nasal bridge. Inheritance Dominant inheritance of phenytoin toxicity was proposed by Vesell (1979).
-
Psoriasis 4, Susceptibility To
Omim
Since psoriasis is considered a polygenic disorder, Capon et al. (1999) investigated the relationship between the HLA-C (142840) and 1q21 loci with respect to their contribution to psoriasis susceptibility. They first demonstrated an association with HLA-Cw6 in a sample of 1q-linked pedigrees by means of the transmission/disequilibrium test. ... In the third aspect of the study, they found a significant increment of the 'weighted' lod score with respect to the baseline lod. This provided the first significant evidence for linkage in the Italian population with the HLA region. ... Screening for K4022X in 441 unrelated Chinese psoriasis cases revealed another 2 patients who were homozygous; the variant was also found in heterozygosity in 29 (6.6%) of the patients and in 15 (3%) of 500 controls. The odds ratio for the dominant model was 2.552 (p = 0.002), suggesting an association of the K4022X variant with the psoriasis/ichthyosis vulgaris phenotype in the Chinese population.
-
Androgen Insensitivity, Partial
Omim
According to Wilson (1976), Morris (1953) first described incomplete testicular feminization and concluded that the complete (AIS; 300068) and incomplete forms never occur in the same family. ... However, Wilson et al. (1984) described well-studied cases that indicated that the Lubs syndrome (Lubs et al., 1959), like classic testicular feminization, is due to mutation in the androgen receptor. The patients were first cousins; their mothers were sisters. In the family described by Rosewater et al. (1965), gynecomastia with hypogonadism occurred in 4 males of 3 sibships in 2 generations connected through females in a pattern consistent with X-linked or autosomal dominant inheritance. None of those affected had hypospadias.

-
Glandular Odontogenic Cyst
Wikipedia
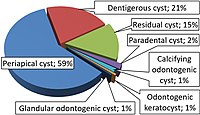
A computed tomography and panoramic x-ray must be undertaken in order to observe the severity of internal complications. [5] These scans allow for the observation of the GOC size, radiolucency, cortical bone, dentition , root, and vestibular zone. [5] In some cases, the dentition may be embedded into the cavity walls of the lesion, depending on the position of expansion at the odontogenic tissue. [13] The diagnosis of a smaller sized GOC is related to the attachment of only two teeth. [6] While, a greater sized GOC develops over two teeth. [6] Presentation of a greater sized lesion usually requires a biopsy for a differential diagnosis and a precise treatment plan. [6] Treatment process [ edit ] The unilocular and multilocular nature is imperative to the determination of treatment style. [6] Local anesthesia is regularly provided as the GOC is embedded within the tissue structure of the jaw and requires an invasive procedure for a safe and accurate extraction. [2] For unilocular GOCs with minimal tissue deterioration, " enucleation , curettage , and marsupialization " is a suitable treatment plan. [6] Notably, the performance of enucleation or curettage as the primary action is linked to an incomplete extraction of the GOC and is only recommended to the less invasive lesions. [6] Multilocular GOCs require a more invasive procedure such as "peripheral ostectomy , marginal resection, or partial jaw resection". [6] GOCs associated with a more severe structural damage are encouraged to undergo marsupialization as either an initial or supplementary surgery. [6] The frequency of reappearance is likely due to the lingering cystic tissue structures that remain after the performance of curettage. [13] The incorporation of a "dredging method i.e. repetition of enucleation and curettage" is also suggested until the remnants of the GOC diminishes for certain. [9] The treatment ensures scar tissue is removed to promote the successful reconstruction of osseous material for jaw preservation. [9] Alongside the main treatments, bone allograft application, cryosurgery , and apicoectomy are available but have not been consistently recommended. [9] [13] [5] Though Carnoy's solution , the chloroform -free version, is recommended with the treatment as it degenerates the majority of the damaged dental lamina . [13] The most effective type of treatment remains unknown due to the lack of detailed data from reported cases. [3] Post-treatment protocols [ edit ] Follow-up appointments