-
Achromatopsia
Wikipedia
CS1 maint: ref=harv ( link ) Ronchi, A. M. (2009). eCulture: cultural content in the digital age . ... Achromatopsia at MedicineNet Achromatopsia at Merriam-Webster Achromatopsia at NCBI Classification D ICD - 10 : H53.5 ICD - 9-CM : 368.54 OMIM : 216900 MeSH : D003117 DiseasesDB : 83 External resources MedlinePlus : 001002 GeneReviews : NBK1418 GARD : ACHM Orphanet : 49382 Achromatopsia v t e Color topics Red Orange Yellow Green Cyan Blue Indigo Violet Purple Magenta Pink Brown White Gray Black Color science Color physics Electromagnetic spectrum Light Rainbow Visible Spectral colors Chromophore Structural coloration Animal coloration Color of chemicals Water On Vision and Colours Metamerism Spectral power distribution Color perception Color vision Color blindness Achromatopsia test Tetrachromacy Color constancy Color term Color depth Color photography Spot color Color printing Web colors Color mapping Color code Color management Chrominance False color Chroma key Color balance Color cast Color temperature Eigengrau The dress Color psychology Color symbolism Color preferences Lüscher color test Kruithof curve Political color National colors Chromophobia Chromotherapy Color philosophy Color space Color model additive subtractive Color mixing Primary color Secondary color Tertiary color (intermediate) Quaternary color Quinary color Aggressive color (warm) Receding color (cool) Pastel colors Color gradient Color scheme Color tool Monochromatic colors Complementary colors Analogous colors Achromatic colors (Neutral) Polychromatic colors Impossible colors Light-on-dark Tinctures in heraldry Color theory Chromaticity diagram Color solid Color wheel Color triangle Color analysis (art) Color realism (art style) Color terms Basic terms Blue Green Red Yellow Pink Purple Orange Black Gray White Brown Cultural differences Linguistic relativity and the color naming debate Blue–green distinction in language Color history Color in Chinese culture Traditional colors of Japan Human skin color Color dimensions Hue Dichromatism Colorfulness (chroma and saturation) Tints and shades Lightness (tone and value) Grayscale Color organizations Pantone Color Marketing Group Color Association of the United States International Colour Authority International Commission on Illumination (CIE) International Color Consortium International Colour Association Lists List of colors: A–F List of colors: G–M List of colors: N–Z List of colors (compact) List of colors by shade List of color palettes List of color spaces List of Crayola crayon colors history Color chart List of RAL colors List of web colors Related Vision Digital image processing Multi-primary color display Quattron Qualia Lighting Local color (visual art) Category Index v t e Physiology of the visual system Vision Accommodation Gaze Intraocular pressure Visual field Color vision Color blindness Achromatopsia Köllner's rule Opponent process Dichromacy Monochromacy Pentachromacy Tetrachromacy