-
Acro–dermato–ungual–lacrimal–tooth Syndrome
Wikipedia
ADULT ectodermal dysplasia syndrome resulting from the missense mutation R298Q in the p63 gene Wiley-Blackwell. doi:10.1111/j.1365-2230.2004.01643.x ^ Propping P, Zerres K (1993). "ADULT-syndrome: an autosomal-dominant disorder with pigment anomalies, ectrodactyly, nail dysplasia, and hypodontia". ... ADULT syndrome caused by a mutation previously associated with EEC syndrome Wiley-Blackwell. doi:10.1111/j.1525-1470.2010.01131.x ^ Rinne T., Spadoni E., Kjaer K. W., Danesino C., Larizza D., Kock M.; et al. (2006).
- Otocephaly Wikipedia
-
Cholestasis, Progressive Familial Intrahepatic, 5
OMIM
Other features include abnormal liver enzymes, low to normal gamma-glutamyl transferase (GGT) activity, increased alpha-fetoprotein, and a vitamin K-independent coagulopathy (summary by Gomez-Ospina et al., 2016). ... INHERITANCE - Autosomal recessive GROWTH Other - Failure to thrive ABDOMEN Liver - Liver failure - Ductal reaction seen on liver biopsy - Intralobular cholestasis - Diffuse giant cell transformation - Ballooning of hepatocytes - Fibrosis - Cirrhosis - Undetectable BSEP expression in bile canaliculi SKIN, NAILS, & HAIR Skin - Jaundice HEMATOLOGY - Vitamin K-independent coagulopathy - Increased INR - Prolonged prothrombin time - Decreased levels of factor V and VII PRENATAL MANIFESTATIONS Amniotic Fluid - Hydrops (1 patient) LABORATORY ABNORMALITIES - Abnormal liver enzymes - GGT is not increased - Increased alpha-fetoprotein - Hypoglycemia - Hyperammonemia MISCELLANEOUS - Onset at birth or in the neonatal period - Rapid progression - Fatal unless liver transplant is performed - Two unrelated families have been reported (last curated July 2016) MOLECULAR BASIS - Caused by mutation in the nuclear receptor subfamily 1, group H, member 4 gene (NR1H4, 603826.0001 ) ▲ Close
-
Cholestasis-Lymphedema Syndrome
OMIM
Nine of these died in early infancy, mainly of bleeding because of unavailability of vitamin K at the time. Two died of cirrhosis in later childhood. ... Nine died in infancy or early childhood, mainly of bleeding before vitamin K treatment was available; 4 died of cirrhosis at ages 2, 7, 8, and 50 years, respectively; and 2 died of unrelated causes in late adulthood.LCS1, CD1A, NOTCH1, S100A1, S100B, SOD1, TP53, ARHGEF5, TIMELESS, HAVCR1, CD207, HAVCR2, TIMD4, CCBE1

-
Nevo Syndrome
Wikipedia
National Institutes of Health. 133A (2): 158-164. ^ a b c Kanemoto N, Kanemoto K, Nishimura G, et al. (December 2005). ... “A Pilot Study for Evaluation of Hypotonia in Children with Neruofibromatosis Type 1”. 30(3): 382-382. ^ Tatton-Brown K, Rahman N. (March 2007). “ Sotos syndrome”. 15 (3) 264-271. ^ Al-Gazali L, Bakalinova D, Varady E, et al. (1997).
-
Histiocytoma (Dog)
Wikipedia
PMID 8623937 . ^ Kaim U, Moritz A, Failing K, Baumgärtner W (2006). "The regression of a canine Langerhans cell tumour is associated with increased expression of IL-2, TNF-alpha, IFN-gamma and iNOS mRNA" . ... Advanstar Communications: 1S–8S. ^ Affolter, Verena K. (2004). "Histiocytic Proliferative Diseases in Dogs and Cats" .