are necessary after the removal of the GOC as there is a high chance of remission, which may be exacerbated in cases dealing with "cortical plate perforation". [13] [5] The GOC has a significant remission rate of 21 to 55% that can potentially develop during the period of 0.5 to 7 years post-surgery. [7] [6] Cases occupied with a lower risk lesion are expected to continue appointments with physicians for up to 3 years post-surgery. [6] A higher risk lesion is encouraged to consistently consult with physicians during a 7 year period after treatment. [13] Remission events require immediate attention and appropriate procedures such as enucleation or curettage. [6] In more damaging cases of remission, tissue resection, and marsupialization may have to be performed. [7] Epidemiology [ edit ] The clinical presentation of the GOC is very low in the population as noted by the 0.12 to 0.13% occurrence rate, extrapolated from a sample size of the 181 individuals. [2] The GOC mainly affects older individuals in the population, especially those that are in their 40 to 60s. [8] However, the GOC can affect younger individuals i.e. 11, and more older individuals i.e. 82 in the population. [2] The age distribution starts at a much lower number for people living in Asia and Africa. [2] Those in their first 10 years of life have not been diagnosed with the GOC. [14] The GOC does present a tendency to proliferate in more males than females. [3] There is no definitive conclusion towards the relevance of gender and its influence on the rate of incidence. [7] References [ edit ] ^ a b Borges, Leandro Bezerra; Fechine, Francisco Vagnaldo; Mota, Mário Rogério Lima; Sousa, Fabrício Bitu; Alves, Ana Paula Negreiros Nunes (March 2012). ... Basic Oral and Maxillofacial Pathology . 1 . v t e Cystic diseases Respiratory system Langerhans cell histiocytosis Lymphangioleiomyomatosis Cystic bronchiectasis Skin stratified squamous: follicular infundibulum Epidermoid cyst and Proliferating epidermoid cyst Milia Eruptive vellus hair cyst outer root sheath Trichilemmal cyst and Pilar cyst and Proliferating trichilemmal cyst and Malignant trichilemmal cyst sebaceous duct Steatocystoma multiplex and Steatocystoma simplex Keratocyst nonstratified squamous: Cutaneous ciliated cyst Hidrocystoma no epithelium: Pseudocyst of the auricle Mucocele other and ungrouped: Cutaneous columnar cyst Keratin implantation cyst Verrucous cyst Adenoid cystic carcinoma Breast cyst Human musculoskeletal system Cystic hygroma Human digestive system oral cavity: Cysts of the jaws Odontogenic cyst Periapical cyst Dentigerous cyst Odontogenic keratocyst Nasopalatine duct cyst liver: Polycystic liver disease Congenital hepatic fibrosis Peliosis hepatis bile duct: Biliary hamartomas Caroli disease Choledochal cysts Bile duct hamartoma Nervous system Cystic leukoencephalopathy Genitourinary system Polycystic kidney disease Autosomal dominant polycystic kidney Autosomal recessive polycystic kidney Medullary cystic kidney disease Nephronophthisis Congenital cystic dysplasia Other conditions Hydatid cyst Von Hippel–Lindau disease Tuberous sclerosis
-
Lujan–fryns Syndrome
Wikipedia
Males are normally hemizygous for the X chromosome, having only one copy. As a result, X-linked dominant disorders usually show higher expressivity in males than females. ... This is because, typically, females have two copies of the X-chromosome, while males have only one copy. The difference between dominant and recessive inheritance patterns also plays a role in determining the chances of a child inheriting an X-linked disorder from their parentage. In LFS, X-linked dominant inheritance was suspected, as boy and girl siblings in one family both exhibited the disorder. [13] [37] A scenario such as this would also be possible with X-linked recessive inheritance, but in this particular case report, the girl was believed to be a manifesting heterozygote [13] [37] carrying one copy of the mutated gene. ... Close attention and specialized follow-up care, including neuropshycological evaluation methods and therapies, and special education, should be given to diagnose and prevent psychiatric disorders and related behavioral problems such as psychosis and outbursts of aggression. [9] Epidemiology [ edit ] Lujan–Fryns syndrome is a rare X-linked dominant syndrome and is more common in males than females. ... "X-linked mental retardation with marfanoid habitus: first report of four Italian patients".

