In the Edwin Smith Papyrus (1700 BC) it was prescribed a treatment based on wine and breast milk to cure this disease. [3] The ancient Greek and Indian civilizations were aware of atrophic rhinitis. [7] See also [ edit ] Empty nose syndrome References [ edit ] ^ Dutt SN, Kameswaran M (November 2005). "The aetiology and management of atrophic rhinitis" .
PMID 29374960 . ^ Belge G, Meyer A, Klemke M, et al. (2008). "Upregulation of HMGA2 in thyroid carcinomas: A novel molecular marker to distinguish between benign and malignant follicular neoplasias" .
Congenital rubella refers to the group of birth defects that occur in an infant whose mother is infected with the virus that causes German measles (rubella) during pregnancy. Congenital rubella occurs when the rubella virus in the mother affects the developing baby in the first 3 months of pregnancy. After the fourth month, if the mother has a rubella infection, it is less likely to harm the developing baby. The most common problems are hearing loss due to damage to the nerve pathways from the inner ear to the brain (sensorineural hearing loss), ocular abnormalities (cataract, infantile glaucoma, and pigmentary retinopathy ) and heart problems. Other symptoms and signs may include intrauterine growth retardation, prematurity , stillbirth, miscarriage, neurological problems (intellectual disability, low muscle tone, very small head), liver and spleen enlargement (hepatosplenomegaly), jaundice, skin problems, anemia, hormonal problems, and other issues.
An infectious embryofetopathy that may present in an infant as a result of maternal infection early in pregnancy and subsequent fetal infection with rubella virus. The disorder can lead to deafness, cataract, and variety of other permanent manifestations including cardiac and neurological defects. Epidemiology Congenital rubella syndrome (CRS) has been reported to affect an estimated 100,000 infants each year, mainly in developing countries. Clinical description Acquired during the first 8-12 weeks of gestation, rubella infection may cause multiple fetal defects (up to 90% of cases) including neurological (microcephaly), ophthalmic (cataracts, microphthalmia, glaucoma, pigmentary retinopathy, chorioretinitis), auditory (sensorineural deafness), cardiac (peripheral pulmonary artery stenosis, patent ductus arteriosus or ventricular septal defects, etc) defects and fetal wastage or stillbirth. Infection occurring later in gestation is associated with a decline in the risk of birth defects.
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a rare neurodegenerative disorder characterized by adult-onset progressive intention tremor and gait ataxia. Epidemiology Prevalence and incidence are unknown. The disease primarily affects males and there is a lifetime cumulated risk for men in the general population of about 1/8,000. Clinical description The age of onset of tremor and/or ataxia in males is about 60 years. The clinical presentation is heterogeneous with variable dominant manifestations including: intention tremor, progressive cerebellar gait ataxia, frontal executive dysfunction, cognitive decline, peripheral neuropathy, and dysautonomia. Other signs include mild parkinsonism and psychiatric manifestations (depression, anxiety, agitation) with possible progression to dementia.
Fragile X-associated tremor/ataxia syndrome (FXTAS) is characterized by problems with movement and thinking ability (cognition). FXTAS is a late-onset disorder, usually occurring after age 50, and its signs and symptoms worsen with age. This condition affects males more frequently and severely than females. Affected individuals have areas of damage in the part of the brain that controls movement (the cerebellum ) and in a type of brain tissue known as white matter, which can be seen with magnetic resonance imaging (MRI). This damage leads to the movement problems and other impairments associated with FXTAS.
Fragile X syndrome is a genetic condition that causes a range of developmental problems including learning disabilities and cognitive impairment. Usually, males are more severely affected by this disorder than females. Affected individuals usually have delayed development of speech and language by age 2. Most males with fragile X syndrome have mild to moderate intellectual disability, while about one-third of affected females are intellectually disabled. Children with fragile X syndrome may also have anxiety and hyperactive behavior such as fidgeting or impulsive actions.
A number sign (#) is used with this entry because fragile X tremor/ataxia syndrome (FXTAS) is caused by an expanded trinucleotide repeat in the FMR1 gene (309550.0004). In FXTAS, the expanded repeats range in size from 55 to 200 repeats and are referred to as 'premutations;' full repeat expansions with greater than 200 repeats results in fragile X syndrome (FXS; 300624) (Jacquemont et al., 2003). Description Jacquemont et al. (2007) provided a review of fragile X syndrome, which they characterized as a neurodevelopmental disorder, and FXTAS, which they characterized as a neurodegenerative disorder. Amiri et al. (2008) provided a review of FXTAS and noted that the pathogenesis of the disorder is distinct from that in fragile X syndrome. FXTAS results form a toxic gain of function of FMR1 RNA, whereas fragile X syndrome results from a loss of FMR1 function.