-
Echopraxia
Wikipedia
References [ edit ] ^ a b c d e f g h i j k l m Ganos C, Ogrzal T, Schnitzler A, Münchau A (September 2012). ... PMID 16540574 . ^ Cho YJ, Han SD, Song SK, Lee BI, Heo K (June 2009). "Palilalia, echolalia, and echopraxia-palipraxia as ictal manifestations in a patient with left frontal lobe epilepsy".
-
Blue Diaper Syndrome
Wikipedia
. ^ Park SY, Kim JK, Kim IJ, Choi BK, Jung KY, Lee S, Park KJ, Chairoungdua A, Kanai Y, Endou H, Kim do K (2005). "Reabsorption of neutral amino acids mediated by amino acid transporter LAT2 and TAT1 in the basolateral membrane of proximal tubule". ... PMID 15918515 . S2CID 2139640 . ^ Kim do K, Kanai Y, Matsuo H, Kim JY, Chairoungdua A, Kobayashi Y, Enomoto A, Cha SH, Goya T, Endou H (2002).

-
Vulvar Vestibulitis
Wikipedia
Laboratory tests are used to exclude bacterial or viral infection, and a careful examination of the vulvovaginal area is conducted to assess whether any atrophy is present Treatment [ edit ] Treatment consists of general advice about hygiene and sexual behaviour, pelvic floor and desensitisation exercises, and psychological treatment by a multidisciplinary team. [5] [6] [7] [8] References [ edit ] ^ a b Bergeron S, Binik YM, Khalifé S, Meana M, Berkley KJ, Pagidas K (1997). "The treatment of vulvar vestibulitis syndrome: Toward a multimodal approach". ... Bergeron S, Binik YM, Khalifé S, Pagidas K (1997). "Vulvar vestibulitis syndrome: a critical review".
-
Polyorchidism
Wikipedia
Prior to advances in ultrasound technology , it was common practice to remove the supernumerary testicle. [3] Several cases have been described where routine follow-up examinations conducted over a period of years showed that the supernumerary testicle was stable. [1] A meta-analysis in 2009 suggested removing non-scrotal supernumerary testicles because of the increased risk of cancer, and regular follow-up in the remaining cases to ensure that the supernumerary testicle remains stable. [1] References [ edit ] ^ a b c d e f g Bergholz, R.; Wenke, K. (2009). "Polyorchidism: A Meta-Analysis". ... Reprod Dom Anim . 50 (1): 172–176. doi : 10.1111/rda.12461 . PMID 25472870 . ^ a b c Leung, A. K. (1988). "Polyorchidism". American Family Physician . 38 (3): 153–156.

-
Hidea Syndrome
Wikipedia
History [ edit ] This condition was first described in 2014. [2] The causative mutation was discovered in 2019. [3] References [ edit ] ^ Kaasinen E, Rahikkala E, Koivunen P, Miettinen S, Wamelink MM, Aavikko M, Palin K, Myllyharju J, Moilanen JS, Pajunen L, Karhu A, Aaltonen LA (2019) Clinical characterization, genetic mapping and whole-genome sequence analysis of a novel autosomal recessive intellectual disability syndrome. Eur J Med Genet 57(10):543-551 ^ Kaasinen E, Rahikkala E, Koivunen P, Miettinen S, Wamelink MM, Aavikko M, Palin K, Myllyharju J, Moilanen JS, Pajunen L, Karhu A, Aaltonen LA (2019) Clinical characterization, genetic mapping and whole-genome sequence analysis of a novel autosomal recessive intellectual disability syndrome.
-
Lymphovascular Invasion
Wikipedia
PMID 21545433 . ^ Schoppmann SF, Bayer G, Aumayr K, Taucher S, Geleff S, Rudas M, et al. ... PMID 16334758 . ^ Moreira LF, Kenmotsu M, Gochi A, Tanaka N, Orita K (1999). "Lymphovascular and neural invasion in low-lying rectal carcinoma".