-
Cystic Kidney Disease
Wikipedia
Renal cysts have been reported in more than 50% of patients over the age of 50. [2] Typically, cysts grow up to 2.88 mm annually and may cause related pain and/or hemorrhage. [2] Of the cystic kidney diseases , the most common is polycystic kidney disease with two sub-types: the less prevalent autosomal recessive and more prevalent autosomal dominant. [1] Autosomal recessive polycystic kidney disease (ARPKD) is primarily diagnosed in infants and young children while autosomal dominant polycystic kidney disease (ADPKD) is most often diagnosed in adulthood. [1] Another example of cystic kidney disease is Medullary sponge kidney . ... The most common subset is polycystic kidney disease (PKD) which is a genetic anomaly with two subsets, autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD). ... Mutations in genes PKD1 and PKD2 are responsible for autosomal dominant polycystic kidney disease (ADPKD). Those genes encode for polycystic proteins and mutations regarding those genes are inherited and responsible for the disorder of autosomal dominant cystic kidney disease. In the United States, more than half a million people have PKD, making it the fourth leading cause of kidney failure. ... PKD Clinic, www.pkdclinic.org/pkd-prognosis/342.html ^ "Autosomal Dominant Polycystic Kidney Disease (ADPKD) - Genitourinary Disorders."TMEM67, UMOD, ANKS6, ALG9, FAT4, SDCCAG8, IFT80, SEC61A1, ALG8, INS, DNAJB11, GLIS2, HNF1B, NEK8, GLIS3, PRKD1, CC2D2A, NPHP3, NPHP1, XPNPEP3, GPSM1, TTC21B, JADE1, HSPB11, DNAAF1, INVS, CYS1, AGT, CPQ, KIF3A, PKD2, COL4A1, CTNNB1, EIF4EBP1, FGF2, MTOR, HIF1A, IL6, PKD1, PKHD1, ARF4, HNF1A, TFAP2A, TNF, TNS1, TSC2, IFT88, OFD1, STX11, MIR17HG

-
Cheilitis
Wikipedia
. ^ "Journal of the American Academy of Dermatology", Volume 56, Issue 2, Pages AB94 – AB94 ^ a b Jeske, Arthur H. ... ISBN 978-1-60327-519-4 . ^ Journal of the American Academy of Dermatology, Volume 54, Issue 2, Pages 336–337 P. Carrington, T. Horn ^ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007).COL7A1, OCRL, IL17F, IL17RC, CLEC7A, SLC39A4, FERMT1, MBTPS2, IL17RA, TRAF3IP2, SLC46A1, MTMR11, LUC7L3, VEGFA, TXN, TP53, STAT3

-
Coronary Artery Anomaly
Wikipedia
Symptoms include chest pain, shortness of breath and syncope , although cardiac arrest may be the first clinical presentation. Several varieties are identified, with a different potential to cause sudden cardiac death . ... Coronary arteries arise from ostia, openings of the aorta (the largest artery in the human body) at the upper third or middle third of the sinuses of Valsalva (the first part of the big pipe coming off the main pumping chamber). ... The posterior descending artery , providing blood flow to the infero-posterior wall of the heart, originates from the RCA in 70-90% of individuals (“right coronary dominance”), whereas in 10-15% cases it originates from the LCx (“left coronary dominance”). ... In most cases, however, coronary artery anomalies are silent for many years and the first clinical manifestation of these pathological entities is sudden cardiac death (e.g. due to malignant arrhythmias such as ventricular fibrillation ) typically after strenuous physical exertion (when arterial compression is more severe, and cardiac work is maximal) such as in young athletes or military recruits.
-
Arthrogryposis, Distal, Type 10
Omim
The transmission pattern was consistent with autosomal dominant inheritance with variable expressivity. Stevenson et al. (2006) reported a 5-generation Utah family in which multiple individuals were affected with plantar flexion contractures in an autosomal dominant pattern of inheritance. The authors termed the disorder distal arthrogryposis type 10 (DA10). ... Mapping In a large 5-generation Utah family with distal arthrogryposis and plantar tendon shortening, originally reported by Stevenson et al. (2006), Stevenson et al. (2006) performed genomewide linkage analysis and obtained a maximum lod score of 3.96 at marker D2S364 on chromosome 2q. INHERITANCE - Autosomal dominant SKELETAL Limbs - Wrist contractures - Hamstring contractures - Elbow contractures Hands - Finger contractures Feet - Plantar flexion contractures LABORATORY ABNORMALITIES - Normal serum creatine kinase MISCELLANEOUS - Onset in early childhood - Variable severity - Toe-walking - Plantar contractures become apparent with onset of ambulation - Contractures other than plantar are less common and less severe ▲ Close
-
Myoclonus And Ataxia
Omim
Cerebrospinal fluid uric acid was elevated in 2. Autosomal dominant inheritance with reduced penetrance was suggested. ... Neumann (1959) described combined degeneration of the globus pallidus and the dentate nucleus, and reports of autosomal dominant inheritance of the combination were referred to by Takahata et al. (1978). ... Neuro - Myoclonus - Cerebellar ataxia - Intention tremor - Occasional tonic-clonic seizures Lab - Lesion of cerebellar dentate nucleus - Degeneration of globus pallidus - Elevated cerebrospinal fluid uric acid - Mitochondrial abnormalities - Ragged-red fibers on muscle biopsy Inheritance - Autosomal dominant - heterogeneous ▲ Close
-
Keratoconus 5
Omim