Trichromacy v t e Diseases of the human eye Adnexa Eyelid Inflammation Stye Chalazion Blepharitis Entropion Ectropion Lagophthalmos Blepharochalasis Ptosis Blepharophimosis Xanthelasma Ankyloblepharon Eyelash Trichiasis Madarosis Lacrimal apparatus Dacryoadenitis Epiphora Dacryocystitis Xerophthalmia Orbit Exophthalmos Enophthalmos Orbital cellulitis Orbital lymphoma Periorbital cellulitis Conjunctiva Conjunctivitis allergic Pterygium Pseudopterygium Pinguecula Subconjunctival hemorrhage Globe Fibrous tunic Sclera Scleritis Episcleritis Cornea Keratitis herpetic acanthamoebic fungal Exposure Photokeratitis Corneal ulcer Thygeson's superficial punctate keratopathy Corneal dystrophy Fuchs' Meesmann Corneal ectasia Keratoconus Pellucid marginal degeneration Keratoglobus Terrien's marginal degeneration Post-LASIK ectasia Keratoconjunctivitis sicca Corneal opacity Corneal neovascularization Kayser–Fleischer ring Haab's striae Arcus senilis Band keratopathy Vascular tunic Iris Ciliary body Uveitis Intermediate uveitis Hyphema Rubeosis iridis Persistent pupillary membrane Iridodialysis Synechia Choroid Choroideremia Choroiditis Chorioretinitis Lens Cataract Congenital cataract Childhood cataract Aphakia Ectopia lentis Retina Retinitis Chorioretinitis Cytomegalovirus retinitis Retinal detachment Retinoschisis Ocular ischemic syndrome / Central retinal vein occlusion Central retinal artery occlusion Branch retinal artery occlusion Retinopathy diabetic hypertensive Purtscher's of prematurity Bietti's crystalline dystrophy Coats' disease Sickle cell Macular degeneration Retinitis pigmentosa Retinal haemorrhage Central serous retinopathy Macular edema Epiretinal membrane (Macular pucker) Vitelliform macular dystrophy Leber's congenital amaurosis Birdshot chorioretinopathy Other Glaucoma / Ocular hypertension / Primary juvenile glaucoma Floater Leber's hereditary optic neuropathy Red eye Globe rupture Keratomycosis Phthisis bulbi Persistent fetal vasculature / Persistent hyperplastic primary vitreous Persistent tunica vasculosa lentis Familial exudative vitreoretinopathy Pathways Optic nerve Optic disc Optic neuritis optic papillitis Papilledema Foster Kennedy syndrome Optic atrophy Optic disc drusen Optic neuropathy Ischemic anterior (AION) posterior (PION) Kjer's Leber's hereditary Toxic and nutritional Strabismus Extraocular muscles Binocular vision Accommodation Paralytic strabismus Ophthalmoparesis Chronic progressive external ophthalmoplegia Kearns–Sayre syndrome palsies Oculomotor (III) Fourth-nerve (IV) Sixth-nerve (VI) Other strabismus Esotropia / Exotropia Hypertropia Heterophoria Esophoria Exophoria Cyclotropia Brown's syndrome Duane syndrome Other binocular Conjugate gaze palsy Convergence insufficiency Internuclear ophthalmoplegia One and a half syndrome Refraction Refractive error Hyperopia Myopia Astigmatism Anisometropia / Aniseikonia Presbyopia Vision disorders Blindness Amblyopia Leber's congenital amaurosis Diplopia Scotoma Color