-
Dysgerminoma
Wikipedia
.; Matsuzaki, Shinya; Klar, Maximilian; Roman, Lynda D.; Sood, Anil K.; Gershenson, David M. (2020-05-29). ... PMID 32485873 . ^ Maoz, Asaf; Matsuo, Koji; Ciccone, Marcia A.; Matsuzaki, Shinya; Klar, Maximilian; Roman, Lynda D.; Sood, Anil K.; Gershenson, David M. (2020-05-29).CTNNB1, MAP3K1, OPCML, AKT1, CDH1, PIK3CA, FGFR2, PRKN, KIT, SRY, IGHV1-12, TSPY1, TP53, RNF139, TSPY10, BRCA1, TSPY3, DDX4, YBX2, PPP1R2C, NUTM1, PADI4, POU5F1P3, POU5F1P4, EFS, HMGA2, VIM, AFP, TSN, TLR4, ZEB1, SLC22A3, PTEN, POU5F1, PGD, KRT7, CYP27B1, CYP19A1, CD14, FOXL2, AMHR2, SOX9

-
Yunnan Sudden Death Syndrome
Wikipedia
Y.; Shi, G. Q.; Fontaine, R.; Wei, K.; Feng, T.; Wang, F.; Wang, G. Q.; Qu, Y.; Li, Z. ... J.; Yang, Z. L.; Zeng, G.; Liu, J. K. (2012). "Evidence for the Natural Toxins from the Mushroom Trogia venenata as a Cause of Sudden Unexpected Death in Yunnan Province, China".
-
Rosah Syndrome
Wikipedia
History [ edit ] This condition was first described in 2019. [1] References [ edit ] ^ a b Williams LB, Javed A, Sabri A, Morgan DJ, Huff CD, Grigg JR, Heng XT, Khng AJ, Hollink IHIM, Morrison MA, Owen LA, Anderson K, Kinard K, Greenlees R, Novacic D, Nida Sen H, Zein WM, Rodgers GM, Vitale AT, Haider NB, Hillmer AM, Ng PC, Shankaracharya, Cheng A, Zheng L, Gillies MC, van Slegtenhorst M, van Hagen PM, Missotten TOAR, Farley GL, Polo M, Malatack J, Curtin J, Martin F, Arbuckle S, Alexander SI, Chircop M, Davila S, Digre KB, Jamieson RV, DeAngelis MM (2019) ALPK1 missense pathogenic variant in five families leads to ROSAH syndrome, an ocular multisystem autosomal dominant disorder.

-
Aminoaciduria
Wikipedia
ISBN 9781444144154 . ^ a b c Mundt, LA; Shanahan, K (2011). "Chapter 7: Urinary and metabolic diseases and related urinalysis findings. ... External links [ edit ] Classification D DiseasesDB : 14901 SNOMED CT : 35912001 External resources MedlinePlus : 003366 v t e Components and results of urine tests Components Albumin Myoglobin hCG Leukocyte esterase Urine pregnancy test Ketone bodies Glucose Urobilinogen Bilirubin Creatinine RBC WBC Urinary casts Chemical properties Urine specific gravity Isosthenuria Urine osmolality Hypersthenuria Urine pH Urine anion gap Abnormal findings Red blood cells Hematuria ( Microscopic hematuria ) White blood cells Eosinophiluria Proteinuria Albuminuria / Microalbuminuria Albumin/creatinine ratio Urine protein/creatinine ratio Myoglobinuria Hemoglobinuria Bence Jones protein Small molecules Glycosuria Ketonuria Bilirubinuria Hyperuricosuria Aminoaciduria Other Bacteriuria Chyluria Crystalluria v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria This article about an endocrine, nutritional, or metabolic disease is a stub .ASL, SUCLA2, PEX1, ATP7B, PEX5, SCO1, SLC1A1, GLYCTK, COX20, TK2, COX14, UMPS, XPA, XPC, KYNU, OCRL, SLC7A7, GRHPR, KMT2B, COA8, ZMPSTE24, SLC35A1, SLC19A2, FTCD, SPINK5, FASTKD2, HIBCH, RRM2B, OTC, OAT, ERCC5, NEU1, BCS1L, CASR, CLCN5, COX6B1, COX8A, COX10, CPS1, CTNS, DDB2, EHHADH, ERCC2, ERCC3, ERCC4, TACO1, FH, GALE, GALT, GCLC, PET100, HGD, HNF4A, LMNA, MARS1, TRNN, TRNS1, NAGA, SLC6A19, CLTRN, PTH, ABCB6, TP53, HNF1A, SLC5A2, LYZ, GPT, LINC01672