. ^ a b c d e f Li, B.; Duysen, E. G.; Carlson, M.; Lockridge, O. (2007). "The Butyrylcholinesterase Knockout Mouse as a Model for Human Butyrylcholinesterase Deficiency".
A number sign (#) is used with this entry because butyrylcholinesterase deficiency (BCHED) can be caused by homozygous or compound heterozygous mutation in the BCHE gene (177400) on chromosome 3q26. Severe cholinesterase deficiency results in postanesthetic apnea. Description Individuals deficient in butyrylcholinesterase (BCHE) appear asymptomatic, apart from a heightened sensitivity to muscle relaxants such as suxamethonium (succinylcholine) and mivacurium, 2 BCHE carboxylester substrates. In individuals with usual BCHE levels, these drugs are rapidly hydrolyzed in plasma and their duration of action is short (less than 10 minutes). BCHE deficiency results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. Prolonged neuromuscular block occurs with BCHE deficiencies of marked severity (impairment over 70%).
Pseudocholinesterase deficiency is a condition that causes increased sensitivity to certain muscle relaxant drugs used during general anesthesia (choline esters). These drugs relax the muscles used for movement, including those used for breathing. Normally, the muscles are able to move again a few minutes after the drugs are given. People with pseudocholinesterase deficiency may not be able to move or breathe on their own for a few hours after these drugs are given. They therefore may need mechanical ventilation until the drugs are cleared from the body.
Butyrylcholinesterase (BChE) deficiency is a metabolic disorder characterised by prolonged apnoea after the use of certain anaesthetic drugs, including the muscle relaxants succinylcholine or mivacurium and other ester local anaesthetics. The duration of the prolonged apnoea varies significantly depending on the extent of the enzyme deficiency. Epidemiology The prevalence of BChE deficiency is highest in the Caucasian population with between 3.4 and 4% of the population displaying a partial enzyme deficiency leading to slightly prolonged apnoea (between 5 minutes and 1 hour) and 1 in 2500 individuals showing a prolongation of more than 1 hour. Individuals with undetectable levels of BChE activity display a severe prolongation lasting more than 8 hours. The prevalence of this severe form is estimated at 1 in 100 000 individuals.
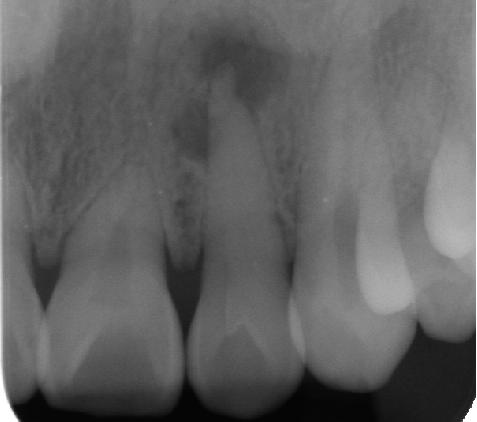
INHERITANCE - Autosomal recessive GROWTH Other - Growth retardation HEAD & NECK Head - Microcephaly Face - Short smooth philtrum (rare) - Retrognathia, mild (rare) Ears - Large ears Eyes - Brittle and sparse eyebrows - Brittle and sparse eyelashes - Nystagmus (rare) - Optic atrophy (rare) - Epicanthal folds (rare) - Microcornea (in some patients) - Microphthalmia (rare) - Nystagmus (in some patients) - Infantile cataract (in some patients) - Decreased best corrected visual acuity (in some patients) - Dry eye syndrome (in some patients) - Ocular surface fluorescein staining (in some patients) - Early onset of age-related macular degeneration (rare) Nose - Short nose (rare) - Large and depressed nasal root (rare) - Thick alae (rare) - Anteverted nares (rare) Mouth - White plaques on tongue Teeth - Thin enamel - Hypoplastic teeth (rare) CARDIOVASCULAR Heart - Ventricular septal defect (rare) GENITOURINARY Internal Genitalia (Male) - Decreased fertility Internal Genitalia (Female) - Decreased fertility SKELETAL - Delayed ossification (rare) SKIN, NAILS, & HAIR Skin - Ichthyosiform areas of skin (rare) Nails - Hypoplastic nails - Spoon-shaped nails - Dysplastic nails - Dyschromic nails Hair - Short, woolly hair - Sparse hair - Brittle hair of scalp, beard, eyebrows, eyelashes, and axillary and pubic areas - Stubby eyebrow hair - Trichorrhexis nodosa - Reduced cystine content of hair - Reduced sulfur content of hair - Loss of normal scale pattern on light and electron microscopy - Irregular, ridged surface on light and electron microscopy - Grooved surface appears twisted on light and electron microscopy - Alternating birefringent pattern on polarization microscopy NEUROLOGIC Central Nervous System - Psychomotor retardation - Hypotonia, severe (rare) - Partial agenesis of the corpus callosum (rare) - Cortical atrophy (rare) MOLECULAR BASIS - Caused by mutation in the M-phase specific PLK1 interacting protein (MPLKIP, 609188.0001 ) ▲ Close