blindness Achromatopsia Dichromacy Monochromacy Nyctalopia Oguchi disease Blindness / Vision loss / Visual impairment Anopsia Hemianopsia binasal bitemporal homonymous Quadrantanopia subjective Asthenopia Hemeralopia Photophobia Scintillating scotoma Pupil Anisocoria Argyll Robertson pupil Marcus Gunn pupil Adie syndrome Miosis Mydriasis Cycloplegia Parinaud's syndrome Other Nystagmus Childhood blindness Infections Trachoma Onchocerciasis Authority control GND : 4242792-7 LCCN : sh85028584 NLI : 000664610
-
Kabuki Syndrome
Wikipedia
Retrieved 2018-04-01 . ^ a b c d e f g h i j k l m "Kabuki syndrome" . Genetics Home Reference, U.S. ... S2CID 24533876 . ^ a b c d e f g Adam MP, Hudgins L, Hannibal M (16 May 2013). Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.).KMT2D, KDM6A, RAP1B, RAP1A, KMT2A, KMT2B, MKKS, MACROD2, ANOS1, HTC2, HNRNPK, MAPK1, MAP2K7, TUBA3D, CHD7, TERF2IP, FZR1, ANKRD11, RABGEF1, TNFRSF13B, ZFPM2, POGZ, KDM1A, SEPTIN9, GH1, MBD2, MYH6, POMC, XIST, UTY, EPHB2, TUBA3C, SOX3, SET, RBPJ, ITGB7, BRAF

-
Eosinophilic Esophagitis
Wikipedia
It is possible that long-standing, untreated disease may result in esophageal remodeling, leading to strictures, Schatzki ring and, eventually, achalasia. [7] History [ edit ] The first case of eosinophilic esophagitis was reported in 1978. [1] In the early 1990s, it became recognized as a distinct disease. [25] See also [ edit ] Eosinophilic gastroenteritis References [ edit ] ^ a b Gómez-Aldana A, Jaramillo-Santos M, Delgado A, Jaramillo C, Lúquez-Mindiola A (August 2019). ... S2CID 25180186 . ^ Nielsen JA, Lager DJ, Lewin M, Rendon G, Roberts CA (April 2014).TSLP, CAPN14, TRIM8, TNK2, STAT6, JAZF1, EMSY, CCL26, IL13, IFFO2, HSF2BP, LINC02588, ANKRD27, CCDC81, MEAK7, SHROOM3, KIF17, IL5, CLEC16A, DCC, TIMP2, P2RX6, LINC02240, XKR6, CAPN5, CEP295NL, TGFB1, POSTN, FLG, TNF, DSG1, IL33, CCL11, FOXP3, ATP12A, ATP4A, IL4, IL6, IL15, EPC2, SPINK7, WDR36, CLDN7, CPA3, SIGLEC8, MID1, IL18, TNFSF10, SOAT1, EPX, CLDN1, DHTKD1, RNASE2, IL9, FFAR3, ALOX15, BDNF, IL18R1, COPD, LOC283710, IL32, IL1RL1, SLC9C2, SERPINA13P, MIR223, SPAG9, NR1I2, OGDHL, DEFB103A, FST, TNFRSF14, MIR375, EMBP1, BECN1, ABCB11, DEFB4B, BANCR, EOS, CBLL2, RSAD2, TRPV1, HBS1L, DEFB103B, CRLF2, ANO1, OXR1, SLC52A1, RHOF, TLR9, BCL11A, KLF13, CPA4, MUL1, LRRC31, CHIA, ACAD8, PANK2, MAPK8IP2, FIP1L1, SYNPO, PTGDR2, SERPINB12, SPINK5, REEP5, AGA, VCAM1, GFER, ESR2, F2RL1, FGF9, FKBP4, FKBP5, GABPA, LRRC32, GJA1, DNM1, GRM5, HIF1A, ICAM1, IFNG, IL1A, IL1B, IL5RA, ESR1, DEFB4A, TYK2, CD1D, ALOX5, ANXA5, CCND1, BID, BNIP3, CALB2, CAMP, CD44, CYP2C19, CEACAM8, CLC, CCR3, CCR8, ABCC2, COL8A2, CSF2, IL9R, IL15RA, ITGAM, CCL18, KLK6, PTGDR, PTGS2, RNASE3, S100A7, SAFB, CCL2, SGSH, KCNJ2, AGXT, SPRR3, STAT1, STC1, TFDP1, TLR3, TNFRSF4, KLK7, PRG2, PTPA, PLG, LALBA, LCT, LOX, LTC4S, SMAD2, KITLG, MIF, MMP2, MMP14, MPG, MYB, NFE2L2, SLC22A18, OSM, PRKN, SLC9A3

-
Neurodevelopmental Disorder, X-Linked, With Craniofacial Abnormalities
OMIM