-
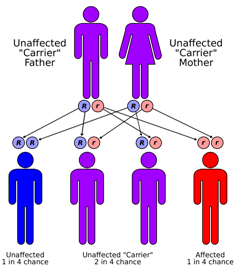
Sanjad-Sakati Syndrome
Wikipedia
. ^ Sanjad, S. A.; Sakati, N. A.; Abu-Osba, Y. K.; Kaddoura, R.; Milner, R. D. (February 1991). ... Nature Genet. 32: 448-452, 2002. ^ Courtens, W., Wuyts, W., Poot, M., Szuhai, K., Wauters, J., Reyniers, E., Eleveld, M., Diaz, G., Nothen, M.
-
Germinal Matrix Hemorrhage
Wikipedia
Consequently, increased arterial blood pressure in these blood vessels leads to rupture and hemorrhage into germinal matrix. [2] Diagnosis [ edit ] Grades [ edit ] Four grades are distinguished (by imaging or histology): [ citation needed ] grade I - hemorrhage is confined to the germinal matrix grade II - intraventricular hemorrhage without ventricular dilatation grade III - intraventricular hemorrhage with ventricular dilatation grade IV - intraventricular rupture and hemorrhage into the surrounding white matter Prevention [ edit ] Antenatal corticosteroids have a role in reducing incidence of germinal matrix hemorrhage in premature infants. [3] Management [ edit ] Stem cell-based therapies may help to treat germinal matrix hemorrhage in preterm babies but there is currently no reliable evidence to support their use. [4] See also [ edit ] Ganglionic eminence , a part of the germinal matrix References [ edit ] ^ Enzmann D, Murphy-Irwin K, Stevenson D, Ariagno R, Barton J, Sunshine P (1985). ... External links [ edit ] Classification D ICD - 10 : P52 ICD - 9-CM : 772.11 External resources eMedicine : radio/305 v t e Conditions originating in the perinatal period / fetal disease Maternal factors complicating pregnancy, labour or delivery placenta Placenta praevia Placental insufficiency Twin-to-twin transfusion syndrome chorion / amnion Chorioamnionitis umbilical cord Umbilical cord prolapse Nuchal cord Single umbilical artery presentation Breech birth Asynclitism Shoulder presentation Growth Small for gestational age / Large for gestational age Preterm birth / Postterm pregnancy Intrauterine growth restriction Birth trauma scalp Cephalohematoma Chignon Caput succedaneum Subgaleal hemorrhage Brachial plexus injury Erb's palsy Klumpke paralysis Affected systems Respiratory Intrauterine hypoxia Infant respiratory distress syndrome Transient tachypnea of the newborn Meconium aspiration syndrome Pleural disease Pneumothorax Pneumomediastinum Wilson–Mikity syndrome Bronchopulmonary dysplasia Cardiovascular Pneumopericardium Persistent fetal circulation Bleeding and hematologic disease Vitamin K deficiency bleeding HDN ABO Anti-Kell Rh c Rh D Rh E Hydrops fetalis Hyperbilirubinemia Kernicterus Neonatal jaundice Velamentous cord insertion Intraventricular hemorrhage Germinal matrix hemorrhage Anemia of prematurity Gastrointestinal Ileus Necrotizing enterocolitis Meconium peritonitis Integument and thermoregulation Erythema toxicum Sclerema neonatorum Nervous system Perinatal asphyxia Periventricular leukomalacia Musculoskeletal Gray baby syndrome muscle tone Congenital hypertonia Congenital hypotonia Infections Vertically transmitted infection Neonatal infection rubella herpes simplex mycoplasma hominis ureaplasma urealyticum Omphalitis Neonatal sepsis Group B streptococcal infection Neonatal conjunctivitis Other Miscarriage Perinatal mortality Stillbirth Infant mortality Neonatal withdrawal