Clinical Features Rabinowitz et al. (1992) studied a family segregating autosomal dominant keratoconus over 3 generations. ... Exclusion Studies In a 3-generation family segregating autosomal dominant keratoconus, Rabinowitz et al. (1992) selected COL6A1 (120220) as a candidate gene and by linkage analysis excluded this specific gene as well as the most telomeric region of chromosome 21 as the site of the mutation. INHERITANCE - Autosomal dominant HEAD & NECK Eyes - Keratoconus - Central steepening of cornea - Stromal thinning - Subepithelial fibrosis - Fragmentation of Bowman membrane - Fleischer ring ▲ Close
-
Schwannomatosis 2
Omim
Inheritance The transmission pattern of schwannomatosis-2 in the families reported by Piotrowski et al. (2014) was consistent with autosomal dominant inheritance and incomplete penetrance. ... The findings suggested that loss of LZTR1 function can predispose to the development of autosomal dominant multiple schwannomas, thus implicating LZTR1 as a tumor suppressor gene. INHERITANCE - Autosomal dominant SKELETAL Spine - Schwannomas Limbs - Schwannomas SKIN, NAILS, & HAIR Skin - Schwannomas NEUROLOGIC Central Nervous System - Spinal tumors NEOPLASIA - Multiple schwannomas MISCELLANEOUS - Incomplete penetrance - Variable expressivity - Germline and somatic mutations contribute to this disorder MOLECULAR BASIS - Caused by mutation in the leucine zipper-like transcriptional regulator 1 gene (LZTR1, 600574.0001 ) ▲ Close
-
Palatopharyngeal Incompetence
Omim
The inability to limit the flow of air-sound through the nose is responsible for the speech defect described as 'hypernasality' or 'nasal speech.' Occasionally dominant inheritance may obtain, with great variability, making this essentially a multifactorial trait. ... Vantrappen et al. (2002) described a kindred in which 10 members of 3 successive generations had isolated velopharyngeal insufficiency in an autosomal dominant pattern including 5 examples of male-to-male transmission. ... Kannu et al. (2003) reported a family in which father-to-son transmission demonstrated autosomal dominant inheritance of velopharyngeal insufficiency.
-
Spondylocarpotarsal Synostosis
Orphanet
Differential diagnosis Differential diagnosis may include isolated Klippel-Feil syndrome and other vertebral dysplasias, such as autosomal dominant spondylocostal dysplasia and multiple synostoses syndrome. Genetic counseling SCT syndrome follows an autosomal recessive inheritance ( FLNB , MYH3 ) or occasionally autosomal dominant inheritance ( MYH3 ). Genetic counseling should be proposed to at risk couples informing them that there is 25% (autosomal recessive) or 50% (autosomal dominant) risk of tranmitting the disease to offspring.
-
Hemochromatosis, Type 5
Omim
Clinical Features Kato et al. (2001) studied a Japanese family segregating autosomal dominant iron overload. The proband was a 56-year-old woman who, during evaluation for early gastric cancer, was found to have low signal intensity of liver, heart, and bone marrow on MRI, indicative of iron deposition. ... Molecular Genetics In 3 affected members of a Japanese family segregating autosomal dominant hemochromatosis, who were negative for hemochromatosis-associated mutations in the HFE (613609) and TFR2 (604720) genes, Kato et al. (2001) identified a heterozygous mutation in the FTH1 gene (134770.0001). ... However, the authors noted because the daughter had just given birth and was breastfeeding, she was likely to exhibit an increased level of physiologic iron loss. INHERITANCE - Autosomal dominant ABDOMEN Liver - Heavy iron deposition in most hepatocytes - Iron deposition in some Kupffer cells - Maximal deposition of iron in Rappaport zones 1 and 2, with relative sparing of zone 3 - Low signal intensity on T1- and T2-weighted abdominal MRI Spleen - Iron deposition in macrophages HEMATOLOGY - No abnormalities LABORATORY ABNORMALITIES - Elevated serum ferritin - Elevated serum iron - Elevated serum transferrin saturation - Elevated total iron-binding capacity MISCELLANEOUS - Based on report of 1 Japanese family (last curated November 2013) MOLECULAR BASIS - Caused by mutation in the ferritin heavy chain-1 gene (FTH1, 134770.0001 ) ▲ Close
-
Myopia 20, Autosomal Dominant
Omim
Description Myopia, or nearsightedness, is a refractive error of the eye. Light rays from a distant object are focused in front of the retina and those from a near object are focused in the retina; therefore distant objects are blurry and near objects are clear (summary by Kaiser et al., 2004). For a discussion of genetic heterogeneity of susceptibility to myopia, see 160700. Mapping Shi et al. (2011) conducted a genomewide association study of 493,947 SNPs in 419 Han Chinese individuals with high myopia and 669 unrelated controls and found that none of the SNPs were significantly associated after correction for multiple testing; however, 34 SNPs in 22 chromosomal regions were associated after adjustment for genomic control, gender, and age, with nominal p values less than 10(-4). Testing those 34 SNPs in a follow-up cohort of 843 Han Chinese individuals with high myopia and 2,525 controls yielded a significant association between SNP rs9318086 at chromosome 13q12.12 and high myopia after adjustment for gender and age (heterozygous odds ratio (OR), 1.40; homozygous OR, 1.68; corrected p = 7.75 x 10 (-5)).