However, patient cells showed cell cycle abnormalities, including increased percentage of G2/M cells and upregulation of genes involved in cell division, mitotic regulation, and DNA replication compared to controls.
-
Thrombotic Microangiopathy
Wikipedia
PMID 16760911 . ^ Ruggenenti P, Noris M, Remuzzi G (September 2001). "Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura".ADAMTS13, VEGFA, C3, NOS3, NOS2, CFH, VWF, CD46, HP, SERPINE1, CFHR1, THBD, F5, CABIN1, DGKE, IL6, STX2, F2, C5, PLG, NT5C1A, CFI, PTX3, REN, C20orf181, ERVK-6, CLEC1B, TWNK, ZFP36, GP6, KLF4, KLF2, TNFSF13B, CFHR3, PDGFC, SH3BP4, POLG, BCR, PIK3CB, PIGA, C5AR1, CAPG, CD59, CPB2, CRP, SLC25A10, CD55, F3, F8, FGG, MTOR, GPI, HLA-DRB1, HMOX1, IFNA1, IFNA13, IFNB1, KDR, KRT5, LCN2, MTHFR, ERVK-32

-
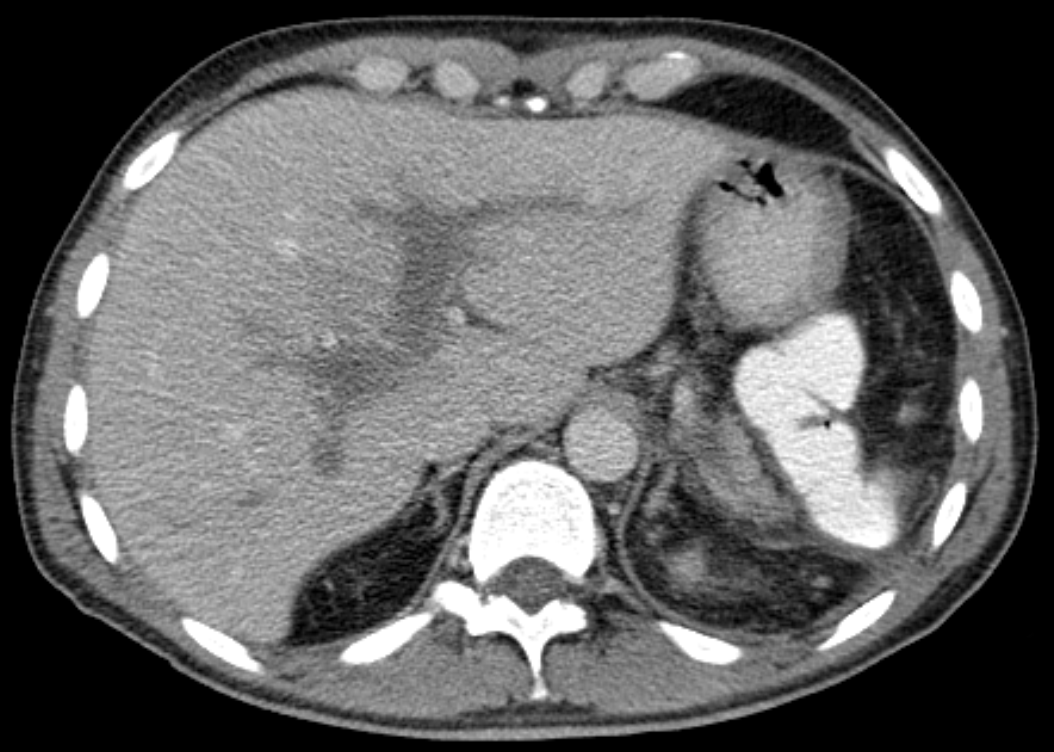
Portal Vein Thrombosis
Wikipedia
PMID 31895720 . ^ Hall TC, Garcea G, Metcalfe M, Bilku D, Dennison AR (November 2011).JAK2, MPL, SERPINC1, PIGM, MET, CTNNB1, F2, TET2, F5, AFP, MTHFR, SERPINE1, ALB, SELP, SLC17A5, VEGFA, COX8A, MIR21, PROS1, SH2B2, AKAP1, VWF, ADAMTS13, ABL1, SMUG1, RPL36, ACAD8, NAAA, GOLM1, ZNF469, MIRLET7C, MIR195, MIR27B, MIR31, LNCRNA-ATB, GP6, MYH9, ADAM17, PVR, APC, KLK3, ARG1, BCS1L, CALR, CDKN2A, CHI3L1, CRP, F2R, F3, F8, F10, FAH, GPT, IGF1R, IL6, LPA, NEK2, PC, H3P10
-
Ovarian Vein Syndrome
Wikipedia
History [ edit ] The entity was first described by Clark in 1964. [8] Following initial scepticism and lively discussion in the medical literature, [9] [10] it is now recognised by many authors. [1] References [ edit ] ^ a b Tourné G, Ducroux A, Bourbon M, Blinding H (September 2002). "The ovarian vein syndrome: eight cases and review of the literature" .

- Hydrosalpinx Wikipedia
- Vertebrobasilar Insufficiency Wikipedia
-
Tic Disorder
Wikipedia
Retrieved on December 29, 2011. ^ Moran, M. "DSM-5 provides new take on neurodevelopment disorders".DRD4, SLC6A3, CD24, PROM1, CD44, SIX1, STAT3, VIM, BECN1, HCN4, SQSTM1, PRKCI, GNA13, HRH3, PNKD, TPH2, MAP2K7, ADAM10, PPARG, ADRA2A, NFATC2, KCNE1, INS, HSPD1, F10, EZH2, ENO2, ECT2, BRCA1, BLM, ATP7B, ALDH1A1, NOS2

-
Aniridia
Wikipedia
Retrieved 2 February 2010 . ^ Lee H, Khan R, O'Keefe M (November 2008). "Aniridia: current pathology and management".PAX6, TRIM44, WT1, FOXC1, ELP4, FOXD3, PITX2, FOXE3, MDH2, SDHA, SDHAF2, REST, RET, KIF1B, SDHB, MAX, SDHC, SDHD, POU6F2, TRIM28, VHL, WNT10B, SEM1, MAFB, SLC25A11, TP63, TRIP13, BTRC, PORCN, COL25A1, DLX5, DLX6, CHN1, FAM111A, EPS15L1, FH, GPC3, BRCA2, DIS3L2, LDHD, H19, TMEM127, ITPR1, DLST, CAT, DEL11P13, ACP1, CYP1B1, PAX3, HRAS, LGR4, SLURP1, DCPS, PHF21A, WT1-AS, RN7SL263P, KIF21A, RIEG2, RASSF7, CXCL8, ALDH1A1, APOB, ARSD, ATP2A2, BCR, CTNNB1, EGF, SERPINB1, FGF2, MTOR, KRT12, VEGFA, NOTCH1, NRAS, GPR143, OCA2, SIX6, OTX2, PAX2, ADH7, CCL3, NR2E1, DEL11P13

-
Persistent Genital Arousal Disorder
Wikipedia
CS1 maint: uses authors parameter ( link ) ^ a b c d e f g h i j k l m n o p q r Brian A. Sharpless (2016).