-
Hawkinsinuria
Wikipedia
Scand . 64 (2): 209–214. doi : 10.1111/j.1651-2227.1975.tb03823.x . PMID 1130176 . ^ Tomoeda K, Awata H, Matsuura T, Matsuda I, Ploechl E, Milovac T, Boneh A, Scott CR, Danks DM, Endo F (2000). ... External links [ edit ] Classification D ICD - 10 : E72.1 OMIM : 140350 MeSH : D020176 DiseasesDB : 29836 v t e Inborn error of amino acid metabolism K → acetyl-CoA Lysine /straight chain Glutaric acidemia type 1 type 2 Hyperlysinemia Pipecolic acidemia Saccharopinuria Leucine 3-hydroxy-3-methylglutaryl-CoA lyase deficiency 3-Methylcrotonyl-CoA carboxylase deficiency 3-Methylglutaconic aciduria 1 Isovaleric acidemia Maple syrup urine disease Tryptophan Hypertryptophanemia G G→ pyruvate → citrate Glycine D-Glyceric acidemia Glutathione synthetase deficiency Sarcosinemia Glycine → Creatine : GAMT deficiency Glycine encephalopathy G→ glutamate → α-ketoglutarate Histidine Carnosinemia Histidinemia Urocanic aciduria Proline Hyperprolinemia Prolidase deficiency Glutamate / glutamine SSADHD G→ propionyl-CoA → succinyl-CoA Valine Hypervalinemia Isobutyryl-CoA dehydrogenase deficiency Maple syrup urine disease Isoleucine 2-Methylbutyryl-CoA dehydrogenase deficiency Beta-ketothiolase deficiency Maple syrup urine disease Methionine Cystathioninuria Homocystinuria Hypermethioninemia General BC / OA Methylmalonic acidemia Methylmalonyl-CoA mutase deficiency Propionic acidemia G→ fumarate Phenylalanine / tyrosine Phenylketonuria 6-Pyruvoyltetrahydropterin synthase deficiency Tetrahydrobiopterin deficiency Tyrosinemia Alkaptonuria / Ochronosis Tyrosinemia type I Tyrosinemia type II Tyrosinemia type III / Hawkinsinuria Tyrosine → Melanin Albinism : Ocular albinism ( 1 ) Oculocutaneous albinism ( Hermansky–Pudlak syndrome ) Waardenburg syndrome Tyrosine → Norepinephrine Dopamine beta hydroxylase deficiency reverse: Brunner syndrome G→ oxaloacetate Urea cycle / Hyperammonemia ( arginine aspartate ) Argininemia Argininosuccinic aciduria Carbamoyl phosphate synthetase I deficiency Citrullinemia N-Acetylglutamate synthase deficiency Ornithine transcarbamylase deficiency / translocase deficiency Transport / IE of RTT Solute carrier family : Cystinuria Hartnup disease Iminoglycinuria Lysinuric protein intolerance Fanconi syndrome : Oculocerebrorenal syndrome Cystinosis Other 2-Hydroxyglutaric aciduria Aminoacylase 1 deficiency Ethylmalonic encephalopathy Fumarase deficiency Trimethylaminuria This genetic disorder article is a stub .

-
Glutaminase Deficiency
Wikipedia
JAMA Neurol ^ van Kuilenburg ABP, Tarailo-Graovac M, Richmond PA, Drögemöller BI, Pouladi MA, Leen R, Brand-Arzamendi K, Dobritzsch D, Dolzhenko E, Eberle MA, Hayward B, Jones MJ, Karbassi F, Kobor MS, Koster J1, Kumari D, Li M, MacIsaac J, McDonald C, Meijer J, Nguyen C, Rajan-Babu IS, Scherer SW, Sim B, Trost B, Tseng LA, Turkenburg M, van Vugt JJFA, Veldink JH, Walia J, Wang Y, van Weeghel M, Wright GEB1, Xu X, Yuen RKC1, Zhang J, Ross CJ, Wasserman WW, Geraghty MT, Santra S, Wanders RJA, Wen XY, Waterham HR, Usdin K, van Karnebeek CDM (2019) glutaminase deficiency caused by short tandem repeat expansion in GLS.