Dubowitz syndrome is a very rare genetic and developmental disorder with a broad range of signs and symptoms. The typical findings of Dubowitz syndrome include growth failure/short stature, characteristic facial features such as a small triangular face, high sloping forehead, drooping eyelid (ptosis), short eyelids, increased distance between eyes (hypertelorism) broad and flat nasal bridge with a prominent and rounded nasal tip, smaller than normal head (microcephaly), intellectual disability, and eczema , especially on the face and behind the knees. Other common findings are behavioral disorders (hyperactivity, and/or autistic features), speech alterations, scanty or absent hair, foot abnormalities, delayed bone age , bone defects of the lower part of the spine (sacrum and coccyx), testicles that are still not located in the scrotum (cryptorchidism), memory and / or learning problems. There may be an increased risk of having cancer such as leukemia , or lymphoma . The diagnosis is made based on the symptoms (specially the facial features), but there is no specific laboratory test.
Clinical Features Dubowitz (1965) reported 4 patients with a malformation syndrome characterized by intrauterine growth retardation, short stature, microcephaly, mild mental retardation with behavior problems, eczema, and unusual and distinctive facies. Various minor malformations, such as pilonidal dimples, submucous clefts, high-pitched voice and sparse hair, were also seen. Two of the 4 cases were full sibs with nonconsanguineous parents. An older sib of Dubowitz's original proband was probably affected but studies were not done at her death. Grosse et al. (1971) added 2 more cases. Features were intrauterine growth retardation (with primordial shortness of stature), microcephaly, variable degrees of eczema and mental retardation, and characteristic facies (blepharophimosis, micrognathia, apparent hypertelorism). Opitz et al. (1973) concluded that eczema may be absent, stature may be normal, and intelligence may also be normal, although head circumference is always below the third percentile.
Dubowitz syndrome (DS) is a rare multiple congenital syndrome characterized primarly by growth retardation, microcephaly, distinctive facial dysmorphism, cutaneous eczema, a mild to severe intellectual deficit and genital abnormalities. Epidemiology The total birth prevalence in Europe has been estimated at 1/500,000. Over 150 cases have been described to date. Clinical description Newborns often have a low birth weight with a small head and body size. Facial appearance is characteristic with narrow or triangular shaped head and high or sloping forehead, flat supraorbital ridge, scanty lateral eyebrows, short palpebral fissures, blepharophimosis, ptosis, abnormally modeled ears, broad and flat nasal bridge, micrognathia and unusual configuration of the mouth. Sumucous cleft palate is common. Other findings include cutaneous ezema, high-pitched or hoarse voice, hypospadias and cryptorchidism.
Familial periodic paralysis is a disease characterized by sudden attacks of weakness and paralysis. Weakness is recurrent, affecting mainly the limbs, and is often brought on by exercising or eating too many or too few carbohydrates. There are 4 forms of familial periodic paralysis: hypokalemic , hyperkalemic , thyrotoxic , and Andersen-Tawil syndrome . In the hypokalemic form, the paralysis is caused by low levels of potassium. In the hyperkalemic form, the paralysis is caused by high levels of potassium in the blood.
. ^ Schwartz's principles of surgery: self assessment and board review, 8th edition, chapter 38, page 257; textbook p.1476 ^ Alpman A, Cogulu O, Akgul M, et al. (March 2008). "Prenatally Diagnosed Turner Syndrome and Cystic Hygroma: Incidence and Reasons for Referrals" .
Description Fetal cystic hygromas are congenital malformations of the lymphatic system appearing as single or multiloculated fluid-filled cavities, most often in the neck. They are thought to arise from failure of the lymphatic system to communicate with the venous system in the neck. They often progress to hydrops and cause fetal death (Chervenak et al., 1983). Clinical Features Bieber et al. (1979) reported a sibship in which the first and third pregnancies ended in stillborn female infants with a 'bag of water' at the nape of the neck. The mother sought genetic counseling in relation to the fourth pregnancy.