-
Genital Trauma
Wikipedia
PMID 17073065 . ^ a b c d e f g Frioux, Sarah M.; Blinman, Thane; Christian, Cindy W.
-
Abortion In South Africa
Wikipedia
In 2001, 27% of abortions were second-trimester. [7] See also [ edit ] Abortion by country Abortion debate Abortion law Law of persons in South Africa Religion and abortion Choice on Termination of Pregnancy Act, 1996 History of Abortion Law Debate Birth control in Africa Literature [ edit ] Susanne M. Klausen: Abortion under Apartheid .

-
Atrophic Vaginitis
Wikipedia
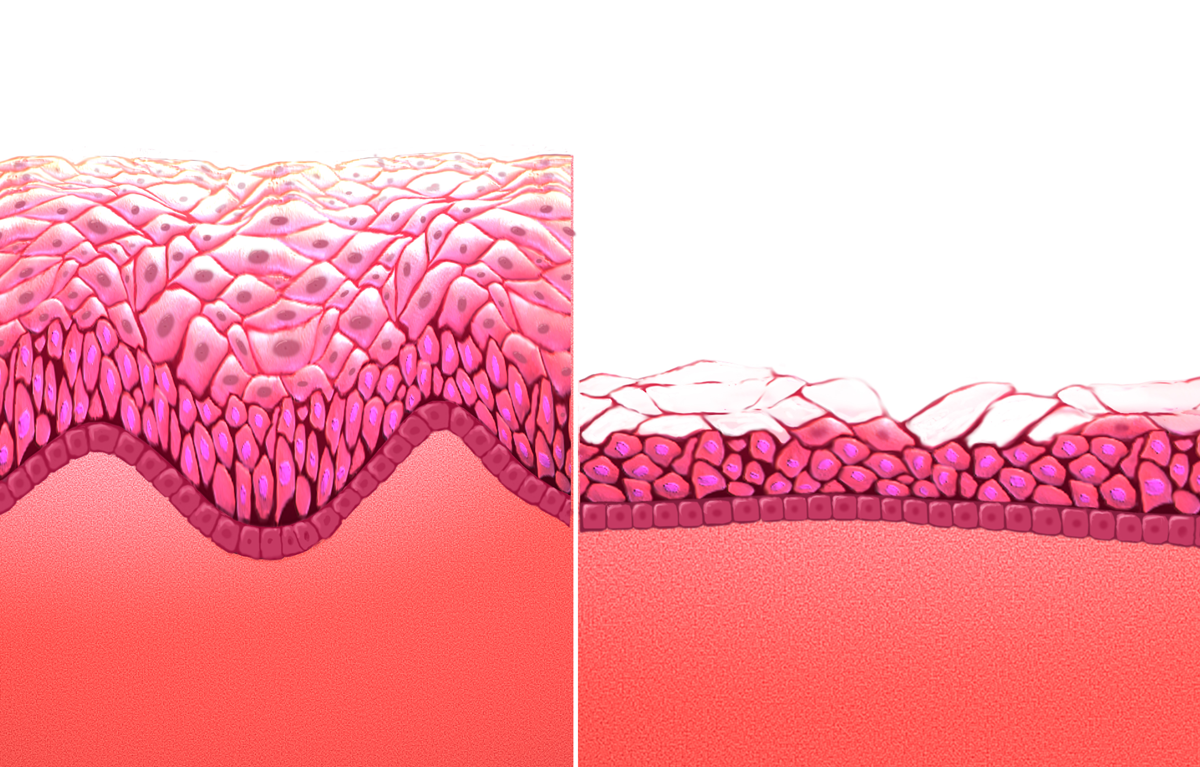
Few adverse effects were noted. [1] In 2018, the FDA issued a warning that lasers and other high energy devices were not approved for this application and it had received multiple reports of injuries. [11] References [ edit ] ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag Faubion, SS; Sood, R; Kapoor, E (December 2017).
- Progressive Bulbar Palsy Wikipedia
-
Penile Fracture
Wikipedia
Peitzman; Michael Rhodes; C. William Schwab; Donald M Yealy; Timothy C Fabian (1 September 2007).

- Hemopericardium Wikipedia
- Brief Psychotic Disorder Wikipedia
-
Rhotacism (Speech Impediment)
Wikipedia
Lambdacism , the equivalent condition with L / l / Perception of English /r/ and /l/ by Japanese speakers References [ edit ] ^ Stinchfield, Sara M (1933). Speech Disorders: A PSYCHOLOGICAL STUDY of the Various Defects of Speech .