A cystic hygroma is a fluid-filled sac that results from a blockage in the lymphatic system. It is most commonly located in the neck or head area, but can be located anywhere in the body. It may be discovered in a fetus during a pregnancy ultrasound, or it may be apparent at birth as a soft bulge under the skin. When it is identified on pregnancy ultrasound, there is an increased risk for miscarriage. In some cases, it is not discovered until a person is older. Symptoms can vary depending on its size and specific location, and it can potentially cause problems with nearby structures or organs.
A number sign (#) is used with this entry because branchiootorenal syndrome-1 (BOR1) is caused by heterozygous mutation in the EYA1 gene (601653) on chromosome 8q13. Description Branchiootorenal syndrome is an autosomal dominant disorder characterized by sensorineural, conductive, or mixed hearing loss, structural defects of the outer, middle, and inner ear, branchial fistulas or cysts, and renal abnormalities ranging from mild hypoplasia to complete absence. Reduced penetrance and variable expressivity has been observed (Fraser et al., 1978). Genetic Heterogeneity of Branchiootorenal Syndrome See also BOR2 (610896), caused by mutation in the SIX5 gene (600963) on chromosome 19q13. Sanchez-Valle et al. (2010) stated that approximately 40% of patients with BOR have mutations in the EYA1 gene and 5% have mutations in the SIX5 gene.
Branchiootorenal syndrome is characterized by birth defects or anomalies of tissues in the neck, malformations of the external ear, hearing loss, and kidney malformations. Symptom and symptom severity can vary greatly from person to person. It can be caused by mutations in the EYA1, SIX1, or SIX5 genes. It is passed through families in an autosomal dominant fashion. Treatment may include surgery to remove the anomalies of the neck (i.e., branchial fistulae or cysts), careful assessment and management of hearing loss, and follow-up by a kidney specialist (nephrologist). In some cases dialysis or kidney transplant may be required.
A number sign (#) is used with this entry because of evidence that branchiootorenal syndrome-2 (BOR2) is caused by heterozygous mutation in the SIX5 gene (600963) on chromosome 19q13. For a phenotypic description and a discussion of genetic heterogeneity of the branchiootorenal syndrome, see BOR1 (113650). Clinical Features Hoskins et al. (2007) reported 5 patients with a clinical diagnosis of BOR who carried heterozygous mutations in the SIX5 gene. One of the patients had bilateral dysplastic kidneys and a right preauricular tag but normal hearing. A second had bilateral cervical fistulas; right-sided hemifacial microsomia, preauricular sinus, and pinna malformation; hearing loss in both ears, with the right side more affected than the left; and bilateral renal dysplasia with diminished renal function.
Branchiootorenal (BOR) syndrome is characterized by branchial arch anomalies (branchial clefts, fistulae, cysts), hearing impairment (malformations of the auricle with pre-auricular pits, conductive or sensorineural hearing impairment), and renal malformations (urinary tree malformation, renal hypoplasia or agenesis, renal dysplasia, renal cysts). Epidemiology Prevalence is 1/40,000. Renal involvement can lead to chronic renal insufficiency. Clinical description The expression of the disease varies widely from one family to another and among individuals of the same family. Some families do not present with renal abnormalities or a urinary tree malformation. Etiology The causative gene, EYA1 , is located on the long arm of chromosome 8.
A rare developmental defect during embryogenesis characterized by hamartomatous intestinal polyposis, lipomas, macrocephaly and genital lentiginosis. Epidemiology The prevalence is unknown, but Bannayan-Riley-Ruvalcaba syndrome (BRRS) is generally considered as a rare disease. Clinical description BRRS shares some of the clinical characteristics of Cowden syndrome (CS;) but with differing frequencies. Unlike CS, the classic presentation of BRRS occurs neonatally or shortly thereafter with macrocephaly, Hashimoto struma, lipomatosis, vascular malformations and speckled lentiginosis of the penis or vulva. Developmental delay and gastrointestinal hamartomatous polyposis occur in a subset of BRRS patients.
Bannayan-Riley-Ruvalcaba syndrome (BRRS) is a genetic condition that leads to the growth of both non-cancerous and cancerous tumors. Symptoms of BRRS may include large head size, increased birth weight, developmental delay, and intellectual disability. Other symptoms include the appearance of non-cancerous tumors in the digestive system, fatty tumors under the skin, and freckles on the penis. People with BRRS have an increased risk of developing breast, thyroid, and uterine cancer. This condition is part of a group of conditions known as the PTEN hamartoma tumor syndromes, which all share similar features and are caused by genetic changes ( DNA variants ) in the PTEN gene.
Sclerosing mesenteritis is one of many terms that describes a spectrum of inflammatory disorders that affect the mesentery. The mesentery is the membrane that anchors the small intestine to the back of the abdominal wall. The condition mostly affects men between their 40s and 60s, but women and children can also be affected. It may result in a variety of symptoms including abdominal pain, nausea and vomiting, constipation or diarrhea, weight loss, and fever. Some people have an abdominal mass. The cause of the condition is poorly understood.
Overview Sclerosing mesenteritis, also called mesenteric panniculitis, occurs when the tissue (mesentery) that holds the small intestines in place becomes inflamed and forms scar tissue. Sclerosing mesenteritis is rare, and it's not clear what causes it. Sclerosing mesenteritis can cause abdominal pain, vomiting, bloating, diarrhea and fever. But some people experience no signs and symptoms and may never need treatment. In rare cases, scar tissue formed by sclerosing mesenteritis can block food from moving through your digestive tract. In this case, you may need surgery. Symptoms Symptoms of sclerosing mesenteritis include pain in your belly, vomiting, bloating, diarrhea and fever.
PMID 8141044 . Allen NB, Cox CC, Cobo M, et al. (March 1990). "Use of immunosuppressive agents in the treatment of severe ocular and vascular manifestations of Cogan's syndrome".
Cogan syndrome is a rare autoimmune disease that affects the eyes and inner ears. Symptoms of the syndrome include irritation and pain in the eyes, decreased vision, hearing loss, and vertigo . Other symptoms may include joint or muscle pain or inflammation of the blood vessels. The exact cause of Cogan syndrome is not well-understood. It is thought that the syndrome is caused by an autoimmune response that causes the immune system to attack the tissues of the eyes and ears. Cogan syndrome is not known to run in families. Diagnosis of Cogan syndrome is based on observing symptoms associated with the syndrome and ruling out other possible causes of the symptoms.
A rare inflammatory/autoimmune disorder of unknown origin characterized by interstitial keratitis (IK) and audiovestibular dysfunctions. Epidemiology Cogan syndrome (CS) prevalence is unknown. To date, approximately 300 cases have been reported. The disease is primarily described in causasians patients with no gender predilection. Clinical description CS mainly affects young adults, with a median age at onset between 20 and 30 years, and occasionally affects children. The syndrome shows a large spectrum of clinical features. Non-syphilitic IK and cochleovestibular symptoms with unilateral or bilateral sensorineural hearing loss, vertigo and tinnitus are typical CS manifestations.
Naegeli syndrome belongs to a group of disorders known as ectodermal dysplasias . This condition is characterized by absent fingerprints, thickening of the palms and soles ( palmoplantar keratoderma ), decreased sweating ( hypohidrosis ), heat intolerance, patches of darker (hyperpigmented) skin, brittle nails, abnormally colored teeth, and early tooth loss. Naegeli syndrome is caused by mutations in the KRT14 gene and inherited in an autosomal dominant manner. While there is no cure for Naegeli syndrome, treatment is based on each individual's symptoms.
A number sign (#) is used with this entry because of evidence that the Naegeli-Franceschetti-Jadassohn syndrome (NFJS) is caused by heterozygous mutation in the keratin-14 gene (KRT14; 148066) on chromosome 17q21. A closely related disorder, dermatopathia pigmentosa reticularis (DPR; 125595), is also caused by heterozygous mutation in the KRT14 gene. Clinical Features Naegeli (1927) described the syndrome in a father and 2 daughters. In a restudy of the original family, Franceschetti and Jadassohn (1954) documented autosomal dominant inheritance. The disorder was earlier confused with incontinentia pigmenti (IP; see 308300).
Naegeli-Franceschetti-Jadassohn (NFJ) syndrome is a rare ectodermal dysplasia that affects the skin, sweat glands, nails, and teeth. Epidemiology Several families with multiple affected members (males and females) from several generations have been reported so far. Prevalence is estimated at 1 in 3 million. Clinical description The cardinal features are absence of dermatoglyphics (fingerprints), reticular cutaneous hyperpigmentation (starting at about the age of 2 years without a preceding inflammatory stage), hypohidrosis with diminished sweat gland function and discomfort provoked by heat, nail dystrophy, tooth enamel defects, and moderate hyperkeratosis of the palms and soles. Diffuse palmoplantar keratoderma may coexist with punctate keratoses that are sometimes accentuated in the creases or exhibit a linear pattern. Congenital misalignment of the great toenails was reported in some patients.